

Abstract
It is well established that BDNF may enhance oligodendrocyte differentiation following a demyelinating lesion, however, the endogenous sources of BDNF that may be harnessed to reverse deficits associated with such lesions are poorly defined. Here, we investigate roles of astrocytes in synthesizing and releasing BDNF. These cells are known to express BDNF following injury in vivo. In culture, they increase BDNF synthesis and release in response to glutamate metabotropic stimulation. Following cuprizone-elicited demyelination in mice, astrocytes contain BDNF and increase levels of metabotropic receptors. The metabotropic agonist, trans-(1S,3R)-1-amino-1,3-cyclopentanedicarboxylic acid (ACPD), was therefore injected into the demyelinating lesion. Increases in BDNF, as well as myelin proteins, were observed. Effects of ACPD were eliminated by coinjection of trkB-Fc to locally deplete BDNF and by deletion of astrocyte-derived BDNF. The data indicate that astrocyte-derived BDNF may be a source of trophic support that can be used to reverse deficits elicited following demyelination.
Keywords: ACPD, astrocytes, BDNF, cuprizone, metabotropic receptors, oligodendrocytes
Introduction
Neurotrophin signaling impacts development and health of oligodendrocyte lineage cells. Brain-derived neurotrophic factor (BDNF) has been of particular interest. BDNF increases DNA synthesis in cultured basal forebrain oligodendrocyte progenitors (Van't Veer et al., 2009) and enhances oligodendrocyte differentiation to myelin protein-expressing cells (Du et al., 2006). Moreover, BDNF deficient mice exhibit deficits in progenitors and myelin protein expression (Vondran et al., 2010) and the conditional knock-out of the BDNF receptor TrkB from mature, MBP+ oligodendrocytes results in reduced myelin thickness in both the spinal cord and the corpus callosum (Wong et al., 2013).
These effects may be relevant to in vivo demyelination. For example, in the cuprizone demyelination model expression of BDNF is decreased in the corpus callosum, and animals deficient in BDNF exhibit a more severe loss of myelin protein in the lesioned corpus callosum than do their wild-type littermates (VonDran et al., 2011). Likewise, the administration of mesenchymal stem cells that overproduce BDNF promotes recovery from experimental autoimmune encephalomyelitis-induced demyelination (Makar et al., 2009), suggesting that restoring this neurotrophin to optimal levels impacts recovery. These data translate well to the pathophysiology of human demyelinating disease. BDNF levels are decreased in patients with relapsing-remitting multiple sclerosis (Azoulay et al., 2005), and the disease modifying agents glatiramer acetate and fingolimod enhance levels of this neurotrophin in vivo (Aharoni et al., 2005; Aktas et al., 2010; Deogracias et al., 2012).
It would seem advantageous, then, to increase BDNF levels after demyelination. However, because of the difficulties associated with the administration of exogenous proteins to the CNS (Poduslo and Curran, 1996), it is important to consider the possibility that endogenous sources of trophic support exist and can be used in the injured CNS. BDNF levels are very low (pg/g) in the unlesioned adult brain (Matsumoto et al., 2008). However, it is well known that glial cells increase expression of a variety of growth factors, including BDNF (Dougherty et al., 2000) following injury. Moreover, glial cells in cultures that are found to mimic those in the ischemic brain (Zamanian et al., 2012) increase BDNF in response to the neurotransmitter glutamate (Wu et al., 2004; Jean et al., 2008), raising the possibility that neurotrophin availability might be regulated by neuronal signals. Further studies indicate that these effects are mediated through the action of metabotropic glutamate receptors (mGluR) and the mGluR-specific agonist ACPD was found to enhance the production (Wu et al., 2004) and release (Jean et al., 2008) of BDNF by cultured astrocytes.
In the present study, we explore the possibility that the metabotropic glutamate receptor agonist ACPD increases BDNF and myelin protein levels in vivo and that this manipulation may prove advantageous to a demyelinating lesion. In particular, using the cuprizone model we find that ACPD enhances production of astrocyte-derived BDNF, which supports the synthesis of myelin proteins in the demyelinated corpus callosum. These studies indicate that reactive astrocytes in a CNS lesion can be a source of trophic support and can be manipulated to support proximate cells, injured after a demyelinating lesion.
Materials and Methods
Experimental animals
Mice were managed by the Rutgers Robert Wood Johnson Animal Facility. Animal maintenance, husbandry, transportation, and use were in compliance with the Laboratory Animal Welfare Act (PL 89-544; PL-91-579) and with NIH guidelines (NIH Manual Chapter 4206).
Wild-type mice on a 129/BALBC/C57 background (The Jackson Laboratory) were used for the majority of these studies. In addition, we used homozygous HA-BDNF mice generated by replacing the endogenous Bdnf coding exon with a murine Bdnf sequence with a C-terminal hemagglutinin (HA) tag (Yang et al., 2009).
The hGFAP-CreERT2-bdnf fl/fl-ROSA26 mouse line was a result of crosses of the hGFAP-CreERT2 mouse (MMRC), the bdnf fl/fl mouse (The Jackson Laboratory), and the ROSA26 mouse (a gift from Michael Matise, Rutgers Robert Wood Johnson Medical School, Piscataway, NJ). Bdnffl/fl-ROSA26 mice lacking cre expression were used as controls. These ROSA26 cre reporter mice, however, although able to detect cre-dependent recombination in nonastrocytic populations, are known to be very inefficient in detecting recombination in astrocytes (Casper and McCarthy, 2006). Therefore, we used another reporter mouse (astrocyte cre reporter; ACR, MMRC). In this mouse GFAP+ cells express β-gal before and eGFP after tamoxifen-elicited recombination (Casper and McCarthy, 2006). The mouse was mated to the hGFAP-CreERT2bdnffl/fl mouse.
Stereotaxic injections
Sterile surgical and postoperative procedures were followed as in the Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals. Stereotaxic injections were made into the corpus callosum of adult male mice, at coordinates from bregma: 1.0 and 0.5 mm lateral to the sagittal sinus. A Hamilton syringe was lowered to a depth of 1.875 mm to target the midcaudal corpus callosum over the fimbria/fornix. Two microliters of 0.5 μm ACPD (Tocris Bioscience) or 0.9% saline vehicle was infused. In some cases TrkB receptor bodies (TrkB-Fc; 0.5 μg/μl; Sigma-Aldrich) were administered along with ACPD or saline.
Cuprizone treatment
Demyelination was initiated by feeding 8-week-old wild-type male mice or HA male mice 0.2% cuprizone (Sigma-Aldrich) milled into mouse feed (Harlan Teklad) or identically processed control feed for 4 weeks. ACPD or saline vehicle was injected after 4 weeks of continuous treatment, then killed and processed for Western blot or immunohistochemistry.
Tamoxifen administration and conditional knock-outs
Eight-week-old hGFAP-CreERT2-bdnf fl/fl-ROSA26 or Bdnffl/fl-ROSA26 mice were injected with tamoxifen (1 mg 2×/d for 5 d) to initiate recombination. Demyelination was then elicited by feeding the animals 0.3% cuprizone (Sigma-Aldrich) for 4 weeks. ACPD or saline was injected into the corpus callosum, and animals were killed after 6 h.
