

Abstract
Cerebellar circuits are patterned into an array of topographic parasagittal domains called zones. The proper connectivity of zones is critical for motor coordination and motor learning, and in several neurological diseases cerebellar circuits degenerate in zonal patterns. Despite recent advances in understanding zone function, we still have a limited understanding of how zones are formed. Here, we focused our attention on Purkinje cells to gain a better understanding of their specific role in establishing zonal circuits. We used conditional mouse genetics to test the hypothesis that Purkinje cell neurotransmission is essential for refining prefunctional developmental zones into sharp functional zones. Our results show that inhibitory synaptic transmission in Purkinje cells is necessary for the precise patterning of Purkinje cell zones and the topographic targeting of mossy fiber afferents. As expected, blocking Purkinje cell neurotransmission caused ataxia. Using in vivo electrophysiology, we demonstrate that loss of Purkinje cell communication altered the firing rate and pattern of their target cerebellar nuclear neurons. Analysis of Purkinje cell complex spike firing revealed that feedback in the cerebellar nuclei to inferior olive to Purkinje cell loop is obstructed. Loss of Purkinje neurotransmission also caused ectopic zonal expression of tyrosine hydroxylase, which is only expressed in adult Purkinje cells when calcium is dysregulated and if excitability is altered. Our results suggest that Purkinje cell inhibitory neurotransmission establishes the functional circuitry of the cerebellum by patterning the molecular zones, fine-tuning afferent circuitry, and shaping neuronal activity.
Keywords: ataxia, circuitry, connectivity, gene expression, inhibition, physiology
Introduction
Cerebellar circuits are wired into a patterned map. The map is organized into parasagittal domains called zones (Apps and Hawkes, 2009; White and Sillitoe, 2013a). While zonal organization has been shown to be critical for motor coordination (Horn et al., 2010; Graham and Wylie, 2012), motor learning (Attwell et al., 1999; Mostofi et al., 2010), and motor circuit degeneration (Welsh et al., 2002; Sarna and Hawkes, 2003; Strømme et al., 2011), we still have a poor understanding of how functional zones are formed. The normal function of zones and their differential sensitivity to disease seem to depend on Purkinje cell molecular phenotype (Wadiche and Jahr, 2005; Paukert et al., 2010; Ebner et al., 2012). Purkinje cells, the only output neurons of the cerebellar cortex, express zonal molecular markers such as zebrinII (Sillitoe and Hawkes, 2013) and they project topographically organized axons to the cerebellar nuclei (Sugihara, 2011). Because climbing and mossy fiber inputs also respect zonal boundaries (Ruigrok, 2011), connectivity around the Purkinje cell forms a cortico-nucleo-olivary loop that lies at the heart of cerebellar function and motor behavior (Angaut and Sotelo, 1987; Lang et al., 1996; Bengtsson et al., 2004; Witter et al., 2013). We know that to form these circuits, genetic and cellular differentiation programs must first establish a prefunctional embryonic map comprised of crude zones (Sotelo, 2004; Apps and Hawkes, 2009; White and Sillitoe, 2013a). How this crude map segregates into precisely wired functional zones remains largely unknown. Here, we tested whether circuit activity influences functional zone formation.
Purkinje cells are inhibitory; they release GABA and modulate the activity of neurons in the cerebellar nuclei (Ito, 1984; Gauck and Jaeger, 2000; Uusisaari and Knöpfel, 2011; Person and Raman, 2012). However, it is not known how Purkinje cell synaptic communication influences zone development. Understanding how zones form is critical for solving how cerebellar circuits develop into functional adult networks. One possibility is that Purkinje cell activity influences zone patterning. Based on this idea, we sought to test the hypothesis that chemical synaptic signaling itself controls the separation of Purkinje cells into molecularly distinct zones, the topographic wiring of afferents into specific zones, and ultimately the firing properties of neurons that are linked into zonal modules. To test our hypothesis, we devised a conditional genetic approach to constitutively block inhibitory synaptic transmission of Purkinje cells, thereby eliminating cerebellar cortical communication with the cerebellar nuclei. This strategy enabled us to investigate whether Purkinje-cerebellar nuclei neurotransmission shapes the formation of adult functional zones and how it affects cerebellar nuclei and Purkinje cell spike-firing properties, and ultimately, motor behavior. To systematically examine these circuit properties we combined our genetic approach with molecular zonal analyses, neural circuit tracing, in vivo electrophysiology, and behavioral paradigms. This study sheds new light on the role of Purkinje cell synaptic transmission for the development of cerebellar circuits and it provides a new approach to test the role of cerebellar corticonuclear communication in motor and non-motor behavior (Brielmaier et al., 2012; Tsai et al., 2012; Reith et al., 2013).
Materials and Methods
Mice.
All the studies were performed under an approved Institutional Animal Care and Use Committee animal protocol according to the institutional guidelines at Baylor College of Medicine. We compared L7Cre;Vgatflox/flox conditional mutant mice to Vgatflox/flox control littermates of either sex throughout the study, and note that L7Cre and L7Cre;Vgatflox/+ mice do not exhibit motor behavior defects (see Results). Noon on the day a vaginal plug was detected was considered embryonic day 0.5 (E0.5). The day of birth was designated as postnatal day 0 (P0), and mice that were P28 or older were considered mature. All the mice used in this study were between 1 and 3 months old. Genotyping for the L7Cre allele was performed using standard Cre primers (Sillitoe et al., 2008, 2010), and genotyping for the Vgatflox allele was performed according to Tong et al. (2008).
Tissue preparation and cutting.
For perfusion fixation, animals were deeply anesthetized with 2, 2, 2-tribromoethanol (Avertin), and then perfused through the heart with 0.1 m PBS (pH 7.2), followed by 4% paraformaldehyde (PFA) diluted in PBS. The brains from the perfused mice were postfixed for 24–48 h in 4% PFA and then cryoprotected stepwise in PBS-buffered sucrose solutions (15 and 30% each time until the brain sunk). Serial 40-μm-thick coronal or sagittal sections were cut on a cryostat, and then collected and processed free floating in PBS.
Body and brain weight measurement and quantification.
Body weights of six control and six L7Cre;Vgatflox/flox mice were measured at age P30. Afterward, they were deeply anesthetized with isoflurane, decapitated, and the brain quickly dissected. The brain was immediately weighed and included anterior brain tissue starting from the olfactory bulbs to brainstem tissue including the medulla—tissue past the flexure at the inferior olive was excluded. The cerebellum was then dissected away from the rest of the brain and weighed separately. Body, whole-brain, and cerebellum weight distributions in control and L7Cre;Vgatflox/flox mice were compared using Student's t test.
Immunohistochemistry.
Immunohistochemistry was performed as described previously (Sillitoe et al., 2003, 2010; Reeber et al., 2011; White and Sillitoe, 2013b). Briefly, tissue sections were thoroughly washed, blocked with 10% normal goat serum (NGS; Sigma) for 1 h at room temperature and then incubated in 0.1 m PBS containing 10% NGS, 0.1% Tween 20, and the primary antibodies (see below for details) for 16–18 h at room temperature, shaking gently. The tissue sections were then washed three times in PBS and incubated in secondary antibodies (see below for details) for 2 h at room temperature, again shaking gently. The tissue was rinsed again and immunoreactivity revealed as described below.
Monoclonal anti-zebrinII (Brochu et al., 1990) was used directly from spent hybridoma culture medium at a concentration of 1:250 (gift from Dr. Richard Hawkes, University of Calgary). ZebrinII recognizes an antigen on the Aldolase C protein (Ahn et al., 1994) and it is a well established marker for Purkinje cell zones. Rabbit polyclonal anti-phospholipase C β 4 (PLCβ4; 1:150; catalog #sc-20760) was purchased from Santa Cruz Biotechnology and it revealed an identical pattern of Purkinje cell zones to what has previously been described (Sarna et al., 2006). Rabbit polyclonal anti-HSP25 (StressGen) was used diluted at 1:500 and our studies using this antibody resulted in a tissue-staining pattern identical to that previously reported in wild-type mice (Armstrong et al., 2000, 2001). Mouse monoclonal anti-vesicular glutamate transporter 2 (VGLUT2; 1:500; catalog #MAB5504) was purchased from Millipore Bioscience Research Reagents and it was used to visualize climbing and mossy fiber terminals (Hisano et al., 2002; Reeber and Sillitoe, 2011; Gebre et al., 2012). Rabbit polyclonal anti-vesicular glutamate transporter 1 (VGLUT1; 1:500; catalog #135 302) was purchased from Synaptic Systems and was used to visualize parallel fibers and mossy fiber terminals (Gebre et al., 2012). Mouse monoclonal anti-calbindin-D28K (calbindin; 1:10,000; catalog #CD38), rabbit polyclonal anti-parvalbumin (1:1000; catalog #PV25), and rabbit polyclonal anti-calretinin (1:500; catalog #CR7699/3H) were purchased from Swant. Rabbit anti-GABARα6 is a granule cell marker that was purchased from Millipore; we used it at a concentration of 1:500 (catalog #AB5610). Mouse monoclonal anti-NFH (also called anti-SMI-32; 1:1500) was purchased from Covance. Anti-SMI-32 recognizes the nonphosphorylated form of NFH (see manufacturer product datasheet for details), which on tissue sections labels the soma, dendrites, and axons of adult Purkinje cells, and also basket cell axons and pinceaux (Demilly et al., 2011). Rabbit anti-neurogranin (1:500) was raised against full-length recombinant rat neurogranin protein (Millipore Bioscience Research Reagents; catalog #AB5620). Neurogranin recognizes Purkinje cells in the developing cerebellum and Golgi cells in the adult cerebellum (Singec et al., 2003; Larouche et al., 2006). Rabbit polyclonal anti-cocaine- and amphetamine-related transcript peptide (CART 55-102; catalog #H-003-62) was used at a concentration of 1:250 to detect climbing fibers (Reeber and Sillitoe, 2011) and it was purchased from Phoenix Pharmaceuticals. The CART signal was amplified using a biotinylated secondary antibody and the Vectastain Elite ABC method from Vector Laboratories (Reeber and Sillitoe, 2011). Rabbit anti-tyrosine hydroxylase was purchased from Calbiochem/Millipore (catalog #657012) and it used at a concentration of 1:500. The rabbit polyclonal antibody against the vesicular GABA transporter (VGAT; 1:500; catalog #PA1-4701) from Pierce/Thermo Scientific and guinea pig polyclonal anti-VGAT (Synaptic Systems; 1:500) were used to mark inhibitory GABAergic cerebellar synapses. We identified Purkinje cell terminal puncta by labeling coronal cut tissue sections with a Purkinje cell marker called carbonic anhydrase VIII (Hirota et al., 2003; Rong et al., 2004; Jiao et al., 2005; CAR8). We used a rabbit-polyclonal CAR8 antibody (1:500) that was purchased from Santa Cruz Biotechnology (catalog #sc-67330).
We visualized immunoreactive complexes either using diaminobenzidine (DAB; 0.5 mg/ml; Sigma) or fluorescent secondary antibodies. For the DAB reaction, we used horseradish peroxidase (HRP)-conjugated goat anti-rabbit and goat anti-mouse secondary antibodies (diluted 1:200 in PBS; DAKO) to bind the primary antibodies. Staining for fluorescent immunohistochemistry was performed using donkey anti-mouse, anti-rabbit, or anti-guinea pig secondary antibodies conjugated to Alexa 488 or 555 fluorophores (Invitrogen), all diluted to 1:1500. Tissues sections were coverslipped using either Entellan mounting media (for DAB; Electron Microscopy Sciences) or FLUORO-GEL with Tris buffer (Electron Microscopy Sciences). We tested the specificity of the secondary antibodies by processing the tissue sections in the absence of primary antibodies. No signal was detected in such control experiments indicating that the staining we observed was not due to nonspecific signals from the Alexa or HRP-conjugated antibodies (data not shown).