Recombination efficiency was determined using ACR mice that were injected with tamoxifen 2×/d for 5 d and then killed 2 weeks after the first injection. The loss of X-gal staining after recombination allowed for determination of recombination efficiency.
Western blot
A mouse brain matrix, which permits coronal cuts to be made in 1 mm segments along the rostral-caudal axis, was used to dissect the midline corpus callosum overlying the fimbria-fornix and rostral hippocampus. Tissue was lysed and protein concentrations were determined using a BCA protein assay kit (Pierce).
Antibodies for Western blot.
Antibodies for Western blots included a mouse monoclonal antibody to MBP (Serotec), as well as rabbit polyclonal antibodies to BDNF (Santa Cruz Biotechnology), MAG (Santa Cruz Biotechnology), mGluR1 (Millipore), mGluR5 (Millipore), and mGluR2/3 (Millipore). Mouse monoclonal antibodies to GAPDH (Biodesign) and beta-tubulin (Sigma-Aldrich) were used as loading controls.
Western blot technique.
For the analysis of BDNF and MBP, protein was run on 12% Bis-Tris gels (Invitrogen). For the analysis of MAG, protein was run on a 4–12% Tris-Glycine gels (Invitrogen). For the analysis of mGluR1, mGluR5, and mGluR2/3, protein was run on 3–8% Tris-acetate gels (Invitrogen). Protein was then transferred to a PVDF membrane (Millipore) and membranes were exposed to anti-mouse, anti-rabbit, or anti-goat primary antibodies, as appropriate. All Western blots were visualized with a chemiluminescence system (GE Healthcare) and data were analyzed using Quantity One V 4.2.1 software (Bio-Rad). Membranes were reprobed with anti-GAPDH or anti-beta-tubulin as loading controls.
Immunohistochemistry
Tissue preparation.
Brains from adult mice perfused with 4% paraformaldehyde (PFA) were postfixed in PFA, dehydrated, and embedded in OCT (Tissue Tek). Fourteen micrometer serial sections were taken in the coronal plane from the basal forebrain to the caudal hippocampus. For mGluR2/3 (Aronica et al., 2001), mice were perfused with 2% PFA and 20% saturated picric acid (Sigma-Aldrich).
Antibodies used for immunohistochemistry.
Antibodies for immunohistochemistry included mouse monoclonal antibodies to CC1 (Calbiochem) MBP (Serotec), Neurofilament light-chain (NF-L; Millipore), and GFAP (Millipore); a goat polyclonal antibody to proteolipid protein (PLP; Santa Cruz Biotechnology); rabbit polyclonal antibodies to BDNF (Alomone Labs), HA (Sigma-Aldrich), MAG (Santa Cruz Biotechnology), mGluR1 (Millipore), mGluR5 (Millipore), and mGluR2/3 (Millipore); a chicken polyclonal antibody to GFAP (Millipore); and a rat monoclonal antibody to CD11b (Abcam).
Visualization of proteins.
MBP, PLP, MAG, and CC1 were visualized using the avidin-biotin complex (Vector Laboratories) and DAB (Sigma-Aldrich). For CC1 visualization sections were microwaved in 0.01 m citrate buffer for antigen retrieval before primary antibody incubation. For immunofluorescence, anti-rabbit AlexaFluor 594, anti-mouse AlexaFluor 488, anti-rat AlexaFluor 488, or anti-chicken AlexaFluor 657 (Invitrogen) were used.
Quantification of CC1+ oligodendrocytes
For counts of CC1+ oligodendrocytes, images of 14 μm serial sections separated by 56 μm were obtained from the midline corpus callosum overlying the fimbria-fornix and rostral hippocampus. Images were analyzed at 100× using a Leica microscope. CC1+ cells were counted in a total of 16 sections for each animal.
Imaging
Colocalization was initially characterized using a Leica fluorescent microscope equipped with an Olympus MagnaFire digital camera and ImagePro image analysis software. Colocalization of the different cellular markers was confirmed by taking 1 μm confocal images on a Zeiss 510LSM confocal microscope running Zeiss image browser software.
Data analysis
For each experiment a control and cuprizone fed mouse, injected with saline or ACPD were compared. In some cases, a control and cuprizone fed mouse, injected with saline or ACPD were also injected with TrkB-FC or vehicle and compared with one another. In other cases, tamoxifen injected mice were fed control or cuprizone food and then injected with saline or ACPD. For the CC1+ cell counts cuprizone treated ACPD mice were compared with vehicle injected mice. Each experiment was repeated ≥3 times. Statistical differences were determined using ANOVA followed by Fisher's protected least significant post hoc test or Student's t test as appropriate. Conditions were considered significant at p < 0.05.
Results
ACPD increases the expression of BDNF and myelin proteins in the demyelinated corpus callosum
In culture, the metabotropic agonist ACPD increases the production and release of BDNF from astrocytes (Wu et al., 2004; Jean et al., 2008). In vivo, injection of metabotropic agonists are reported to reverse deficits associated with a model of Parkinson's disease (Agari et al., 2008) and in another report to increase expression of BDNF in the intact mouse brain (Di Liberto et al., 2010), suggesting that metabotropic agonists may enhance trophic effects in the brain.
To evaluate the effects of ACPD in the cuprizone model, wild-type mice treated with cuprizone received a single stereotaxic injection of either ACPD (0.5 μm) or saline vehicle into the midcaudal corpus callosum. After 6 h, mice were killed and processed for Western blot. Cuprizone treatment results in a significant decrease in BDNF, MBP, and MAG protein levels in the vehicle-injected group. However, ACPD elevated BDNF protein (both the pro- and mature forms) to unlesioned control levels in the cuprizone-treated group. Moreover, the expression of MBP and MAG protein was also significantly elevated (Fig. 1a,b).
Figure 1.

Cuprizone-lesioned mice exhibit an increase in BDNF and myelin protein levels 6 h after a single stereotaxic injection of ACPD. a, Western blots demonstrate BDNF, MBP, and MAG protein levels in the corpus callosum of wild-type mice subjected to a 4 week cuprizone lesion and injected with ACPD or 0.9% saline vehicle. GAPDH is shown as a loading control. b, Graphs represent a densitometric analysis of Western blots normalized to GAPDH and are presented as percentage saline-injected control. Levels of mature BDNF are indicated in this analysis. c, Immunohistochemical analysis of MBP, MAG and PLP reactivity in the corpus callosum reveals strong staining intensity in the intact corpus callosum that is decreased following exposure to cuprizone. This MBP, MAG, and PLP deficit is reversed in cuprizone-lesioned animals 6 h after the administration of ACPD. Western blot data analyzed by ANOVA; ***significantly different from saline-injected control at p < 0.0005; *significantly different at p < 0.02. Each Western blot lane is from the corpus callosum of a single mouse within one experiment. Each experiment was repeated eight times. Scale bar, 100 μm. d, Quantification of CC1+ oligodendrocytes in the lesioned corpus callosum revealed that ACPD injection does not affect CC1+ cell numbers; N = 3.
Western blot studies were complemented with immunohistochemical examination of MBP, MAG, and PLP protein in the cuprizone-lesioned corpus callosum of animals that had been injected with either ACPD or saline. As expected, MBP, MAG, and PLP staining is prevalent in the myelin-rich corpus callosum of unlesioned animals. However, cuprizone-treated mice that received saline vehicle exhibit markedly reduced myelin protein staining intensity, and this reduction appears to be at least partially reversed in animals that were injected with ACPD (Fig. 1c). When numbers of CC1+ oligodendrocytes in the cuprizone group were counted following ACPD, no differences were noted (Fig. 1d). Thus, although both BDNF and myelin protein expression are decreased in the corpus callosum in response to cuprizone, a single injection of ACPD can promote the recovery of these proteins/ cell to control levels within 6 h.
We have reported that cuprizone treatment results in an ∼50% decrease in CC1+ oligodendrocytes extending over a 6 week period (VonDran et al., 2011). Importantly these results indicate that the remaining CC1+ oligodendrocytes can respond to a trophic influence and increase their myelin proteins in response to ACPD, suggesting the potential therapeutic utility of this glutamate metabotropic agonist.
The ACPD effect on expression of myelin proteins is dependent on BDNF signaling
The ACPD-mediated increase in myelin protein expression is associated with an increase in BDNF, suggesting that the effect may be dependent upon the enhanced availability of neurotrophic support in the lesion site. This hypothesis is consistent with the known effects of neurotrophin signaling on the health of oligodendrocyte lineage cells, both in culture (Du et al., 2006) and in vivo (Makar et al., 2009). To more directly explore the role of BDNF in mediating the effects of ACPD in this model of demyelination, TrkB-Fc were administered along with the agonist or saline. TrkB-Fc are fusion proteins encoding the extracellular domain of the TrkB receptor fused to a histidine-tagged tail region of human IgG1, and have been used by others to inhibit BDNF signaling through endogenously expressed TrkB receptors (Huang et al., 2010). As seen previously, decreases in MBP and MAG protein in the corpus callosum of cuprizone-treated animals are partially reversed by ACPD. However, the ACPD effect is blocked when the interaction of BDNF with endogenous receptors is inhibited by the coadministration of TrkB-Fc (Fig. 2).
Figure 2.

The ACPD effect on the expression of myelin proteins in the demyelinated corpus callosum is inhibited by TrkB-Fc. Wild-type mice were fed control or cuprizone-laden food for 4 weeks, then stereotaxically injected with either 0.9% saline or ACPD, along with 1 μg TrkB-Fc or vehicle. a, Western blots demonstrate MBP and MAG protein levels in the corpus callosum of wild-type mice subjected to a 4 week cuprizone lesion. GAPDH is shown as a loading control. b, Graphs represent a densitometric analysis of Western blots normalized to GAPDH and are presented as percentage saline-injected control. Data analyzed by ANOVA; *significantly different from saline-injected control at p < 0.0002; ***significantly different at p < 0.0005. Each Western blot lane is from the corpus callosum of a single mouse within one experiment. Each experiment was repeated four times.
Metabotropic glutamate receptors are present on astrocytes following cuprizone
To identify the cells responsible for increases in BDNF, our next experiments examined metabotropic receptors that bind ACPD. ACPD binds to both Group I and II metabotropic receptors. Therefore, protein expression of these receptors was monitored in the lesioned and unlesioned brain. Although Group I (mGluR1, mGluR5) and Group II, (mGluR2/3) receptors are expressed to some degree in the intact corpus callosum, the expression of each of these receptors is significantly upregulated after 4 weeks of treatment with cuprizone. However, treatment with ACPD does not itself impact the expression of any of these receptors (Fig. 3a,b).
Figure 3.
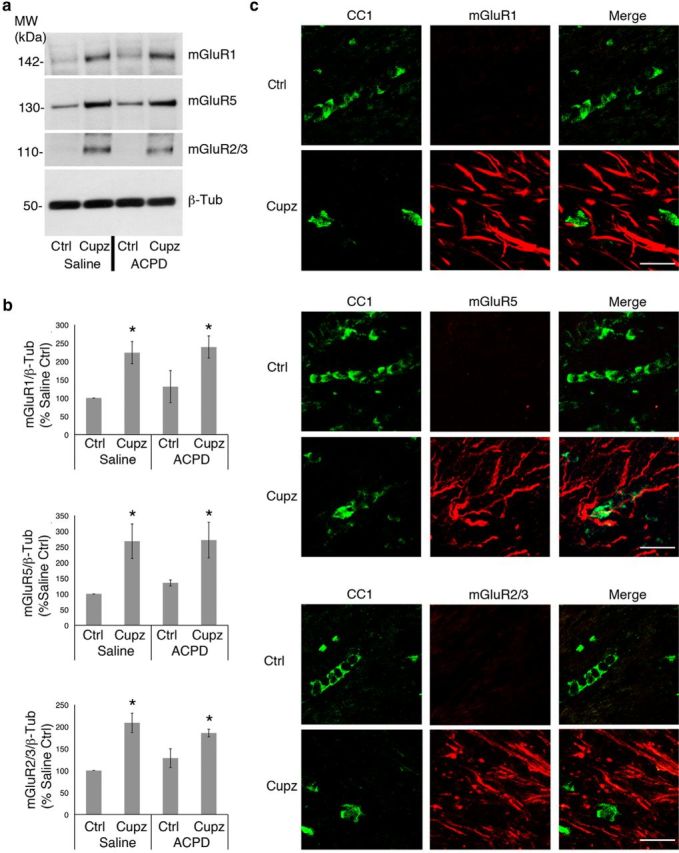
Group I and Group II metabotropic glutamate receptor expression is increased in the cuprizone-lesioned corpus callosum. Wild-type mice were fed control or cuprizone-laden food for 4 weeks, then stereotaxically injected with either saline vehicle or ACPD. a, Western blots demonstrate mGluR1, mGluR5, and mGluR2/3 protein levels in the control and cuprizone-lesioned corpus callsoum. Beta-tubulin is shown as a loading control. b, Graphs represent a densitometric analysis of Western blots normalized to GAPDH and are presented as percentage saline-injected control. c, Immunohistochemical visualization of mGluRs and CC1+ oligodendrocytes. Western blot data analyzed by ANOVA; *significantly different from saline-injected control at p < 0.05. Each Western blot lane is from the corpus callosum of a single mouse within one experiment. Each experiment was repeated four times. Scale bar, 20 μm.
To determine whether effects of metabotropic stimulation are directly on the oligodendrocytes, immunohistochemistry was used to colocalize Group I or Group II receptors to CC1+ oligodendrocytes. Neither Group I (mGluR1 and mGluR5), nor the Group II (mGluR2/3) receptors were detected on CC1+ oligodendrocytes of the intact or cuprizone-lesioned corpus callosum, suggesting that this population is not receptive to metabotropic stimulation with ACPD following cuprizone (Fig. 3c). Moreover, neither the mGluR1, mGluR5 nor the mGluR2/3 receptors are observed on microglia, which are dramatically increased in the lesion site (data not shown).