In situ hybridization.
Mice were anesthetized with isoflurane, the brains rapidly removed from the skull and immersed in OCT, and then they were immediately flash frozen by placing the tissue molds into liquid nitrogen. Sagittal sections (25 μm) were cut through the cerebellum and the sectioned slices placed directly onto electrostatically coated glass slides (Probe On Plus Fisher Brand; Fisher Scientific). The tissue was probed with a Vgat (SLC32A1) mRNA probe using an automated in situ hybridization procedure as described previously (Yaylaoglu et al., 2005; see also genepaint.org).
Fluorescent Nissl histology.
Free-floating tissue sections (40 μm) were washed in PBS and 0.1% Tween 20 for 10 min, and then washed for 5 min twice in PBS. The tissue was then incubated in NeuroTrace fluorescent Nissl stain (Invitrogen) diluted to 1:1000 in PBS for 20 min. Sections were then washed for 5 min twice in PBS, then for 10 min in PBS and 0.1% Tween 20 and again twice in PBS before being washed overnight at 4°C in PBS. We mounted the stained tissue sections with FLUORO-GEL mounting media (Electron Microscopy Sciences) before imaging. For dual staining of calbindin and NeuroTrace, we added NeuroTrace to the secondary antibody mixture.
Neural tracing.
Anterograde tracing was performed according to previous protocols (Sillitoe et al., 2010; Reeber et al., 2011). Approximately 50 nl of a 2% solution of wheat germ agglutinin (WGA) conjugated to Alexa Fluor 555 (catalog #W32464, Invitrogen) was pressure injected into the lower thoracic upper lumbar spinal cord of adult mice (n = 5 for each genotype). After a 48 h survival period the mice were anesthetized as described above and then perfused with 4% PFA (described above). After the perfusion, the WGA-Alexa traced tissue was cut and mounted for imaging using the Zeiss Apotome.2 acquisition system (see below). The tissue was imaged on the same day it was cut to capture the brightest signal.
Behavioral testing.
Gait abnormalities were assessed using footprint analysis (Girirajan et al., 2008). The hindfeet of the mice were painted with red and blue nontoxic acrylic paint (Americana) before they walked on a strip of white paper taped along the floor of a 50 cm custom-made Plexiglas tunnel. The distances between footprints were measured to obtain “stride,” “sway,” and “stance” lengths (Girirajan et al., 2008). The measures were averaged for each mouse, and compared using Student's t test. The p values for each measure are reported in the results. A total of 10 Vgatflox/flox control mice and 10 L7Cre;Vgatflox/flox mice were tested.
The mice were also tested on an accelerating rotarod (model 47600; Ugo Basile Biological Research Apparatus), which was set to accelerate from 4 to 40 rpm over a period of 300 s. Each mouse was monitored for latency to fall (or three consecutive rotations if they gripped and rode around on the rod). A total of 19 Vgatflox/flox control mice and 20 L7Cre;Vgatflox/flox mutant mice were tested using a paradigm of three trials per day, conducted on 3 consecutive days, with 10 min rest time given in their home cages between trials. Between-group differences were statistically evaluated by Student's t test. Between-trial differences were statistically evaluated with repeated-measures ANOVA.
In vivo electrophysiology.
In preparation for in vivo electrophysiology, the mice were anesthetized with an intraperitoneal injection of ketamine/dexmedetomidine (50 and 0.5 mg/kg, respectively) and then during the recording session maintained with ∼0.25% isoflurane. Anesthetized mice were placed into a stereotaxic frame and a small craniotomy (∼2 mm) was performed over the cerebellum. Single-unit recordings were attained with 2–5 MΩ Tungsten electrodes (Thomas Recording) that are controlled by a motorized micromanipulator (MP-225; Sutter Instrument).
The signals were bandpass filtered at 0.3–13 kHz, amplified with an ELC-03XS (NPI) amplifier, and digitized into Spike2 (CED). Analysis of the raw traces was performed with Spike2, Excel, and MATLAB. Purkinje cells were identified by the presence of both simple and complex spikes, the latter of which are a characteristic action potential that results exclusively from excitatory climbing fiber input and causes a subsequent pause of ∼20 ms in simple spikes. Simple and complex spikes were sorted independently and analyzed for frequency (Hz = spikes/s) and for the interspike interval (ISI) coefficient of variance [CV = (SD of ISIs)/(mean of ISIs)]. Cerebellar nuclei neurons were identified by the absence of complex spikes and by stereotaxic coordinates (see below for details). The CV2 (CV2 = 2|ISIn+1 − ISIn|/(ISIn+1 + ISIn; Holt et al., 1996) was also calculated for mutant and control cells. All numerical results are reported as mean ± SEM. Statistical analyses were performed with unpaired, two-tailed Student's t tests. Significance is indicated in graphs for *p < 0.05, **p < 0.01, or ***p < 0.001, respectively. The number of cells in each analysis is indicated with “n = XX.”
Electrophysiological recording and anatomically marking the cerebellar nuclei.
In this study, we used stereotaxic coordinates to record from the middle and lateral cerebellar nuclei (Paxinos and Watson, 2004; Sillitoe et al., 2011; also called the interposed and dentate, respectively). Within a radius of 0.50 mm around a central point relative to bregma (−6.4 mm anteroposterior, +1.3 mm lateral, and 2.5–3 mm deep) we made multiple electrode tracks to record from both nuclei. After recording, we used a Nanoject II (Drummond) to pressure inject 25 nl of 2% WGA conjugated to Alexa fluorophore 555 tracer (WGA-Alexa 555) into the recording spot using the exact same coordinates but this time using a pulled glass pipette. WGA-Alexa 555 is rapidly taken up by neurons at the injection spot and therefore only requires 3–6 h to intensely mark local neuronal populations (Reeber et al., 2011, 2012). We found this method to be ideal for reliably marking the cerebellar nuclei as a way of mapping the recording sites. Marked cerebellar nuclei were analyzed using 40-μm-thick tissue sections that were coverslipped with FLUORO-GEL. The WGA-Alexa 555 was viewed using a Zeiss CY3 filter set as previously described (Reeber et al., 2012).
Imaging and data quantification.
Photomicrographs of tissue sections were captured using Zeiss AxioCam MRm (fluorescence) and AxioCam MRc5 (DAB-reacted tissue sections) cameras mounted on a Zeiss Axio Imager.M2 microscope. Images of tissue sections were acquired and analyzed using either Zeiss AxioVision software (release 4.8) or Zeiss ZEN software (2012 edition). After imaging, the raw data were imported into Adobe Photoshop CS5 and corrected for brightness and contrast levels. Schematics were drawn in Adobe Illustrator CS5.
For the number of zebrinII-expressing cells in lobule VIII, we counted Purkinje cells in the P1+, P1−, and P2+ zones, which is ∼400 μm cerebellar cortex on either side of the midline. Only cells in the rostral half of lobule VIII were counted, and the number of zebrinII cells was normalized to the total number of Purkinje cells located within zebrinII + PLCβ4 zones. The results are reported as a percentage, and we used the unpaired Student's t test for significance. Differences between mutants and controls were considered significant if p < 0.05. Three mutants and three controls were used for these cell counts.
Molecular layer thickness was measured within the primary fissure (between lobules V and VI) on sagittal sections. Molecular layer thicknesses from five sections separated by ∼200 μm around the midline were analyzed using an unpaired Student's t test to compare three mice of each genotype. Differences between mutants and controls were considered significant if p < 0.05.
Images for the quantification of VGAT-immunoreactive terminals were acquired using the ApoTome, a commercially available form of structured illumination microscopy by Zeiss (Weigel et al., 2009). High-resolution z-stacks were acquired with a constant interstack interval and maximum projections were generated using the Zeiss ZEN software. Quantification of VGAT puncta was performed from the projections using built-in functions in the image processing software Fiji (Schindelin et al., 2012). Briefly, a minimum fluorescence intensity threshold was applied consistently across groups to normalize background signals. The corrected images were converted from a 32-bit format into a binary format, to assign each VGAT puncta a value of 1. Particles in close proximity to each other were resolved using a built-in Watershed algorithm. Density of VGAT puncta was then quantified using an Analyze Particles algorithm. To determine whether the loss of VGAT expression occurred specifically in Purkinje cell terminals, double immunolabeling for CAR8 and VGAT was performed. Images were acquired with high power using a Zeiss Axio Imager microscope with ApoTome, as described earlier. Regions showing CAR8 and VGAT-immunopositive puncta surrounding large cerebellar nuclear neurons were cropped and isolated as regions of interest. After background subtraction, colocalization between CAR8 and VGAT-immunopositive terminals was determined by measuring the Pearson's correlation coefficient (R) with the Coloc2 plugin in Fiji. We compared the number of terminals that coexpressed VGAT + CAR8 in control versus mutant mice. Three control mice and four mutants were used for this analysis.
Results
We blocked Purkinje cell synaptic signaling by preventing the loading of neurotransmitters into synaptic vesicles by crossing L7Cre mice (Lewis et al., 2004) with a conditional “floxed” allele of Vgat (Tong et al., 2008). VGAT is widely expressed in GABAergic and glycinergic neurons in the brain and it is essential for loading GABA into presynaptic vesicles for inhibitory neurotransmission (McIntire et al., 1997; Chaudhry et al., 1998; Fujii et al., 2007; Saito et al., 2010). To establish our model, we first investigated whether only Purkinje cells are targeted by the approach, and then we assessed whether losing Purkinje cell VGAT function perturbs cerebellar patterning, motor behavior, and in vivo cell function.
L7Cre;Vgatflox/flox mice do not express Vgat mRNA or VGAT protein in Purkinje cells
Endogenous L7 is expressed in Purkinje cells starting from approximately E14 in mouse (Oberdick et al., 1993). The L7Cre transgenic mouse line used in this study exhibits Cre-induced reporter recombination by approximately E17 (Lewis et al., 2004). Vgat transcript expression appears by approximately E10.5 (Oh et al., 2005) and VGAT protein at approximately E15 (Boulland and Chaudhry, 2012). Purkinje cells are born between E10 and E13 in mouse (Miale and Sidman, 1961). We postulated that crossing the L7Cre line to a Vgatflox allele should eliminate Vgat expression early starting from late embryogenesis, and adult Purkinje cells should completely lack Vgat gene expression. To test this prediction, we compared conditional L7Cre;Vgatflox/flox mutant mice to Vgatflox/flox control littermates (these genotypes were compared throughout the study). In situ hybridization conducted in 4-week-old mice revealed that Vgat mRNA is abolished from adult L7Cre;Vgatflox/flox Purkinje cells, while Vgatflox/flox Purkinje cells strongly express the RNA (Fig. 1). We confirmed the cellular specificity of our approach by demonstrating that the absence of Vgat mRNA in Purkinje cells is accompanied by a persistence of expression in stellate cells, baskets cells, and Golgi cells, which are all cerebellar cortical interneurons that use VGAT for GABAergic signaling (stellate/basket cells = expression in the molecular layer, and Golgi cells = arrows in the granular layer of Fig. 1C,D).
Figure 1.

Purkinje cell-specific removal of Vgat. A, In control animals (Vgatflox/flox), all vermis Purkinje cells (pcl) express Vgat. Inset, Vgat expression in the hemispheres. B, L7Cre deletes Vgat from Purkinje cells. Inset, Loss of Purkinje cell Vgat expression in the hemispheres. The arrows point to the Purkinje cell layer. C, Higher power image from the box in A. D, Higher power image from the box in B. L7Cre;Vgatflox/flox mutant mice lack Vgat mRNA expression specifically in Purkinje cells. In L7Cre;Vgatflox/flox mice, Vgat mRNA is still expressed in Golgi cells (open arrows) in the granular layer (gl) and in stellate and basket cell interneurons in the molecular layer (ml). Lobules are labeled with Roman numerals (Larsell, 1970). Scale bars: A, B, 200 μm; C, D, 20 μm.