In contrast, NF-L+ axons running in the corpus callosum do colocalize extensively with mGluR2/3, although not mGluR1 or mGluR5 (Fig. 4). Moreover, astrocytes, which express increased levels of GFAP following cuprizone, colocalize extensively with receptors mGluR1 and mGluR5, but not with mGluR2/3 (Fig. 5). These observations raise the possibility that axons and astrocytes may respond to ACPD and support the recovery of oligodendrocyte lineage cells from demyelinating injury.
Figure 4.
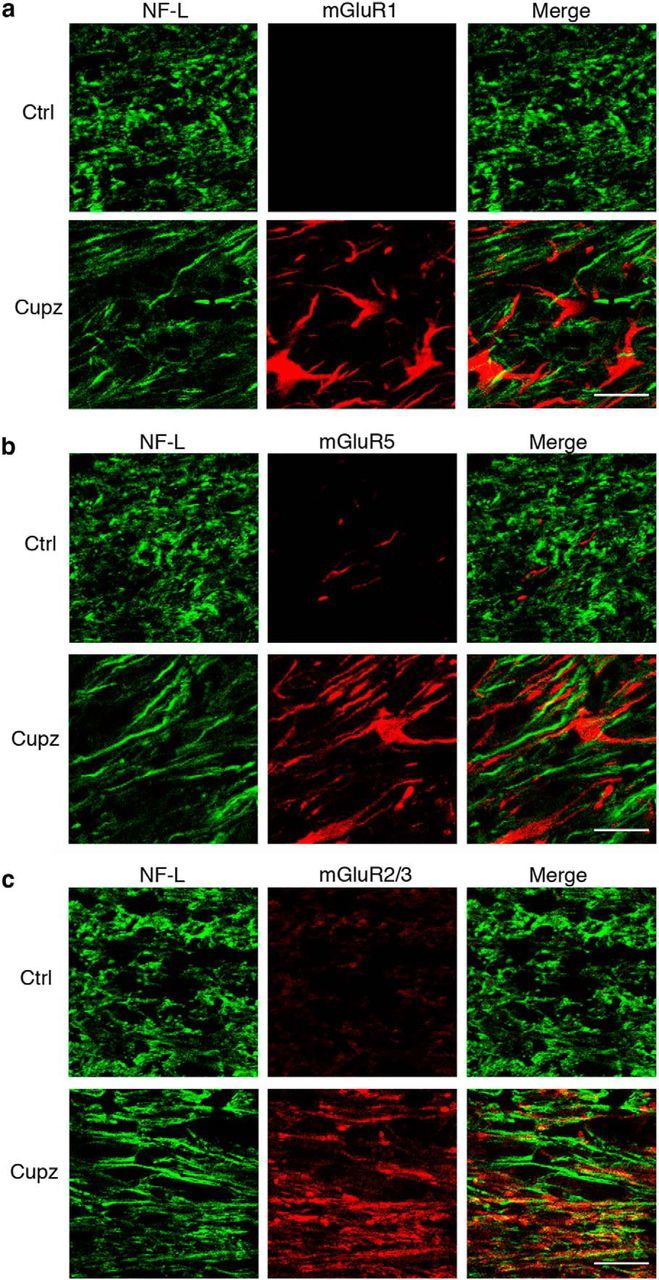
NF-L+ axonal tracts colocalize with the Group II metabotropic glutamate receptors mGluR2/3 following a demyelinating cuprizone lesion. Wild-type mice were fed control or cuprizone-laden food for 4 weeks. NF-L+ axons fail to colocalize with (a) mGluR1, or (b) mGluR5. However, there is extensive colocalization of NF-L+ axonal tracts with mGluR2/3 immunoreactivity after cuprizone (c). These are representative images seen in four independent experiments. Scale bar, 20 μm.
Figure 5.
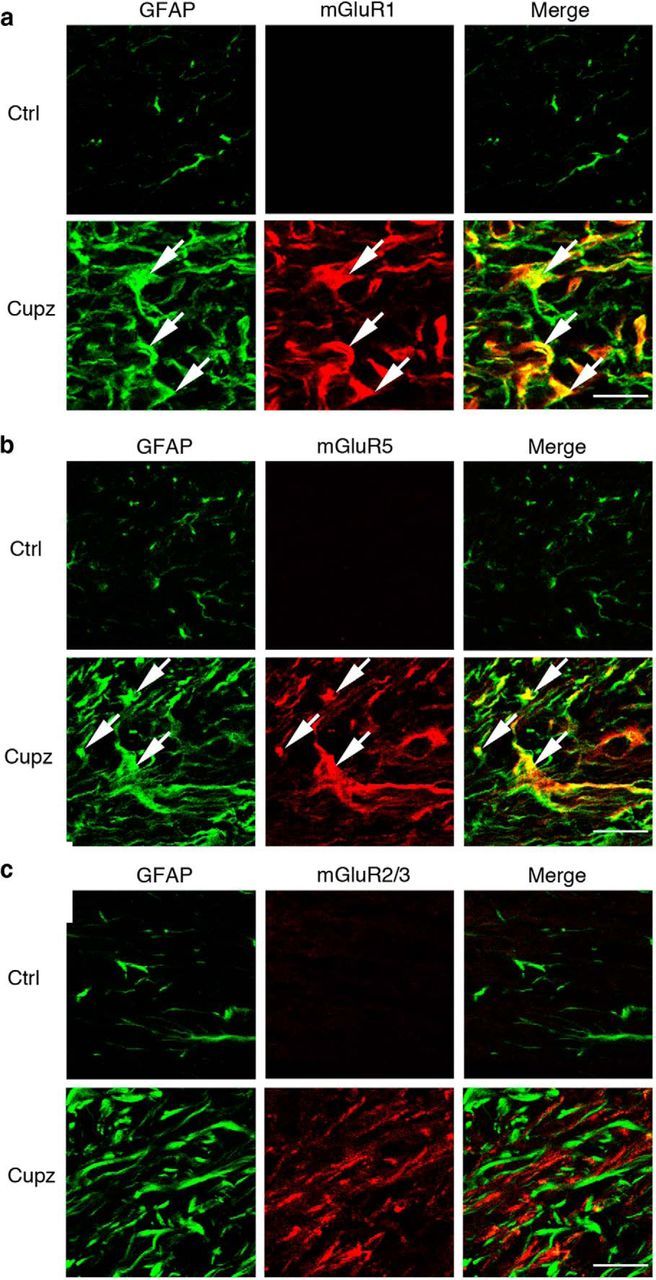
GFAP+ astrocytes express the Group I metabotropic glutamate receptors mGluR1 and mGluR5 in the cuprizone-lesioned corpus callosum. Wild-type mice were fed control or cuprizone-laden food for 4 weeks. There is an increase in the number of GFAP+ astrocytes at this stage of demyelination. These GFAP+ cells colocalize extensively with (a) mGluR1, and (b) mGluR5, but fail to express mGluR2/3 (c). These are representative images seen in four independent experiments. Scale bar, 20 μm.
Astrocytes express BDNF in the cuprizone-lesioned corpus callosum
Although both axons and astrocytes are potentially receptive to ACPD stimulation, it is currently not known whether either of these populations is a source of neurotrophins following a demyelinating injury. Cortical neurons, including those that project through the corpus callosum, are known to express BDNF (Canals et al., 2001). The GFAP+ astrocytic population has also been demonstrated to produce BDNF following injury to the CNS (Dougherty et al., 2000), and as mentioned previously, the production and release of this neurotrophin in culture is enhanced by metabotropic stimulation (Jean et al., 2008). Cuprizone treated mice were therefore evaluated for BDNF in the astrocyte and axonal populations. GFAP+ astrocytes colocalize with BDNF immunoreactivity, while NF-L+ axons are not reactive for this neurotrophin (Fig. 6a). Therefore, in addition to expressing the relevant metabotropic receptors, GFAP+ astrocytes are the only population in the demyelinated corpus callosum to also express BDNF, suggesting their importance as a source of trophic support after injury.
Figure 6.
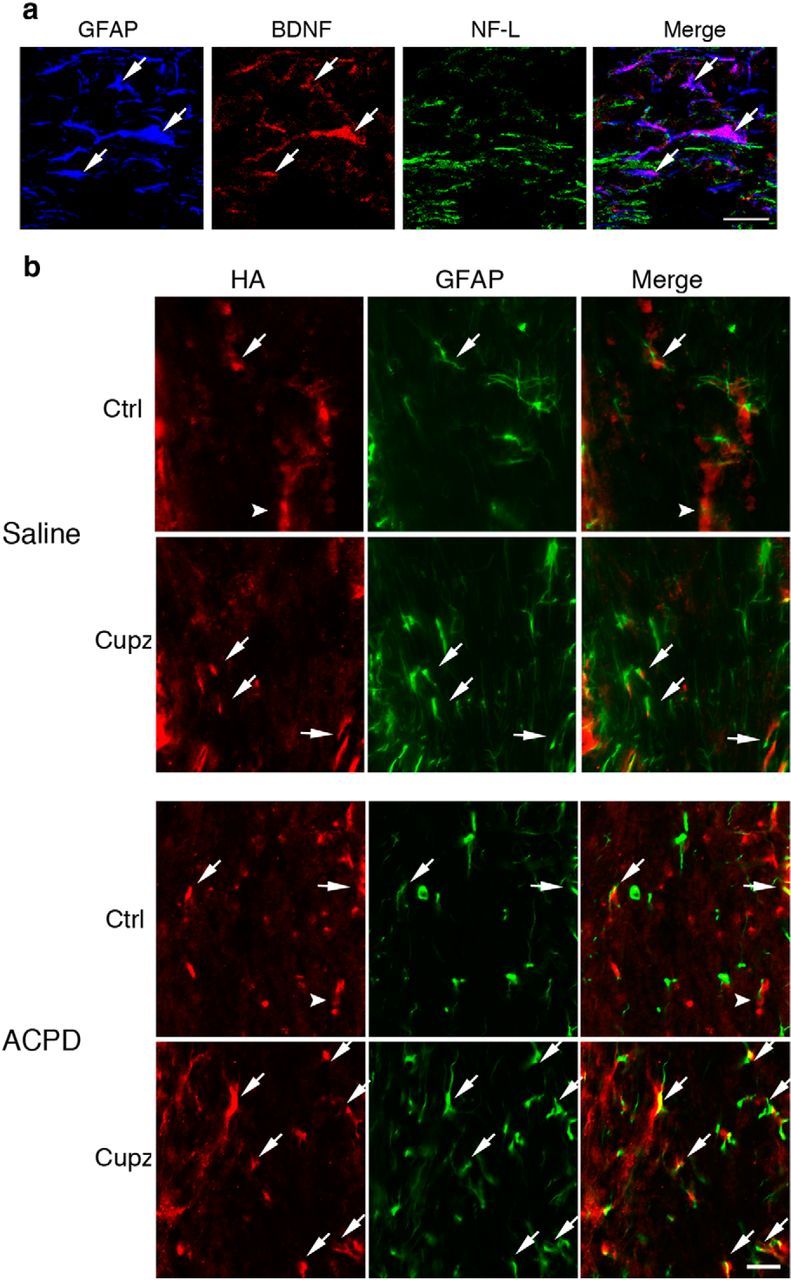
GFAP+ astrocytes express BDNF in the cuprizone-lesioned corpus callosum. Numbers of GFAP+ cells increase after ACPD. a, GFAP+ astrocytes in the cuprizone-lesioned corpus callosum colocalize with BDNF immunoreactivity. NF-L+ axonal tracts are not reactive for BDNF. b, HA-BDNF mice fed control or cuprizone feed for 4 weeks exhibit GFAP and HA immunoreactivity in only a small subset of the GFAP+ cells (arrows) in control mice or those fed cuprizone, whereas HA is predominantly found in non-GFAP+ cells (arrowhead) in controls. After cuprizone, the HA+ non-GFAP+ cells are greatly reduced. When cuprizone-treated mice are injected with ACPD, there is an increase in HA immunoreactivity associated with GFAP+ cells. These are representative images seen in three independent experiments. Scale bar, 20 μm.
To confirm the astrocytes as sources of BDNF, subsequent studies used an bdnfHA/bdnfHA targeted mouse in which the endogenous Bdnf coding exon was replaced with a murine sequence with a C-terminal HA tag (Yang et al., 2009). This permitted the cellular localization of BDNF in the corpus callosum using a highly specific antibody to HA. As was the case with wild-type mice, a subpopulation of GFAP+ astrocytes expresses HA under control conditions. Interestingly, however, the predominant population is composed of non-GFAP+ cells that exhibit BDNF. After cuprizone, the GFAP population remains colocalized, at least in small part, with HA immunoreactivity while HA immunoreactivity in the non-GFAP+ population is reduced. After treatment with ACPD, however, GFAP+ astrocytes increase levels of immunoreactivity for HA. (Fig. 6b). Thus, only a subgroup of astrocytes appears to express BDNF during control and cuprizone conditions. However, following ACPD treatment, this subgroup expands.
Astrocyte-derived BDNF promotes recovery from cuprizone-induced demyelination
To determine whether astrocyte-derived BDNF is the mediator of the ACPD effect, we took advantage of a tamoxifen-inducible mouse model that eliminates BDNF (Rios et al., 2001) from GFAP+ cells (Casper et al., 2007). These hGFAP-CreERT2-bdnf fl/fl-ROSA26 mice express tamoxifen-inducible cre recombinase under the control of the human GFAP promoter. Following tamoxifen administration, cre recombinase enters the nucleus, where it recognizes loxP sites and excises the BDNF coding exon. The mice have been shown by others to exhibit no evidence of nonastrocytic recombination in tamoxifen-induced adult mice (Casper et al., 2007). Moreover, an independently generated Cre-ERT2 mouse line that used an hGFAP promoter sequence, also exhibited recombination primarily in GFAP+ astrocytes and only <1% of oligodendrocytes exhibited recombination in the adult corpus callosum (Chow et al., 2008). In our hands, analysis of an astrocyte cre reporter mouse revealed a level of 49% recombination (percentage GFAP cells exhibiting recombination/total GFAP+ cells) in astrocytes following tamoxifen treatment.