We next tested for protein expression using immunohistochemistry, and we found that VGAT was diminished in terminals surrounding the neurons of the cerebellar nuclei, the projection targets of Purkinje cell axons (Fig. 2). Quantification of VGAT-immunoreactive puncta confirmed that L7Cre;Vgatflox/flox mutant mice have significantly fewer VGAT-immunopositive terminals within the cerebellar nuclei compared with the control littermates (control = 13.05 puncta/100 μm2 ± 4.15 and mutant = 4.18 puncta/100 μm2 ± 2.78; p = 4.68 × 10−5; Fig. 2). To determine whether the remaining VGAT-immunoreactive puncta in the cerebellar nuclei belonged to Purkinje cells, we colabeled coronal cut tissue sections with the Purkinje cell marker CAR8 and VGAT. We found that in control Vgatflox/flox mice CAR8-immunoreactive Purkinje cell terminals had colocalized expression with VGAT (Fig. 2E; mean Pearson's correlation coefficient, r = 0.40), in contrast to the L7Cre;Vgatflox/flox mice, which showed distinct puncta for those expressing CAR8 compared with the remaining puncta that expressed VGAT (Fig. 2F; r = 0.07, and the difference between controls and mutants was significant, p < 0.0001). The remaining VGAT puncta in the cerebellar nuclei of L7Cre;Vgatflox/flox mice likely belong to local GABAergic inhibitory interneurons, and not to Purkinje cells. In accordance with the Vgat in situ data (Fig. 1), VGAT protein remained heavily expressed in the GABAergic terminals of cortical inhibitory interneurons in the molecular layer (control = 19.07 puncta/100 μm2 ± 7.38 and mutant = 23.85 puncta/100 μm2 ± 7.05; p = 0.27; Fig. 2). In addition, VGAT-expressing basket cell pinceaux remained prominent on the base of Purkinje cells in the mutant mice (Fig. 2G,H, asterisks). These data demonstrate that Vgat mRNA is deleted exclusively from Purkinje cells in L7Cre;Vgatflox/flox mutant mice and, accordingly, VGAT protein expression is eliminated from Purkinje cell terminals that contact neurons in the cerebellar nuclei. The other GABAergic neurons in the cerebellar cortex are not affected in the mutants as they still express VGAT in their terminals.
Figure 2.
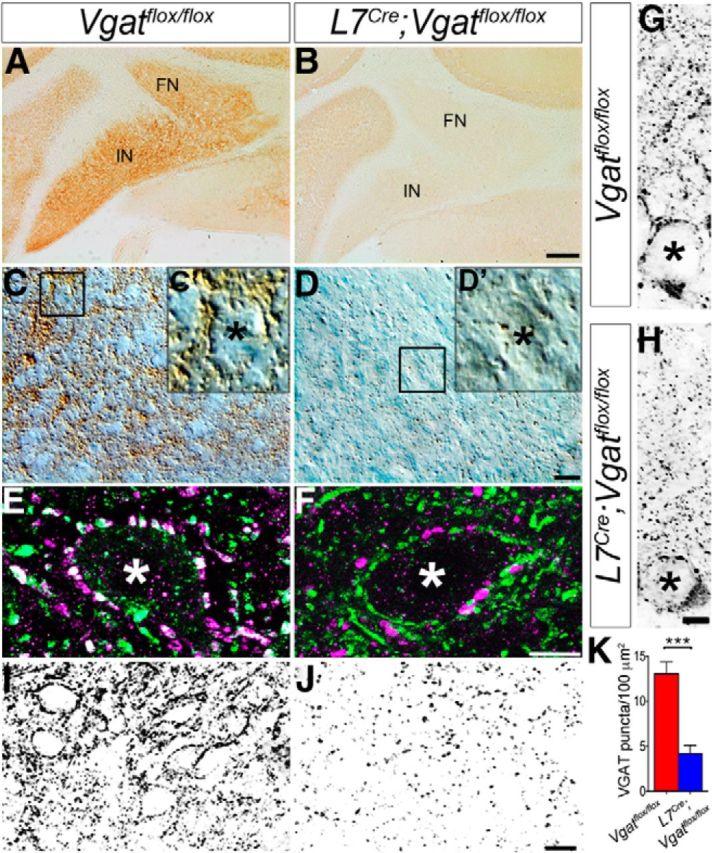
The number of VGAT-expressing axon terminals is reduced in the cerebellar nuclei. A, Coronal section of a Vgatflox/flox cerebellum showing VGAT expression in the cerebellar nuclei. B, Coronal section of an L7Cre;Vgatflox/flox cerebellum showing loss of VGAT expression in the cerebellar nuclei. FN, fastigial nucleus; IN, interposed nucleus. Scale bar, 100 μm. C, High-magnification differential interference microscopic (DIC) image of immunoperoxidase-stained cerebellar nuclei from Vgatflox/flox mice. D, High-magnification DIC image of the cerebellar nuclei from L7Cre;Vgatflox/flox mice. Scale bar, 10 μm. C′, Enlarged image from boxed region in C, showing cerebellar nuclei somata surrounded by VGAT-immunoreactive terminals. D′, Enlarged image from boxed region in D, showing reduced VGAT expression around the somata of the mutant cerebellar nuclei. E, Control cerebellum stained with the Purkinje cell marker CAR8 (green) and VGAT (magenta), showing colocalized puncta surrounding the soma of a cerebellar nuclear neuron. F, In the mutants, CAR8 and VGAT do not overlap. Scale bar, 5 μm. The asterisks in C′, D′, E, and F mark the somata of cerebellar nuclei neurons. G, Grayscale image of a Purkinje cell with VGAT-immunoreactive synapses in the molecular layer of Vgatflox/flox mice. H, Grayscale image of a Purkinje cell with VGAT-immunoreactive synapses in the molecular layer of L7Cre;Vgatflox/flox mice. The asterisks in G and H mark Purkinje cell somata. Scale bar, 5 μm. I, J, Grayscale images of immunostained cerebellar nuclei used for quantification of puncta in Vgatflox/flox (I) and L7Cre;Vgatflox/flox (J) mice. Each puncta was counted as a VGAT-immunoreactive synaptic terminal. Scale bar, 10 μm. K, Summary plot showing VGAT synapse density in the cerebellar nuclei of L7Cre;Vgatflox/flox mice compared with control Vgatflox/flox mice.
L7Cre;Vgatflox/flox mutant mice have impaired motor behavior
Defects in Purkinje cell development and survival cause severe impairments in motor function (Gerlai et al., 1996; Dusart et al., 2006; Gold et al., 2007; Lalonde and Strazielle, 2007). We characterized the basic motor behavior of our mutants because we anticipated that loss of Vgat and the resulting loss of Purkinje cell synaptic transmission would cause motor dysfunction. Open field visual inspection revealed that the mutant mice have difficulties maintaining a straight line of locomotion, and have poor balance, often falling to one side (Movie 1). In fact, in many instances they overcompensate and then fall to the other side. When they do this, both limbs on one of side of the body simultaneously extend outward. We observed the disequilibrium-like and limb extension phenotypes in 100% of the L7Cre;Vgatflox/flox mutant mice that were analyzed (n = 45). None of the L7Cre and L7Cre;Vgatflox/+ mice showed these behavioral defects (Movie 2).
A control Vgatflox/flox mouse and an L7Cre;Vgatflox/flox mutant mouse. Note the extended limbs of the mutant when it falls to its side.
Control L7Cre;Vgatflox/+ and L7Cre mice. Neither mouse exhibits motor behavior deficits.
To quantitatively assess the extent and severity of motor dysfunction in L7Cre;Vgatflox/flox mutant mice, we used two motor behavior paradigms: footprint analysis and accelerating rotarod. We found that L7Cre;Vgatflox/flox mice had significantly different stride lengths compared with control littermates (Vgatflox/flox = 5.449 ± 0.084 cm; L7Cre;Vgatflox/flox = 2.927 ± 0.063; p < 0.0001; n = 10 mice for each genotype; Fig. 3B). They also had significantly different stance widths (Vgatflox/flox = 3.94 ± 0.039 cm; L7Cre;Vgatflox/flox = 3.25 ± 0.044; p = 0.0006; n = 10 mice for each genotype; Fig. 3B). However, L7Cre;Vgatflox/flox mice do not have significantly different sway length (Vgatflox/flox = 2.71 ± 0.032 cm; L7Cre;Vgatflox/flox = 2.79 ± 0.0384; p = 0.5826; n = 10 mice for each genotype; Fig. 3B). We also found that L7Cre;Vgatflox/flox mutant mice performed poorly on an accelerating rotarod paradigm [Vgatflox/flox = 241.44 ± 11.81 s (n = 19) and L7Cre;Vgatflox/flox = 72.50 ± 4.47 s (n = 20); p = 4.5 × 10−6; Fig. 3C]. But interestingly, both control and mutant mice showed significant improvement over the nine trials as measured by comparing the differences using a repeated-measures ANOVA(Vgatflox/flox = F(8,144) = 15.69, p < 0.0001; L7Cre;Vgatflox/flox = F(8,152) = 5.202, p < 0.0001; Fig. 3C). We next examined whether “zonal” circuit structure is affected and how the in vivo firing properties of Purkinje cells and their target cerebellar nuclei neurons are altered in mice that do not have Purkinje cell neurotransmission.
Figure 3.

L7Cre;Vgatflox/flox mice exhibit severe motor impairments. A, Sample footprints from Vgatflox/flox and L7Cre;Vgatflox/flox mice. B, Quantification of footprints reveals significantly shorter stride lengths and stance widths in L7Cre;Vgatflox/flox mice compared with the control littermates. Sway length was not significantly different. C, Accelerating rotarod performance was significantly diminished in L7Cre;Vgatflox/flox mice compared with the control littermates; ***p < 0.001.
ZebrinII, HSP25, and afferent zones are mispatterned in L7Cre;Vgatflox/flox mutant mice
The cerebellum is compartmentalized into parasagittal compartments called zones (Fig. 4; Apps and Hawkes, 2009). The precision of zonal connectivity provides a structural framework for understanding how circuits operate during ongoing motor function and motor learning (Attwell et al., 1999; Wadiche and Jahr, 2005; Horn et al., 2010; Mostofi et al., 2010; Cerminara and Apps, 2011; Graham and Wylie, 2012). Although there has been progress in understanding how zones are genetically controlled during development (Croci et al., 2006; Sillitoe et al., 2008), there is still limited data about how activity in the cerebellar circuit influences the formation of zones into precisely wired functional circuits. To address this gap in our knowledge, we used zebrinII zonal patterning as a readout for how silencing Purkinje cell GABAergic neurotransmission might affect compartmental architecture. We found that adult L7Cre;Vgatflox/flox mutant mice had the correct number of zebrinII zones (Figs. 4, 5; n = 4 controls and 6 mutants). However, compared with control littermates, the boundaries of the zones in the mutant mice were poorly defined (Fig. 4). ZebrinII zonal boundaries were defective in both the posterior (Fig. 4) and anterior lobules (Fig. 5). In lobule VIII of the posterior zone, the medial and lateral edges of the P2+ and P3+ zones are not sharply delineated, and they blend into the neighboring zebrinII P1− and P2− zones (Fig. 4). This is especially apparent when the sections are stained with both zebrinII and PLCβ4, which labels zebrinII-negative zones (Fig. 4). In the mutants some Purkinje cells express both markers (Fig. 4E,F). As for zebrinII, the PLCβ4 zone boundaries are poorly defined (Fig. 4C–E). Moreover, the overall level of zebrinII and PLCβ4 expression is diffuse in L7Cre;Vgatflox/flox mutants compared with control littermates (Fig. 4). The alteration in zebrinII patterning is less severe in the anterior lobules. For example, in lobule III of the anterior zone the zebrinII P2+ zones lack crisp lateral edges, although similar to control mice the center-to-center distance between P1+ and P2+ is ∼500 μm (Fig. 5). Of a total of six stained L7Cre;Vgatflox/flox mice, we found only one with mild posterior zebrinII zonal defects. Cell counts of zebrinII-expressing cells revealed that within the P1+, P1−, and P2+ zones of lobule VIII, 67.9 ± 2.6% of Purkinje cells expressed zebrinII in control mice, whereas in the mutants 86.4 ± 6.4% expressed zebrinII (p = 0.0498). These results show that despite the presence of a zebrinII map, the zone boundaries within the map are not all sharply refined, especially in the posterior lobules where there are more zebrinII-expressing cells in the mutant mice. Phenotypic differences between anterior and posterior lobules are likely due to differences in the onset of L7Cre expression (Lewis et al., 2004), which reaches higher levels earlier in the posterior lobules (for further discussion about the differences see below and also Fig. 6).
Figure 4.
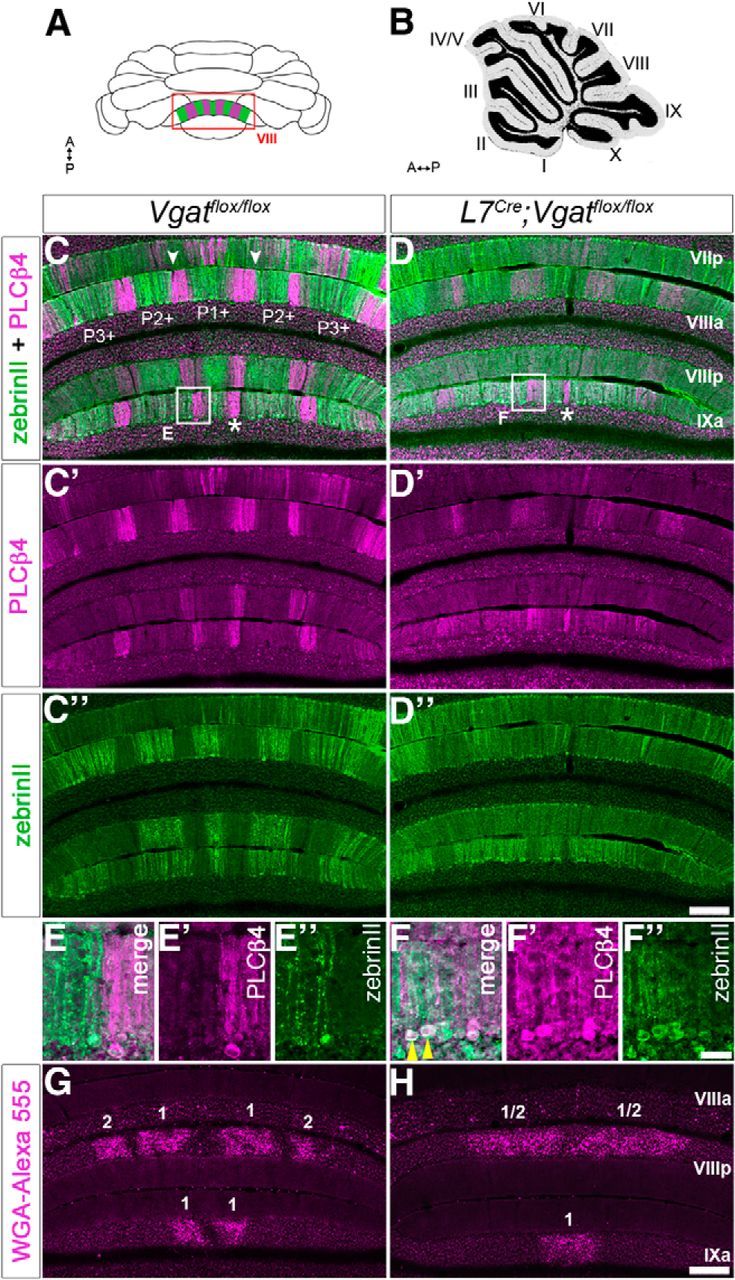
Zonal organization is altered in L7Cre;Vgatflox/flox mice (posterior lobules). A, Schematic whole-mount diagram of the cerebellum showing zebrinII and PLCβ4 expression patterns in lobules VIII and IX. A, anterior; P, posterior. B, Sagittal schematic of the cerebellum. C, Coronal section of a Vgatflox/flox cerebellum showing zebrinII (green) and PLCβ4 (magenta) expression. C′, C″, Separated panels of PLCβ4 and zebrinII, respectively, from C. ZebrinII-positive zones are marked as P1+, P2+, and P3+ using the standard zebrinII zone nomenclature. Examples of well defined zone boundaries are marked with arrowheads. D, ZebrinII and PLCβ4 zones in the L7Cre;Vgatflox/flox mice. D′, D″, Separated panels of PLCβ4 and zebrinII, respectively, from D. Note that some zones are obvious in the mutant, as indicated by the asterisks in C and D. Scale bar, 200 μm. E, High-power image of the box in C. E′, E″, Separated panels of PLCβ4 and zebrinII, respectively, from E. F, High-power image of the box in D. F′, F″, Separated panels of PLCβ4 and zebrinII, respectively, from F. Scale bar, 20 μm. G, Coronal section of a Vgatflox/flox cerebellum showing the zonal organization of spinocerebellar mossy fiber inputs to the granular layer of posterior lobules VIII and anterior IX. Mossy fiber terminal bands are numbered. H, Coronal section of a L7Cre;Vgatflox/flox cerebellum showing poor zonal organization of mossy fiber afferents in the posterior lobules. Scale bar, 200 μm.
Figure 5.
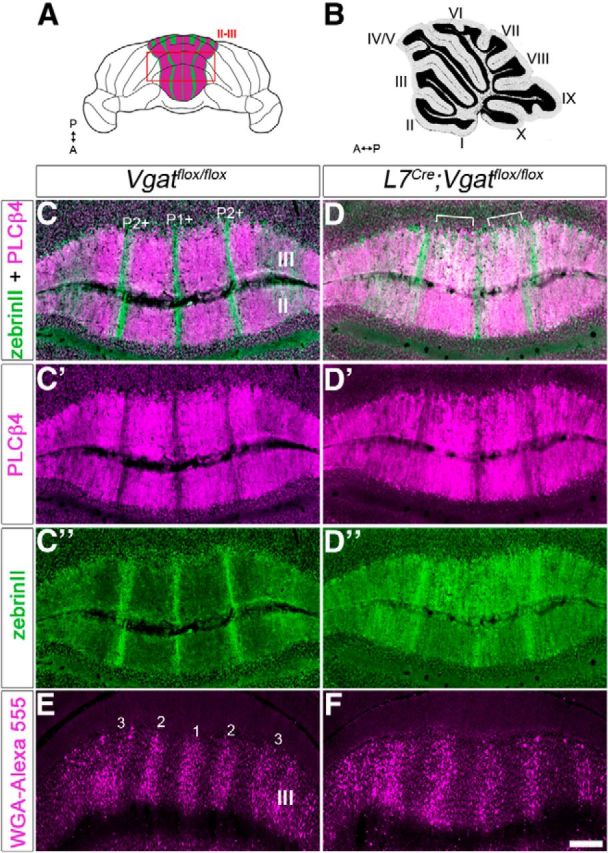
Zonal organization is altered in L7Cre;Vgatflox/flox mice (anterior lobules). A, Schematic whole-mount diagram of the cerebellum showing zebrinII and PLCβ4 expression in the anterior lobules. A, anterior; P, posterior. B, Sagittal schematic of the cerebellum. C, Coronal section cut through a Vgatflox/flox cerebellum showing zones in the anterior lobules II–III. ZebrinII-positive zones are marked as P1+ and P2+. D, Coronal section cut through an L7Cre;Vgatflox/flox cerebellum showing poor zonal boundaries. “White” staining represents overlapping zebrinII/ PLCβ4 expression (marked with brackets). C′, C″, D′, D″, Separated panels from C and D. E, Coronal section of a Vgatflox/flox cerebellum showing the zonal organization of spinocerebellar mossy fiber inputs in the anterior lobules. Mossy fiber terminal bands are numbered. F, Coronal section showing spinocerebellar zones in an L7Cre;Vgatflox/flox mutant mouse. Scale bar, 250 μm.
Figure 6.
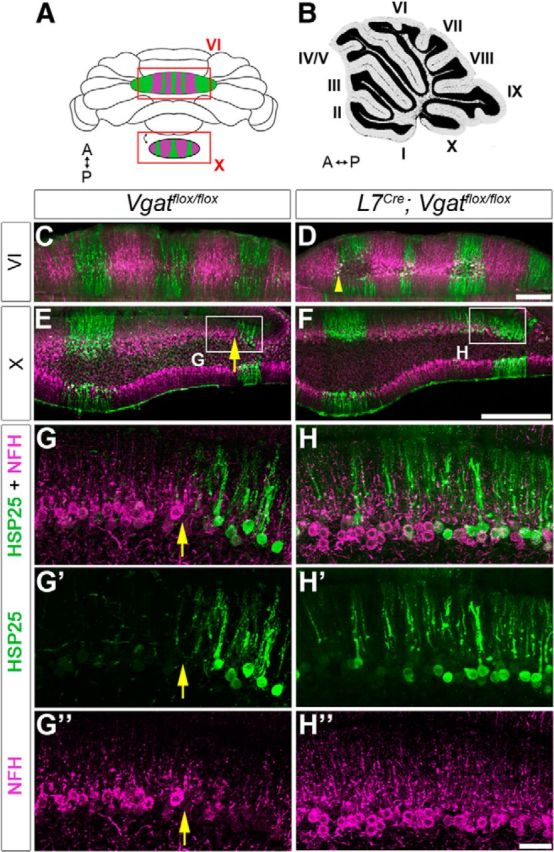
Zonal organization is altered in L7Cre;Vgatflox/flox mice (central and nodular lobules). A, B, Whole-mount (A) and sagittal (B) schematics of the cerebellum. A, anterior; P, posterior. C, E, Coronal tissue sections from a control mouse cut through lobule VI (C) and lobule X (E) and stained with HSP25 (green) and NFH (magenta). D, F, Coronal tissue sections from an L7Cre;Vgatflox/flox mutant mouse cut through lobule VI (D) and lobule X (F). Scale bars: D, F, 200 μm. G, H, Enlarged images from the boxes in E and F. G′, G″, H′, H″, Separated channels for NFH and HSP25 in lobule X. The boundaries of HSP25 zones are poorly defined in the mutant, compared with the obvious boundary in control mice (yellow arrows). Scale bar, 50 μm.
ZebrinII is uniformly expressed in lobules VI–VII and IX–X. Therefore, to determine whether loss of Purkinje cell neurotransmission alters zone refinement in all lobules we used HSP25 as a marker. In control mice, HSP25 reveals zones in the vermis of lobules VI–VII and IX–X (Armstrong et al., 2000, 2001), and these zones are complementary to the expression of NFH (Demilly et al., 2011). In L7Cre;Vgatflox/flox mutants, HSP25 and NFH zones in lobule VI are only subtly affected (Fig. 6C,D, arrowhead) compared with lobule X (Fig. 6E,F). In lobule X of L7Cre;Vgatflox/flox mutants, we found that the boundaries of HSP25 zones were not sharply defined (arrows in Fig. 6 mark the boundary in controls; n = 6 mice for each genotype) and although the width of the zones varies upon location in both control and mutants, it is significantly increased after loss of Purkinje cell neurotransmission [Vgatflox/flox = 191.38 ± 37.33 μm (n = 3) and L7Cre;Vgatflox/flox = 469.92 ± 176.42 μm (n = 4); p = 0.047; Fig. 6]. NFH zones were also extended beyond their normal boundaries (Fig. 6G″, H″). The regional differences between central and nodular lobules likely arise because, temporally, the cellular maturation program in the central lobules lags behind other regions of the cerebellum (Altman and Bayer, 1997; White and Sillitoe, 2013a,2013b). In this particular mouse line L7Cre expression during late embryogenesis is more intense posteriorly than it is anteriorly, and then during postnatal development the anterior lobules start to express the transgene more intensely (Lewis et al., 2004). The central lobules are least affected in our data because of late morphogenesis whereas the anterior lobules are not severely altered because of late Cre onset. Therefore, the severity in zone defects = posterior/nodular lobules > anterior lobules > central lobules. Our data also indicate that regardless of the molecular identity of a Purkinje cell zone, the sharpness of zones throughout the cerebellum relies on intact Purkinje cell inhibitory neurotransmission. Next we decided to examine whether the topography of the cerebellar input pathways is altered since the patterning of the target domains, the Purkinje cell zones, is disrupted.