Both hGFAP-CreERT2-bdnf fl/fl-ROSA26 mice and control cre recombinase negative bdnffl-ROSA26 mice were injected with tamoxifen and then fed cuprizone-laden food. After 4 weeks of treatment, the expression of BDNF in both cre− and cre+ animals subjected to a demyelinating cuprizone lesion and injected with ACPD was monitored using Western blot. As expected, cuprizone treatment induces a significant decrease in BDNF that is reversed within 6 h of ACPD injection in control cre-mice. However, ACPD is unable to enhance the expression of BDNF protein in mice deficient in bdnf in astrocytes (hGFAP-CreERT2-bdnf fl/fl-ROSA26; Fig. 7a,b). Consistent with this observation, ACPD- treated mice exhibited a reduction in BDNF+ GFAP+ astrocyte profiles in the cre+ mice injected with tamoxifen (Fig. 7c). These data suggest that the ACPD effect is dependent upon the ability of GFAP+ astrocytes to synthesize BDNF in the lesion site.
Figure 7.

ACPD-elicited increases in BDNF protein are reduced in cuprizone-treated animals after cre recombination. Cre recombinase + GFAPcreERT2-floxBDNF-ROSA26 mice and Cre recombinase − floxBDNF-ROSA26 controls were injected with tamoxifen and fed cuprizone-laden food for 4 weeks before they received a single stereotaxic injection of either ACPD or saline vehicle. a, Western blots demonstrate BDNF protein in the corpus callosum of these animals. GAPDH is shown as a loading control. b, Graph represents a densitometric analysis of a Western blot of mature BDNF normalized to GAPDH and presented as percentage Cre− saline-injected control. c, Cre recombination results in a decrease in elevations of ACPD-elicited BDNF. Immunohistochemical analysis demonstrates that BDNF is eliminated from ACPD-treated GFAP+ astrocytes in the corpus callosum of cuprizone-treated Cre+ GFAPcreERT2-floxBDNF-ROSA26 mice. Western blot data were analyzed by ANOVA; *significantly different from saline-injected, cre− controls at p < 0.01; ***significantly different at p < 0.01. Each Western blot lane is from the corpus callosum of a single mouse within one experiment. Each experiment was repeated four times. Scale bar, 20 μm.
We hypothesized that the ACPD-stimulated production of astrocyte-derived BDNF increases myelin proteins that were decreased after cuprizone. While treatment with ACPD increases both MBP and MAG expression in cuprizone-lesioned, cre− control mice, the effect is completely eliminated when BDNF is reduced in GFAP+ cells of cre+ animals (Fig. 8a,b). These data demonstrate that ACPD requires the production of BDNF by GFAP+ astrocytes to mediate its effects on expression of myelin proteins in the demyelinated corpus callosum.
Figure 8.

The ACPD effect on myelin protein expression is blocked when astrocyte-derived BDNF is reduced. Cre recombinase + GFAPcreERT2-floxBDNF-ROSA26 mice and Cre recombinase − floxBDNF-ROSA26 controls were injected with tamoxifen and fed cuprizone-laden food for 4 weeks before they received a single stereotaxic injection of either ACPD or saline vehicle. a, Western blots demonstrate MBP and MAG protein in the corpus callosum of these mice. GAPDH is shown as a loading control. b, Graph represents a densitometric analysis of Western blots normalized to GAPDH and are presented as percentage cre- saline-injected control. Western blot data analyzed by ANOVA; *significantly different from saline-injected, cre- controls at p < 0.01; ***significantly different at p < 0.01. Each Western blot lane is from the corpus callosum of a single mouse within one experiment. Each experiment was repeated four times.
Discussion
Previous work defined BDNF as a trophic factor that enhances expression of differentiated traits MBP and MAG following a demyelination lesion. To explore the possibility that small molecules can elevate endogenous sources of BDNF to reverse deficits following a lesion, this work evaluated effects of the metabotropic agonist, ACPD. In the cuprizone model, expression of both the Group I and Group II metabotropic glutamate receptors was found to be elevated, a finding reminiscent of other lesion models (Fazio et al., 2008) and what occurs in multiple sclerosis (Fazio et al., 2008; Geurts et al., 2003). Moreover, subsets of GFAP+ astrocytes colocalize with mGluR1 and mGluR5 as well as BDNF, making them candidate cellular sources for trophin support. To determine effects of metabotropic stimulation a general metabotropic agonist ACPD was injected into the lesion site. ACPD increased both BDNF and myelin protein levels in the demyelinated corpus callosum. This effect was found to be dependent upon the production of BDNF by GFAP+ astrocytes, suggesting that the astrocyte population can be targeted to elaborate neurotrophic support that promotes recovery from demyelination.
Roles of metabotropic receptors on astrocytes
The concept that glial cells may be responsive to metabotropic stimulation is not new. The presence of mGluR5 mRNA transcripts has previously been detected in cultured astrocytes, and these cells respond to stimulation with Group I selective agonists with PKC-dependent oscillations in intracellular calcium concentrations (Balázs et al., 1997; Nakahara et al., 1997). Many groups are investigating the role of these receptors on astrocytes as regulators of extracellular glutamate levels, as well as regulators of other gliotransmitters (Halassa and Haydon, 2010).
Previous work from our lab has demonstrated that glutamate, through the metabotropic glutamate receptors, also supports the production and release of BDNF from cultured basal forebrain astrocytes (Jean et al., 2008), and others have demonstrated that cultured mouse cortical astrocytes respond to metabotropic stimulation by increasing the synthesis and release of TGF-β (Bruno et al., 1998) and NGF (Ciccarelli et al., 1999), as well as IL-1β stimulated release of IL-6 (Aronica et al., 2005). Our work adds to this literature by establishing that metabotropic glutamate agonist stimulated astrocytes may serve as sources of BDNF in vivo. Although previous studies have indicated that group I mGluRs can reverse deficits associated with degenerative disease models (Agari et al., 2008), to the best of our knowledge, this is the first evidence that metabotropic receptors enhance BDNF in astrocytes in vivo in a manner that has functional consequences and may explain the reversal of deficits noted previously.
The effect of ACPD occurs rapidly
We note an increase in BDNF, as well as myelin proteins 6 h following the ACPD injection. This effect of ACPD is consistent with previous work in our laboratory (Wu et al., 2004) indicating that ACPD is able to increase BDNF mRNA within 4 h, suggesting that elevation in BDNF is a rapidly regulated event. Similarly, increases in MBP mRNA in response to signaling, can occur within a relatively short time frame (Hashimoto et al., 2011), as can increases in MBP protein including in response to glutamate stimulation (Wake et al., 2011). The mechanisms underlying increases in BDNF, as well as in the myelin proteins, will be the subject of further investigation.