Mossy fibers carry sensory signals to the cerebellum from more than three dozen brainstem and spinal cord nuclei (Fu et al., 2011). Among the largest of these pathways is the spinocerebellar tract, which emerges from all levels of the spinal cord to supply the cerebellum with proprioceptive and fine touch signals (Bosco and Poppele, 2001). Spinocerebellar mossy fibers terminate in zones that respect the zebrinII pattern (Gravel and Hawkes, 1990; Reeber et al., 2012). During embryonic and early postnatal development, mossy fibers directly contact Purkinje cells (Mason and Gregory, 1984; Takeda and Maekawa, 1989; Manzini et al., 2006; Kalinovsky et al., 2011). Although the basic topography of the mossy fiber terminal projection plan is shaped by a genetic code (Sillitoe et al., 2010), and perhaps under the direct control of Purkinje cells (Sotelo, 2004), it remains unknown how the adult afferent zones attain mature patterns with clearly segregated terminal field domains (Reeber et al., 2011). We wondered whether Purkinje cell neurotransmission might also play a role in defining the precision of zonal connectivity of mossy fiber projections. To test this, we injected WGA-Alexa 555 into the lower thoracic upper lumbar spinal cord of adult L7Cre;Vgatflox/flox mutants and littermate controls to map the topography of the spinocerebellar tract. The spinocerebellar tract is an ideal model for understanding zonal topography, because its map has a relatively simple organization that has been extensively described (Sillitoe et al., 2010; Reeber et al., 2011). Compared with controls, the mutant map was poorly resolved such that individual clusters were not properly segregated. Spinocerebellar fibers were therefore ectopically located in zones that are normally occupied by adjacent forelimb cuneocerebellar terminals (Fig. 4; Gebre et al., 2012). However, the anterior–posterior targeting, which is normally restricted to lobules I–V and VIII–IX, was intact (Sillitoe et al., 2010; Reeber et al., 2011; Gebre et al., 2012; Fig. 4). The lack of clear afferent zones is reminiscent of the lack of a clear zebrinII pattern, providing support for our hypothesis that intact Purkinje cell synaptic transmission is essential for afferent map formation. Moreover, in accordance with the zebrinII phenotype, the anterior termination domain (lobules I–V; Fig. 5) was less affected than the posterior domain (lobules VIII–IX). This result indicates that the patterning of early arriving anterior axons is subtly altered, whereas the later arriving posterior axons are severely mispatterned in the absence of GABAergic Purkinje cell activity (Arsénio Nunes and Sotelo, 1985).
The defective patterning of Purkinje cell zones and mossy fiber topography, and their apparent temporal sensitivity, made us wonder whether other fundamental anatomical features, such as the molecular distribution patterns in different cell types and lobule morphology, develop properly when Purkinje cell inhibitory neurotransmission is blocked.
Purkinje cell-specific deletion of Vgat does not alter the gross morphology or the molecular expression patterns in different cell types of the cerebellum
To examine the gross anatomy of the L7Cre;Vgatflox/flox mutant mice, we began by recording the whole-brain weight and cerebellar weight, as well as body weight (Fig. 7). There was a slight but significant difference in body weight in mutant mice (Vgatflox/flox = 20.783 ± 0.452 g and L7Cre;Vgatflox/flox = 17.904 ± 0.607 g; p = 0.0055; n = 6 mice for each condition; Fig. 7). There was no statistical difference in overall brain weight (Vgatflox/flox = 0.4795 ± 0.0067 g and L7Cre;Vgatflox/flox = 0.4716 ± 0.0051 g; p = 0.375; n = 6 mice for each condition; Fig. 7) or cerebellar weight (Vgatflox/flox = 0.0588 ± 0.0005 g and L7Cre;Vgatflox/flox = 0.0578 ± 0.0007 g; p = 0.2435; n = 6 mice for each condition; Fig. 7). We next examined cerebellar lobule patterning using fluorescent Nissl staining on sagittal tissue sections to compare all lobules in the same cutting plane. The normal mouse cerebellum, like all mammals, contains 10 major lobules (Larsell, 1952, 1970) that are temporally generated (Sudarov and Joyner, 2007). Similar to littermate controls, all 10 lobules were present in adult L7Cre;Vgatflox/flox mutant mice, and their shape and size were unaltered (Fig. 7). We next tested whether loss of Vgat might induce more subtle alterations in the architecture of the cerebellum. We measured the thickness of the molecular layer, which is a sensitive measure for developmental and disease-associated defects that disrupt Purkinje cell dendrite size and/or the placement of Purkinje cells within a perfect monolayer (Hansen et al., 2013). We did not detect any difference in the molecular layer thickness (Vgatflox/flox = 150.316 ± 2.119 μm and L7Cre;Vgatflox/flox = 152.334 ± 4.165 μm; p = 0.669; n = 3 mice for each genotype; Fig. 7). These data suggest that Purkinje cell inhibitory neurotransmission is dispensable for establishing the gross architecture and pattern of the lobules, and it is not required for the proper layering or the apical-basal expansion of the three layers of the cerebellar cortex.
Figure 7.

The gross morphology of the cerebellum is unaltered after blocking Purkinje cell GABAergic neurotransmission. A, A mid-sagittal section from a control cerebellum. Purkinje cells are labeled with calbindin (magenta) and the granular layer is particularly highlighted by the fluorescent Nissl stain (green). B, A mid-sagittal section from a mutant cerebellum. Scale bars: A, B, 200 μm. C, An example image showing how the cerebellar cortical measurements were taken from within the primary fissure. Scale bar, 200 μm. D, Quantification of body weight, brain weight, cerebellar weight, and molecular layer thickness. Ml, molecular layer; pcl, Purkinje cell layer; gl, granular layer; ***p < 0.01.
Since each of the major cerebellar cell types is generated in a spatial and temporal manner (Sudarov et al., 2011), and because the molecular expression of specific cell populations might be affected more than others (Figs. 4, 5), we wondered whether loss of circuit activity could negatively impact the molecular expression profile across the cerebellar circuit. To determine whether the major cerebellar cell types could be identified based on their molecular expression, we performed an in-depth immunohistochemical analysis in control mice and L7Cre;Vgatflox/flox mutant mice to detect whether blocking GABA neurotransmission induces generalized patterning defects. Using a panel of cell-specific antibodies we found that the major cell types in the mutants had a protein expression profile identical to controls and both mossy and climbing fiber afferent terminals projected extensively in the mutants (Fig. 8). Moreover, the expression of calbindin, which is localized to all Purkinje cells in the normal cerebellum (De Camilli et al., 1984; Celio, 1990), was indistinguishable between control and mutant mice suggesting that loss of Vgat does not cause Purkinje cell degeneration (Fig. 8A,A′). Given that L7Cre;Vgatflox/flox mice contain all the major cell types with expression patterns showing a relatively normal tri-laminar distribution (Fig. 8), we next tested how activity in these cells is altered in adult mice.
Figure 8.

Cellular expression patterns are unaltered in L7Cre;Vgatflox/flox mutant mice. A, A′, Staining with calbindin reveals normal Purkinje cell organization and a lack of degeneration. B, B′, Parvalbumin is expressed by inhibitory neurons in the cerebellum. C, C′, NFH is expressed in Purkinje cells and basket cells. D, D′, VGLUT1 is expressed by mossy fibers and parallel fibers. E, E′, VGLUT2 is expressed in climbing fibers that terminate in the molecular layer and mossy fibers that terminate in the granular layer. F, F′, CART is heavily expressed by climbing fibers in lobule X. G, G′, GABARα6 is expressed by granule cells. H, H′, Neurogranin is expressed by Golgi cells in the adult cerebellum. I, I′, Calretinin is expressed by unipolar brush cells in lobule X. J, All parts are taken from midline sagittal sections. K, The images were captured from regions within the primary fissure between lobule V and VIa (upper box) except for F, F′, I, and I′, which are from lobule X (lower box). Scale bar, 20 μm applies to all parts. Ml, molecular layer; pcl, Purkinje cell layer; gl, granular layer.
Deletion of Purkinje cell Vgat induces abnormal firing activity in the cerebellar nuclei
Purkinje cells project to two target cell populations: the cerebellar nuclei in the core of the cerebellum and the vestibular nuclei in the dorsal brainstem (Sillitoe et al., 2011; Fig. 9). We predicted that loss of Purkinje cell GABA neurotransmission might cause an increase in the firing rate of the target cells. To test this prediction, we measured cerebellar nuclei activity by recording the spike-firing properties of neurons in the interposed and dentate nuclei in vivo (Fig. 9). Using extracellular recordings, we demonstrate that, indeed, loss of GABA neurotransmission at Purkinje cell to cerebellar nuclei synapses causes an increase in firing frequency, but only in 8/37 recorded neurons (rate increases >2 SDs above the control mean; Fig. 9; 21.6% of the total cells recorded). Interestingly, although on average the overall frequency as a population was not significantly increased [Vgatflox/flox = 34.603 ± 3.433 Hz (n = 21) and L7Cre;Vgatflox/flox = 39.645 ± 3.571 Hz (n = 37); p = 0.239; Fig. 9], separating the mutant cells based on firing pattern unveiled specific firing defects. We found that mutant cerebellar nuclei neurons could be divided into two groups based on CV, which is a measure of firing regularity. An overall comparison of CV between control cells and mutant cells reveals a significant CV increase in the mutants, which indicates a decrease in regularity [Vgatflox/flox = 0.457 ± 0.053 (n = 8) and L7Cre;Vgatflox/flox = 1.05 ± 0.264 (n = 7); p = 0.0338; Fig. 9]. In mutants, 10/37 nuclear neurons showed an increase in irregularity in spiking >2 SDs of the control mean CV (Fig. 9). When spike-firing rate and regularity were directly compared within the mutant cell population, we resolved a low CV group with a high firing frequency [Vgatflox/flox = 34.603 ± 3.433 Hz (n = 21) and Low CV L7Cre;Vgatflox/flox = 47.531 ± 3.733 Hz (n = 27); p = 0.0142; Fig. 9] and a high CV group with a low firing frequency [Vgatflox/flox = 34.603 ± 3.433 Hz (n = 21) and High CV L7Cre;Vgatflox/flox = 21.516 ± 5.017 Hz (n = 10); p = 0.0454; Fig. 9]. The CV of the Low CV and High CV groups are each significantly different from the control [Vgatflox/flox = 0.457 ± 0.053 (n = 21) and Low CV L7Cre;Vgatflox/flox = 0.266 ± 0.038 Hz (n = 10); p = 0.0047; Fig. 9] and [Vgatflox/flox = 0.457 ± 0.053 (n = 21) and High CV L7Cre;Vgatflox/flox = 3.159 ± 0.583 (n = 27); p = 0.0012; Fig. 9]. Analysis of the firing pattern with CV2, which measures the similarity of neighboring ISIs, revealed an intrinsic regularity of the mutant's spike activity, with neighboring ISIs tending to be more similar than in control mice [CV2 Vgatflox/flox = 0.396 ± 0.028 (n = 21) and CV2 L7Cre;Vgatflox/flox = 0.177 ± 0.015 (n = 37); p = 1.17 × 10−7; Fig. 9], regardless of mutant cell grouping [CV2 Vgatflox/flox = 0.396 ± 0.028 (n = 21) and CV2 Low CV L7Cre;Vgatflox/flox = 0.152 ± 0.014 (n = 10); p = 1.52 × 10−8; Fig. 9] and [CV2 Vgatflox/flox = 0.396 ± 0.028 (n = 21) and CV2 High CV L7Cre;Vgatflox/flox = 0.246 ± 0.031 (n = 27); p = 0.00148; Fig. 9]. The lower CV2 suggests that the cerebellar nuclear cells in the mutants have a stronger tendency for rhythmic firing than cells of control mice. This was confirmed by autocorrelation analysis, which shows a flat histogram for the spike train autocorrelation of control mice but multiple side peaks for the spike trains of mutant mice (Fig. 9E). The first side peaks in mutant autocorrelation histograms are centered at ∼15 ms, signifying a high-frequency occurrence of ISIs of that duration, corresponding to a preferred spike rhythm of ∼66 Hz.