Effects of other astrocyte-derived molecules following a demyelinating lesion
Astrocytes are a well characterized source for other biologically active molecules, both in a culture dish and in vivo. For example, IGF-1 (Komoly et al., 1992), IL1β (Guo et al., 2001), and ΤΝFα (Chung and Benveniste, 1990) have all been detected in astrocytes and appear to be regulators of the myelin repair process. Thus, transgenic mice overexpressing IGF-1 are protected from the cuprizone-induced loss of mature oligodendrocytes (Mason et al., 2000), whereas mice lacking the Type 1 IGF receptor do not remyelinate efficiently when cuprizone is removed from the diet (Mason et al., 2003). Similarly, transgenic mice lacking IL-1β exhibit impaired remyelination compared with wild-type littermates (Mason et al., 2001), and in TNF-α knock-out animals remyelination is inefficient and incomplete (Arnett et al., 2001). It remains to be determined whether enhanced expression by metabotropic agonists may elicit recovery. Interestingly, overexpression of PDGF-A in astrocytes does result in the increase of PDGFRα+ cells following cuprizone-elicited demyelination (Woodruff et al., 2004), providing additional evidence that astrocytes are sources of multiple trophic factors that may be harnessed to reverse deficits.
The cuprizone-elicited lesion site is complex
It is well known that the lesion site in the corpus callosum contains numerous cells and is a complex environment, including with respect to BDNF. Thus oligodendrocytes in the corpus callosum (Dai et al., 2003) express the neurotrophin and cultured as well in vivo astrocytes are known to express the neurotrophin (Dougherty et al., 2000; Wu et al., 2004; Jean et al., 2008). Interestingly, the nonastrocyte derived BDNF appears to dominate overall protein levels of BDNF seen in the unlesioned control since this population is reduced following cuprizone, coincident with a decrease in BDNF as defined by Western blot.
The response of astrocytes with respect to BDNF is also of interest. Following cuprizone astrocytes increase expression of GFAP. However, their expression of BDNF is minimal, with only a subpopulation of astrocytes exhibiting the trophin. Nevertheless, the astrocytes play an important role when stimulated specifically by ACPD. Only astrocytes express BDNF and the metabotropic glutamate receptors that are affected by ACPD. The role of this astrocytic source of BDNF is critical after ACPD treatment, as both the increases in BDNF and the ACPD effects on myelin proteins are largely eliminated after cre-mediated recombination in GFAP+ cells. It appears that astrocytes are important factories of molecules that may be harnessed to enhance repair. Future studies will continue to explore and identify other critical factors.
In sum, we have demonstrated that ACPD is able to reverse deficits in myelin proteins following a cuprizone lesion. We show further that it does so by eliciting increases in BDNF in reactive astrocytes. We suggest that metabotropic agonists may be useful agents to modulate the lesion environment to enhance recovery from a demyelinating injury and possibly other degenerative conditions in the CNS.
Footnotes
This work was supported by NIH RO1 NS036647, NIH RO1 NS030687, NIH P01 HD023315; The National Multiple Sclerosis Society; and the NIH funded Biomedical Science Education Postdoctoral Training Program 5K12GM093854-05. We thank Lauren D. Lercher, Jean Z. Honeywell, and Shir Miretzky for expert technical assistance; Dr. Ken D. McCarthy for assistance in obtaining the h-GFAP-Cre-ERT2 and ACR mice; and Dr. Michael Matise for sharing the ROSA26 mice used in this study.
The authors declare no competing financial interests.
References
- Agari T, Yasuhara T, Matsui T, Kuramoto S, Kondo A, Miyoshi Y, Shingo T, Borlongan CV, Date I. Intrapallidal metabotropic glutamate receptor activation in a rat model of Parkinson's disease: behavioral and histological analyses. Brain Res. 2008;1203:189–196. doi: 10.1016/j.brainres.2008.01.051. [DOI] [PubMed] [Google Scholar]
- Aharoni R, Eilam R, Domev H, Labunskay G, Sela M, Arnon R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc Natl Acad Sci U S A. 2005;102:19045–19050. doi: 10.1073/pnas.0509438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktas O, Küry P, Kieseier B, Hartung HP. Fingolimod is a potential novel therapy for multiple sclerosis. Nat Rev Neurol. 2010;6:373–382. doi: 10.1038/nrneurol.2010.76. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Aronica E, Catania MV, Geurts J, Yankaya B, Troost D. Immunohistochemical localization of group I and II metabotropic glutamate receptors in control and amyotrophic lateral sclerosis human spinal cord: upregulation in reactive astrocytes. Neuroscience. 2001;105:509–520. doi: 10.1016/S0306-4522(01)00181-6. [DOI] [PubMed] [Google Scholar]
- Aronica E, Gorter JA, Rozemuller AJ, Yankaya B, Troost D. Activation of metabotropic glutamate receptor 3 enhances interleukin (IL)-1beta-stimulated release of IL-6 in cultured human astrocytes. Neuroscience. 2005;130:927–933. doi: 10.1016/j.neuroscience.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Azoulay D, Vachapova V, Shihman B, Miler A, Karni A. Lower brain-derived neurotrophic factor in serum of relapsing remitting MS: reversal by glatiramer acetate. J Neuroimmunol. 2005;167:215–218. doi: 10.1016/j.jneuroim.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Balázs R, Miller S, Romano C, de Vries A, Chun Y, Cotman CW. Metabotropic glutamate receptor mGluR5 in astrocytes: pharmacological properties and agonist regulation. J Neurochem. 1997;69:151–163. doi: 10.1046/j.1471-4159.1997.69010151.x. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Casabona G, Copani A, Caciagli F, Nicoletti F. Neuroprotection by glial metabotropic glutamate receptors is mediated by transforming growth factor-beta. J Neurosci. 1998;18:9594–9600. doi: 10.1523/JNEUROSCI.18-23-09594.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals JM, Checa N, Marco S, Akerud P, Michels A, Pérez-Navarro E, Tolosa E, Arenas E, Alberch J. Expression of brain-derived neurotrophic factor in cortical neurons is regulated by striatal target area. J Neurosci. 2001;21:117–124. doi: 10.1523/JNEUROSCI.21-01-00117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper KB, McCarthy KD. GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol Cell Neurosci. 2006;31:676–684. doi: 10.1016/j.mcn.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Casper KB, Jones K, McCarthy KD. Characterization of astrocyte-specific conditional knockouts. Genesis. 2007;45:292–299. doi: 10.1002/dvg.20287. [DOI] [PubMed] [Google Scholar]
- Chow LM, Zhang J, Baker SJ. Inducible Cre recombinase activity in mouse mature astrocytes and adult neural precursor cells. Transgenic Res. 2008;17:919–928. doi: 10.1007/s11248-008-9185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung IY, Benveniste EN. Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1beta. J Immunol. 1990;144:2999–3007. [PubMed] [Google Scholar]