Figure 9.
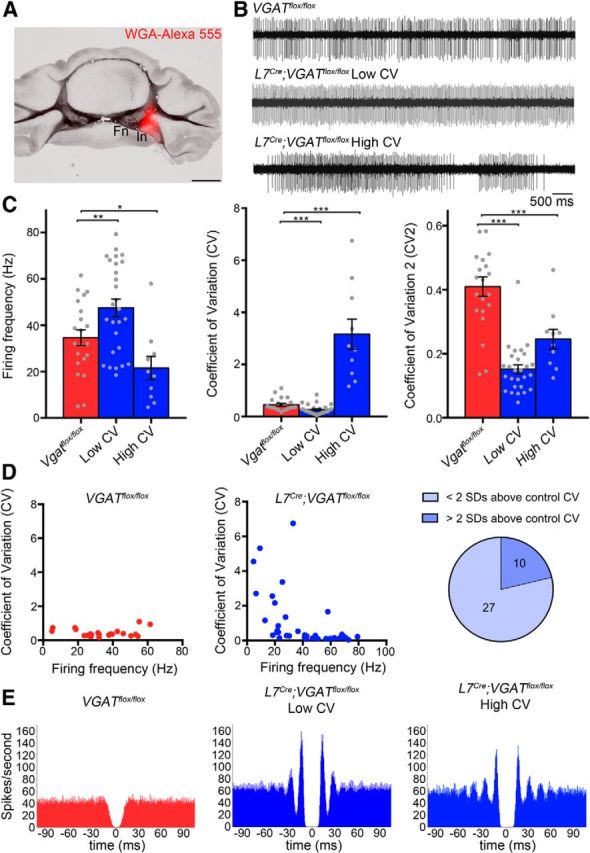
L7Cre;Vgatflox/flox mutant mice exhibit abnormal firing of cerebellar nuclei neurons. A, An example of a cerebellar nuclei recording site marked by tracer injection. B, Sample raw in vivo electrophysiology traces from control and mutant cerebellar nuclei neurons. C, Quantification of firing frequency, CV and CV2 of control and mutant cerebellar nuclei neurons. D, Correlation between CV and firing frequency. The pie graph shows the percentage of L7Cre;Vgatflox/flox cells either less than or greater than 2 SDs above the control CV. E, Autocorrelograms of control and mutant nuclear cell spike trains reveal a flat autocorrelation histogram for control mice but multiple, equidistant side peaks with the primary peak centered at ∼15 ms for mutant mice; *p < 0.05, **p < 0.01, and ***p < 0.001.
Purkinje cell spike activity is altered in L7Cre;Vgatflox/flox mutant mice
The cerebellar circuit consists of a tri-synaptic loop from the Purkinje cell to cerebellar nuclei neurons to inferior olive neurons and back to Purkinje cells (the cortico-nucleo-olivary circuit; Fig. 10D, Libster and Yarom, 2013). This loop is thought to maintain an equilibrium in the circuit (Kenyon et al., 1998) in that decreased Purkinje cell firing should result in a disinhibition of the inhibitory nucleo-olivary pathway and decrease olivary neuron firing, resulting in decreased complex spike firing in Purkinje cells. Based on recent data (Chaumont et al., 2013; Witter et al., 2013), we predicted that eliminating GABAergic signals from Purkinje cells would feed back onto Purkinje cells and as a consequence alter their own firing properties. To test this possibility, we measured Purkinje cell firing frequency and Purkinje cell firing patterns by recording from single units in vivo. We first analyzed simple spike-firing frequency and found that compared with control mice the overall firing rate was unchanged in L7Cre;Vgatflox/flox mutant mice (Vgatflox/flox = 44.416 ± 2.70 Hz, n = 27 and L7Cre;Vgatflox/flox = 47.019 ± 3.37 Hz, n = 27; p = 0.522; Fig. 10B). However, 12/27 Purkinje cells from L7Cre;Vgatflox/flox mice had a significant increase in the ISI CV (calculated as at least 2 SDs above the control CV), indicating that in 44% of the recorded mutant cells the regularity in simple spike firing was decreased compared with the more regular spike firing seen in the cells of control mice (Vgatflox/flox = 0.419 ± 0.043 and L7Cre;Vgatflox/flox = 1.942 ± 0.497; p = 0.0055; Fig. 10B). Analysis of Purkinje cell CV2 revealed no significant difference between control and mutant littermates (Vgatflox/flox = 0.376 ± 0.022 and L7Cre;Vgatflox/flox = 0.408 ± 0.025; p = 0.355; Fig. 10B). Therefore, despite the increased prevalence of long pauses in Purkinje cells from L7Cre;Vgatflox/flox mice that cause the increase in CV, the intrinsic regularity (CV2) and rhythmicity were unchanged. Unlike the High CV and Low CV groups of the cerebellar nuclei, the High CV and Low CV Purkinje cell groups do not show any significant differences between any measures other than CV. The CV is statistically different between the two groups (L7Cre;Vgatflox/flox Low CV = 0.352 ± 0.0458 and L7Cre;Vgatflox/flox High CV = 3.922 ± 0.818; p = 0.00112). However, simple spike Frequency is not statistically different (L7Cre;Vgatflox/flox Low CV = 50.005 ± 5.232 Hz and L7Cre;Vgatflox/flox High CV = 43.288 ± 3.819 Hz; p = 0.310).
Figure 10.
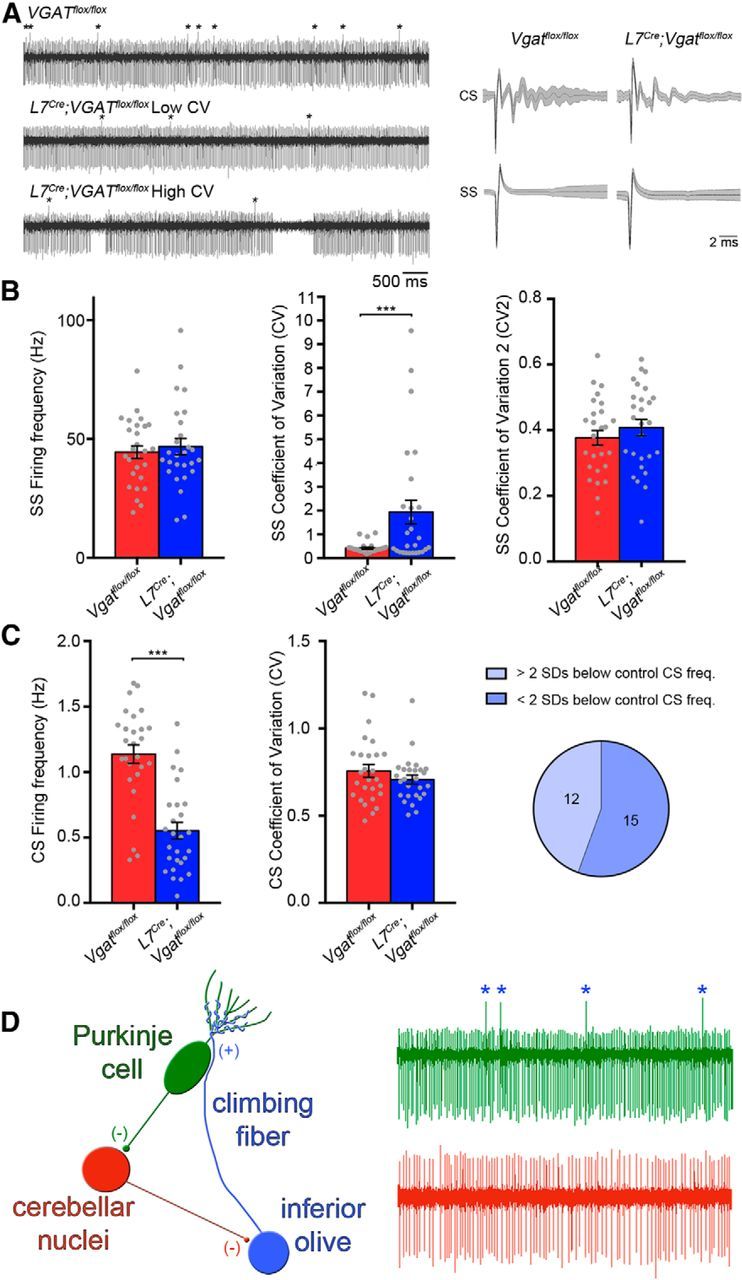
L7Cre;Vgatflox/flox mutant mice exhibit abnormal Purkinje cell activity. A, Sample raw in vivo electrophysiology traces from control and mutant Purkinje cells. Complex spikes(CS) are indicated with asterisks. Right column shows examples of averaged complex and simple spikes (SS) from control and mutant cells, demonstrating the reliability in the Purkinje cell signal for measuring spikes in vivo. B, Quantification of firing frequency, CV, and CV2 of simple spikes. The pie graph shows the percentage of cells either less than or greater than 2 SDs above the control CV. C, Quantification of firing frequency and CV of complex spikes. The pie graph shows the percentage of cells either greater than or less than 2 SDs below the control frequency. D, A schematic of the tri-synaptic loop and the spike waveforms that reflect each cellular component; ***p < 0.001.
In contrast to simple spike firing, the average complex spike-firing frequency was significantly decreased (Vgatflox/flox = 1.14 ± 0.071 Hz and L7Cre;Vgatflox/flox = 0.540 ± 0.066 Hz; p = 5.552 × 10−8; Fig. 10C) with 12/27 cells firing complex spikes at a frequency of >2 SDs below the control mean (Fig. 10C). The complex spike CV, however, was not significantly different between control and L7Cre;Vgatflox/flox mutant mice (Vgatflox/flox = 0.756 ± 0.036 Hz and L7Cre;Vgatflox/flox = 0.770 ± 0.068 Hz; p = 0.864; Fig. 10C). The High CV and Low CV Purkinje cell groups do not show any significant differences between any complex spike measures. Complex spike frequency is not statistically different (L7Cre;Vgatflox/flox Low CV = 0.533 ± 0.107 Hz and L7Cre; Vgatflox/flox High CV = 0.549 ± 0.071 Hz; p = 0.906) and neither is complex spike CV (L7Cre;Vgatflox/flox Low CV Purkinje cell = 0.853 ± 0.124 and L7Cre;Vgatflox/flox High CV Purkinje cell = 0.699 ± 0.027; p = 0.248).