- Ciccarelli R, Di Iorio P, Bruno V, Battaglia G, D'Alimonte I, D'Onofrio M, Nicoletti F, Caciagli F. Activation of A(1) adenosine or mGlu3 metabotropic glutamate receptors enhances the release of nerve growth factor and S-100beta protein from cultured astrocytes. Glia. 1999;27:275–281. doi: 10.1002/(SICI)1098-1136(199909)27:3<275::AID-GLIA9>3.0.CO%3B2-0. [DOI] [PubMed] [Google Scholar]
- Dai X, Lercher LD, Clinton PM, Du Y, Livingston DL, Vieira C, Yang L, Shen MM, Dreyfus CF. The trophic role of oligodendrocytes in the basal forebrain. J Neurosci. 2003;23:5846–5853. doi: 10.1523/JNEUROSCI.23-13-05846.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deogracias R, Yazdani M, Dekkers MP, Guy J, Ionescu MC, Vogt KE, Barde YA. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2012;109:14230–14235. doi: 10.1073/pnas.1206093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Liberto V, Bonomo A, Frinchi M, Belluardo N, Mudò G. Group II metabotropic glutamate receptor activation by agonist LY379268 treatment increases the expression of brain derived neurotrophic factor in the mouse brain. Neuroscience. 2010;165:863–873. doi: 10.1016/j.neuroscience.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Dougherty KD, Dreyfus CF, Black IB. Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiol Dis. 2000;7:574–585. doi: 10.1006/nbdi.2000.0318. [DOI] [PubMed] [Google Scholar]
- Du Y, Lercher LD, Zhou R, Dreyfus CF. Mitogen-activated protein kinase pathway mediates effects of brain-derived neurotrophic factor on differentiation of basal forebrain oligodendrocytes. J Neurosci Res. 2006;84:1692–1702. doi: 10.1002/jnr.21080. [DOI] [PubMed] [Google Scholar]
- Fazio F, Notartomaso S, Aronica E, Storto M, Battaglia G, Vieira E, Gatti S, Bruno V, Biagioni F, Gradini R, Nicoletti F, Di Marco R. Switch in the expression of mGlu1 and mGlu5 metabotropic glutamate receptors in the cerebellum of mice developing experimental autoimmune encephalomyelitis and in autoptic cerebellar samples from patients with multiple sclerosis. Neuropharmacology. 2008;55:491–499. doi: 10.1016/j.neuropharm.2008.06.066. [DOI] [PubMed] [Google Scholar]
- Geurts JJ, Wolswijk G, Bö L, van der Valk P, Polman CH, Troost D, Aronica E. Altered expression patterns of group I and II metabotropic glutamate receptors in multiple sclerosis. Brain. 2003;126:1755–1766. doi: 10.1093/brain/awg179. [DOI] [PubMed] [Google Scholar]
- Guo L, Sawkar A, Zasadzki M, Watterson DM, Van Eldik LJ. Similar activation of glial cultures from different rat brain regions by neuroinflammatory stimuli and downregulation of the activation by a new class of small molecule ligands. Neurobiol Aging. 2001;22:975–981. doi: 10.1016/S0197-4580(01)00295-0. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Udagawa J, Kagohashi Y, Matsumoto A, Hatta T, Otani H. Direct and indirect effects of neuropeptide Y and neurotrophin 3 on myelination in the neonatal brains. Brain Res. 2011;1373:55–66. doi: 10.1016/j.brainres.2010.12.027. [DOI] [PubMed] [Google Scholar]
- Huang YT, Lai PC, Wu CC, Hsu SH, Cheng CC, Lan YF, Chiu TH. BDNF mediated TrkB activation is a survival signal for transitional cell carcinoma cells. Int J Oncol. 2010;36:1469–1476. doi: 10.3892/ijo_00000633. [DOI] [PubMed] [Google Scholar]
- Jean YY, Lercher LD, Dreyfus CF. Glutamate elicits release of BDNF from basal forebrain astrocytes in a process dependent on metabotropic receptors and the PLC pathway. Neuron Glia Biol. 2008;4:35–42. doi: 10.1017/S1740925X09000052. [DOI] [PubMed] [Google Scholar]
- Komoly S, Hudson LD, Webster HD, Bondy CA. Insulin-like growth factor I gene expression is induced in astrocytes during experimental demyelination. Proc Natl Acad Sci U S A. 1992;89:1894–1898. doi: 10.1073/pnas.89.5.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makar TK, Bever CT, Singh IS, Royal W, Sahu SN, Sura TP, Sultana S, Sura KT, Patel N, Dhib-Jalbut S, Trisler D. Brain-derived neurotrophic factor gene delivery in an animal model of multiple sclerosis using bone marrow stem cells as a vehicle. J Neuroimmunol. 2009;210:40–51. doi: 10.1016/j.jneuroim.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Mason JL, Ye P, Suzuki K, D'Ercole AJ, Matsushima GK. Insulin-like growth factor-1 inhibits mature oligodendrocyte apoptosis during primary demyelination. J Neurosci. 2000;20:5703–5708. doi: 10.1523/JNEUROSCI.20-15-05703.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1beta promotes repair of the CNS. J Neurosci. 2001;21:7046–7052. doi: 10.1523/JNEUROSCI.21-18-07046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JL, Xuan S, Dragatsis I, Efstratiadis A, Goldman JE. Insulin-like growth factor (IGF) signaling through type 1 IGF receptor plays an important role in remyelination. J Neurosci. 2003;23:7710–7718. doi: 10.1523/JNEUROSCI.23-20-07710.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, Barde YA. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–133. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- Nakahara K, Okada M, Nakanishi S. The metabotropic glutamate receptor mGluR5 induces calcium oscillations in cultured astrocytes via protein kinase C phosphorylation. J Neurochem. 1997;69:1467–1475. doi: 10.1046/j.1471-4159.1997.69041467.x. [DOI] [PubMed] [Google Scholar]
- Poduslo JF, Curran GL. Permeability at the blood–brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res Mol Brain Res. 1996;36:280–286. doi: 10.1016/0169-328X(95)00250-V. [DOI] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Van't Veer A, Du Y, Fischer TZ, Boetig DR, Wood MR, Dreyfus CF. Brain-derived neurotrophic factor effects on oligodendrocyte progenitors of the basal forebrain are mediated through trkB and the MAP kinase pathway. J Neurosci Res. 2009;87:69–78. doi: 10.1002/jnr.21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vondran MW, Clinton-Luke P, Honeywell JZ, Dreyfus CF. BDNF± mice exhibit deficits in oligodendrocyte lineage cells of the basal forebrain. Glia. 2010;58:848–856. doi: 10.1002/glia.20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonDran MW, Singh H, Honeywell JZ, Dreyfus CF. Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J Neurosci. 2011;31:14182–14190. doi: 10.1523/JNEUROSCI.6595-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Lee PR, Fields RD. Control of local protein synthesis and initial events in myelination by action potentials. Science. 2011;333:1647–1651. doi: 10.1126/science.1206998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AW, Xiao J, Kemper D, Kilpatrick TJ, Murray SS. Oligodendroglial expression of TrkB independently regulates myelination and progenitor cell proliferation. J Neurosci. 2013;33:4947–4957. doi: 10.1523/JNEUROSCI.3990-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff RH, Fruttiger M, Richardson WD, Franklin RJ. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol Cell Neurosci. 2004;25:252–262. doi: 10.1016/j.mcn.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Wu H, Friedman WJ, Dreyfus CF. Differential regulation of neurotrophin expression in basal forebrain astrocytes by neuronal signals. J Neurosci Res. 2004;76:76–85. doi: 10.1002/jnr.20060. [DOI] [PubMed] [Google Scholar]
- Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, Chen ZY, Mark W, Tessarollo L, Lee FS, Lu B, Hempstead BL. Neuronal release of proBDNF. Nat Neurosci. 2009;12:113–115. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]