The overall variability in L7Cre;Vgatflox/flox Purkinje cell firing may be explained in part by the normal variability in intrinsic Purkinje cell firing (Kim et al., 2012; Snow et al., 2014). Despite the loss of Vgat in all Purkinje cells, only certain populations have significant firing defects, whereas other populations are either less affected or even statistically indistinguishable from the control cells. We next sought to determine whether the changes in Purkinje excitability and the mispatterning of Purkinje cell zones are linked by a common underlying defect.
Ectopic expression of tyrosine hydroxylase in zones of Purkinje cells indicates calcium dysregulation in L7Cre;Vgatflox/flox mutant mice
We examined tyrosine hydroxylase expression to determine whether a relationship exists between zonal mispatterning and defective Purkinje cell firing. During normal mouse development, tyrosine hydroxylase is transiently expressed in Purkinje cells (Hess and Wilson, 1991), but as circuit formation finalizes, it is downregulated, coincident with emerging zebrinII zones that mark pattern maturity (Armstrong and Hawkes, 2000). Mutations in calcium channels, specifically, result in zones of ectopic tyrosine hydroxylase expression (Fureman et al., 1999, 2003; Jeong et al., 2001; Sawada et al., 2010), and also a failure to sharpen zebrinII zones (Miyazaki et al., 2012). Tyrosine hydroxylase is a measure of altered Purkinje cell excitability (Fureman et al., 1999), which depends heavily on calcium, and without proper calcium control complex spike firing is obstructed and ataxia ensues (Gao et al., 2012; Todorov et al., 2012). Strikingly, in ataxic L7Cre;Vgatflox/flox mice, we also found strong tyrosine hydroxylase expression in zones of ectopic Purkinje cells (Fig. 11). Interestingly, consistent with the zebrinII mispatterning and the mistargeting of posterior mossy fibers, we observed the most intense and widespread tyrosine hydroxylase expression in the posterior lobules (Fig. 11), and mainly within zebrinII-expressing zones (Fig. 11G,H). Although in the anterior lobules, ectopic expression was similarly restricted to narrow zebrinII-like zones (Fig. 11). The restriction of ectopic tyrosine hydroxylase expression to zebrinII zones, even though other molecular zones are altered (Fig. 6), is reminiscent of several disease models that have preferential degeneration of particular zonal patterns (Sarna and Hawkes, 2003). Our data are also intriguing because tyrosine hydroxylase zones are used as a zonal molecular readout for calcium dysregulation (Sawada and Fukui, 2001), which could be one mechanism that contributes to poor zone refinement in the absence of Purkinje cell Vgat.
Figure 11.
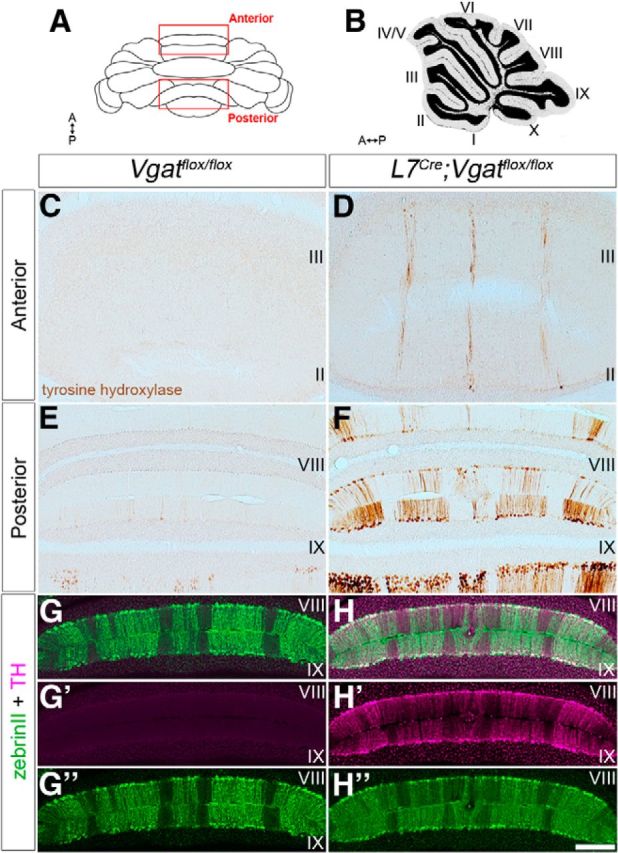
L7Cre;Vgatflox/flox mutant Purkinje cells express ectopic tyrosine hydroxylase (TH). A, Schematic diagram of the cerebellum shown in whole mount. A, anterior; P, posterior. B, Sagittal schematic of the cerebellum. The lobules are numbered with Roman numerals. C, Tyrosine hydroxylase expression was not detected in the anterior lobules of control mice. D, L7Cre;Vgatflox/flox mutant mice ectopically express tyrosine hydroxylase in narrow zebrinII-like zones. E, The posterior lobules of control mice have weak tyrosine hydroxylase expression in a subset of Purkinje cells. F, L7Cre;Vgatflox/flox mice heavily express tyrosine hydroxylase throughout the posterior cerebellum in a striking array of zones. G, H, ZebrinII and tyrosine hydroxylase merged images from control and mutant mice, respectively. G′, H′, Tyrosine hydroxylase expression in control and L7Cre;Vgatflox/flox mutant mice, respectively. G″, H″, ZebrinII expression in control and L7Cre;Vgatflox/flox mutant mice, respectively. Scale bar, 200 μm.
Discussion
We used the cell-specific expression of the L7 gene to drive Cre recombinase expression in Purkinje cells to remove exon II of the Vgat gene (Tong et al., 2008). This deletion selectively blocks Purkinje cell GABA neurotransmission and causes ataxia. Analysis of the conditional mutant mice revealed that Purkinje cell inhibitory function is essential for patterning Purkinje cell molecular zones and spinocerebellar afferent topography. Our data also show that tyrosine hydroxylase is upregulated in Purkinje cells, suggesting that altered excitability after blocking Purkinje cell signaling is in part zonally restricted. Because Purkinje cell function is central to the pathogenesis of several diseases including ataxia (Orr, 2012), dystonia (Calderon et al., 2011; Wilson and Hess, 2013), and tuberous sclerosis (Tsai et al., 2012; Reith et al., 2013), our approach presents a viable strategy for dissecting the normal function of Purkinje cells and their pathophysiology in motor and non-motor developmental disorders (D'Angelo and Casali, 2012).
Previous studies have demonstrated essential roles for genes such as engrailed1 (Sillitoe et al., 2008), engrailed2 (Kuemerle et al., 1997; Baader et al., 1999; Sillitoe et al., 2008), and Ebf2 (Croci et al., 2006) during zone formation. In contrast, in vitro assays suggested that neural activity does not influence zebrinII compartmentation (Seil et al., 1995). Our current data suggest that at least some aspects of zone patterning are dependent on synaptic neurotransmission in vivo. We found that in L7Cre;Vgatflox/flox mice zones failed to form refined boundaries with sharp edges that clearly differentiate Purkinje cells of different molecular phenotypes. The pattern of mossy fiber termination zones was also poorly separated into defined compartments. Interestingly, the defective mossy fiber patterns were reminiscent of the wild-type early postnatal pattern (Sillitoe et al., 2010; Reeber et al., 2011), suggesting that neural activity is essential for zonal patterning to progress during postnatal development. It is interesting that our data are consistent with the results of Tolbert et al. (1994). They found that chronically blocking cerebellar cortical activity with the NMDA antagonist APV or the GABA agonist muscimol, both of which would reduce Purkinje cell neurotransmission, prevented spinocerebellar zone refinement (Tolbert et al., 1994). We propose that genetic cues are essential for establishing the correct pattern of zones, whereas inhibitory Purkinje cell neurotransmission shapes zonal connectivity by delineating zone boundaries.
Support for a role of neural activity in zonal patterning has also been shown in mice containing a Purkinje cell-specific deletion of the CaV2.1 P/Q-type Ca2+ channel, which causes defects at zebrinII zonal boundaries (Miyazaki et al., 2012). CaV2.1 is a major source of Ca2+ transients that contribute to the firing and waveform shape of the Purkinje cell action potential. Similar to our mutants its loss results in poorly defined zebrinII boundaries and ectopic tyrosine hydroxylase expression (Figs. 4, Fig. 11). Our data, however, show that the ectopic tyrosine hydroxylase expression can be triggered by feedback defects at the circuit level and not only by channel mutations within the Purkinje cell. At the cellular level, in tottering mutants ectopic tyrosine hydroxylase expression was originally attributed to an increase in Ca2+ levels (Sawada and Fukui, 2001). In contrast, in our L7Cre;Vgatflox/flox mice the lower complex spike activity may be associated with low Ca2+ levels. But, since Purkinje cells in gain-of-function and loss-of-function tottering alleles express ectopic tyrosine hydroxylase (Miki et al., 2008), either an increase or a decrease in Ca2+ can lead to ectopic tyrosine hydroxylase expression.
There are several possible mechanisms for how activity might alter zones. The loss of, or the disruption of, proper Purkinje cell firing may affect the development of sagittal zone patterning by eliminating traveling waves through the developing cerebellum (Watt et al., 2009). In this case, direct Purkinje cell-to-Purkinje cell communication could influence zone formation through inhibitory GABA function, or potentially even through an early excitatory GABA connection (although the excitatory to inhibitory switch in Purkinje cell GABA function is still poorly understood; Sotelo, 2008). But, in addition to a tri-synaptic loop effect through the inferior olive (Chaumont et al., 2013; Witter et al., 2013; Fig. 10D; and see below), Purkinje cell zonation may be shaped by mossy fiber activity, for instance, via the interposed nucleus to red nucleus connection, which loops back to the cerebellum by way of the rubrocerebellar tract. And since mossy fibers directly contact developing Purkinje cells (Mason and Gregory, 1984; Takeda and Maekawa, 1989; Kalinovsky et al., 2011), the alterations in simple spike CV that we observed (Fig. 10) could be the result of early defects in mossy fiber input.
The electrophysiological consequences of selectively blocking inhibition in all Purkinje cells have not been examined in vivo, although one prediction is that cerebellar nuclear neurons would increase their firing rate in response to loss of inhibition (Shakkottai et al., 2004). Our data support this prediction as a subset of recorded cerebellar nuclear neurons did fire at a significantly higher frequency (Fig. 9). We also observed cells with a significantly higher CV and a lower overall firing rate. The firing characteristics of mutant neurons were distributed over a wider than normal range but with similar mean values to cells in control mice (Fig. 9C). This could be caused by different types of cerebellar nuclear neurons showing cell type-specific differences in response to loss of Purkinje cell inputs. The cerebellar nuclei are in fact heterogeneous clusters consisting of three main cell types: glutamatergic projection neurons, inhibitory projection neurons, and inhibitory GABAergic/glycinergic interneurons (Uusisaari and Knöpfel, 2011). Notably, direct contact from Purkinje cells only exists on the two classes of projection neurons but not on the interneurons (Uusisaari and De Schutter, 2011). It is possible that the diversity in responses we observed reflects the connectivity between pairs of Purkinje cells and cerebellar nuclear neurons (McDevitt et al., 1987). Also, the additional inputs from mossy fiber and climbing fiber collaterals that some cerebellar nuclei neurons receive (Shinoda et al., 1992; Sugihara, 2005) may well contribute to the diversity in responses. Therefore, because Purkinje cells are not always the dominant input to a nuclear neuron, loss of GABA neurotransmission may unlock different responses because of persistent afferent inputs and differences in neuronal identity.
Loss of Purkinje cell GABAergic function also disrupted the firing of Purkinje cells themselves (Fig. 10). This is likely because Purkinje cells project to the cerebellar nuclei, which project to the inferior olive, which projects back to Purkinje cells to form a closed loop circuit (Fig. 10). Strikingly, simple spike-firing frequency was unaltered whereas complex spike-firing frequency was decreased. In contrast, the regularity in spike firing was decreased for simple spikes and unchanged for complex spikes. These data suggest that loss of Purkinje cell inhibition obstructed feedback from the inferior olive, but surprisingly the decrease in complex spike frequency did not affect the modulation of resting state simple spike activity. However, our data are consistent with the normal simple spike firing and altered regularity in the tottering mouse, an extensively studied ataxic mutant that lacks CaV2.1 function (Hoebeek et al., 2005). The findings from our study suggest that the lower complex spike rate in the L7Cre;Vgatflox/flox mice is sufficient to maintain a basal level of simple spike firing. However, the lack of an increased simple spike frequency is somewhat in contrast to previous work in which reversible inactivation of the inferior olive with lignocaine or destruction of the inferior olive with a microlesion (which decreases complex spike firing) caused an increase in simple spike firing and a switch to an irregular pattern (Cerminara and Rawson, 2004). Moreover, lesioning the inferior olive alters simple spike modulation during vestibular function (Barmack and Yakhnitsa, 2003). The main difference between our genetic approach and these previous manipulations is that our method alters the cerebellar circuit starting during development. Thus, complex spike–simple spike interactions may be altered throughout life and the defective Purkinje cell responses that we detected may reflect a combination of developmental and compensatory changes in circuit function. Our results are partly in agreement with previous work since we did detect a significant decrease in simple spike regularity in a subset of Purkinje cells (Fig. 10), which fits with the increased oscillatory discharge of simple spikes seen after long-term inactivation of the inferior olive (Cerminara and Rawson, 2004).
Loss of Purkinje cell GABA neurotransmission caused cerebellar nuclear neurons and Purkinje cells to fire irregularly at either a low or a high CV. The magnitude of change may be dependent upon the zone that is analyzed, since Purkinje cell zones are topographically connected to discrete cohorts of cerebellar nuclear neurons (Sugihara and Shinoda, 2007; Sugihara et al., 2009). Alternatively, Purkinje cell zones plus their glutamatergic versus GABAergic cerebellar nuclear phenotypes may influence the magnitude of high versus low CV defects. Our study examined cells in the anterior and central lobules of the vermis, and in CrusI and II of the hemispheres, but likely from zebrinII-positive and -negative cells. In future studies it will be important to use a combination of electrophysiology, juxtacellular labeling, and zonal marker analysis to resolve these possibilities. Based on our current data we can conclude that neuronal activity is essential for patterning the cerebellum into sharp zonal compartments.
Notes
Supplemental material for this article is available at https://www.bcm.edu/departments/pathology-and-immunology/sillitoe/index.cfm?PMID=22682. This material has not been peer reviewed.
Footnotes
This work was supported by funds from Baylor College of Medicine and Texas Children's Hospital (Houston, TX). R.V.S. is supported by the Bachmann-Strauss Dystonia and Parkinson Foundation Inc., the Caroline Wiess Law Fund for Research in Molecular Medicine, a BCM IDDRC Project Development Award, and by BCM IDDRC Grant Number 5P30HD024064 from the Eunice Kennedy Shriver National Institute Of Child Health and Human Development and by Grant Number C06RR029965 from the National Center For Research Resources. The IDDRC RNA In Situ Hybridization Core (Jan and Dan Duncan Neurological Research Institute of Texas Children's Hospital, Houston, TX) performed the in situ hybridization experiments and the Neuropathology Core performed the histology.
The authors declare no competing financial interests.
References
- Ahn AH, Dziennis S, Hawkes R, Herrup K. The cloning of zebrin II reveals its identity with aldolase C. Development. 1994;120:2081–2090. doi: 10.1242/dev.120.8.2081. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer S. Development of the cerebellar system: in relation to its evolution, structure, and functions. Boca Raton, FL: CRC; 1997. [Google Scholar]
- Angaut P, Sotelo C. The dentato-olivary projection in the rat as a presumptive GABAergic link in the olivo-cerebello-olivary loop. An ultrastructural study. Neurosci Lett. 1987;83:227–231. doi: 10.1016/0304-3940(87)90090-5. [DOI] [PubMed] [Google Scholar]
- Apps R, Hawkes R. Cerebellar cortical organization: a one-map hypothesis. Nat Rev Neurosci. 2009;10:670–681. doi: 10.1038/nrn2698. [DOI] [PubMed] [Google Scholar]
- Armstrong CL, Hawkes R. Pattern formation in the cerebellar cortex. Biochem Cell Biol. 2000;78:551–562. doi: 10.1139/o00-071. [DOI] [PubMed] [Google Scholar]
- Armstrong CL, Krueger-Naug AM, Currie RW, Hawkes R. Constitutive expression of the 25-kDa heat shock protein Hsp25 reveals novel parasagittal bands of purkinje cells in the adult mouse cerebellar cortex. J Comp Neurol. 2000;416:383–397. doi: 10.1002/(SICI)1096-9861(20000117)416:3<383::AID-CNE9>3.0.CO%3B2-M. [DOI] [PubMed] [Google Scholar]
- Armstrong CL, Krueger-Naug AM, Currie RW, Hawkes R. Expression of heat-shock protein Hsp25 in mouse Purkinje cells during development reveals novel features of cerebellar compartmentation. J Comp Neurol. 2001;429:7–21. doi: 10.1002/1096-9861(20000101)429:1<7::AID-CNE2>3.0.CO%3B2-Q. [DOI] [PubMed] [Google Scholar]
- Arsénio Nunes ML, Sotelo C. Development of the spinocerebellar system in the postnatal rat. J Comp Neurol. 1985;237:291–306. doi: 10.1002/cne.902370302. [DOI] [PubMed] [Google Scholar]
- Attwell PJ, Rahman S, Ivarsson M, Yeo CH. Cerebellar cortical AMPA-kainate receptor blockade prevents performance of classically conditioned nictitating membrane responses. J Neurosci. 1999;19:RC45. doi: 10.1523/JNEUROSCI.19-24-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baader SL, Vogel MW, Sanlioglu S, Zhang X, Oberdick J. Selective disruption of “late onset” sagittal banding patterns by ectopic expression of engrailed-2 in cerebellar Purkinje cells. J Neurosci. 1999;19:5370–5379. doi: 10.1523/JNEUROSCI.19-13-05370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmack NH, Yakhnitsa V. Cerebellar climbing fibers modulate simple spikes in Purkinje cells. J Neurosci. 2003;23:7904–7916. doi: 10.1523/JNEUROSCI.23-21-07904.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson F, Svensson P, Hesslow G. Feedback control of Purkinje cell activity by the cerebello-olivary pathway. Eur J Neurosci. 2004;20:2999–3005. doi: 10.1111/j.1460-9568.2004.03789.x. [DOI] [PubMed] [Google Scholar]
- Bosco G, Poppele RE. Proprioception from a spinocerebellar perspective. Physiol Rev. 2001;81:539–568. doi: 10.1152/physrev.2001.81.2.539. [DOI] [PubMed] [Google Scholar]
- Boulland JL, Chaudhry FA. Ontogenetic changes in the distribution of the vesicular GABA transporter VGAT correlate with the excitation/inhibition shift of GABA action. Neurochem Int. 2012;61:506–516. doi: 10.1016/j.neuint.2012.03.018. [DOI] [PubMed] [Google Scholar]
- Brielmaier J, Matteson PG, Silverman JL, Senerth JM, Kelly S, Genestine M, Millonig JH, DiCicco-Bloom E, Crawley JN. Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS One. 2012;7:e40914. doi: 10.1371/journal.pone.0040914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochu G, Maler L, Hawkes R. Zebrin II: a polypeptide antigen expressed selectively by Purkinje cells reveals compartments in rat and fish cerebellum. J Comp Neurol. 1990;291:538–552. doi: 10.1002/cne.902910405. [DOI] [PubMed] [Google Scholar]
- Calderon DP, Fremont R, Kraenzlin F, Khodakhah K. The neural substrates of rapid-onset dystonia-Parkinsonism. Nat Neurosci. 2011;14:357–365. doi: 10.1038/nn.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-H. [DOI] [PubMed] [Google Scholar]
- Cerminara NL, Apps R. Behavioural significance of cerebellar modules. Cerebellum. 2011;10:484–494. doi: 10.1007/s12311-010-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerminara NL, Rawson JA. Evidence that climbing fibers control an intrinsic spike generator in cerebellar Purkinje cells. J Neurosci. 2004;24:4510–4517. doi: 10.1523/JNEUROSCI.4530-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Reimer RJ, Bellocchio EE, Danbolt NC, Osen KK, Edwards RH, Storm-Mathisen J. The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J Neurosci. 1998;18:9733–9750. doi: 10.1523/JNEUROSCI.18-23-09733.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumont J, Guyon N, Valera AM, Dugué GP, Popa D, Marcaggi P, Gautheron V, Reibel-Foisset S, Dieudonné S, Stephan A, Barrot M, Cassel JC, Dupont JL, Doussau F, Poulain B, Selimi F, Léna C, Isope P. Clusters of cerebellar Purkinje cells control their afferent climbing fiber discharge. Proc Natl Acad Sci U S A. 2013;110:16223–16228. doi: 10.1073/pnas.1302310110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croci L, Chung SH, Masserdotti G, Gianola S, Bizzoca A, Gennarini G, Corradi A, Rossi F, Hawkes R, Consalez GG. A key role for the HLH transcription factor EBF2COE2, O/E-3 in Purkinje neuron migration and cerebellar cortical topography. Development. 2006;133:2719–2729. doi: 10.1242/dev.02437. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Casali S. Seeking a unified framework for cerebellar function and dysfunction: from circuit operations to cognition. Front Neural Circuits. 2012;6:116. doi: 10.3389/fncir.2012.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P, Miller PE, Levitt P, Walter U, Greengard P. Anatomy of cerebellar Purkinje cells in the rat determined by a specific immunohistochemical marker. Neuroscience. 1984;11:761–817. doi: 10.1016/0306-4522(84)90193-3. [DOI] [PubMed] [Google Scholar]
- Demilly A, Reeber SL, Gebre SA, Sillitoe RV. Neurofilament heavy chain expression reveals a unique parasagittal stripe topography in the mouse cerebellum. Cerebellum. 2011;10:409–421. doi: 10.1007/s12311-010-0156-y. [DOI] [PubMed] [Google Scholar]
- Dusart I, Guenet JL, Sotelo C. Purkinje cell death: differences between developmental cell death and neurodegenerative death in mutant mice. Cerebellum. 2006;5:163–173. doi: 10.1080/14734220600699373. [DOI] [PubMed] [Google Scholar]
- Ebner TJ, Wang X, Gao W, Cramer SW, Chen G. Parasagittal zones in the cerebellar cortex differ in excitability, information processing, and synaptic plasticity. Cerebellum. 2012;11:418–419. doi: 10.1007/s12311-011-0347-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Tvrdik P, Makki N, Paxinos G, Watson C. Precerebellar cell groups in the hindbrain of the mouse defined by retrograde tracing and correlated with cumulative Wnt1-cre genetic labeling. Cerebellum. 2011;10:570–584. doi: 10.1007/s12311-011-0266-1. [DOI] [PubMed] [Google Scholar]
- Fujii M, Arata A, Kanbara-Kume N, Saito K, Yanagawa Y, Obata K. Respiratory activity in brainstem of fetal mice lacking glutamate decarboxylase 65/67 and vesicular GABA transporter. Neuroscience. 2007;146:1044–1052. doi: 10.1016/j.neuroscience.2007.02.050. [DOI] [PubMed] [Google Scholar]
- Fureman BE, Campbell DB, Hess EJ. L-type calcium channel regulation of abnormal tyrosine hydroxylase expression in cerebella of tottering mice. Ann N Y Acad Sci. 1999;868:217–219. doi: 10.1111/j.1749-6632.1999.tb11289.x. [DOI] [PubMed] [Google Scholar]
- Fureman BE, Campbell DB, Hess EJ. Regulation of tyrosine hydroxylase expression in tottering mouse Purkinje cells. Neurotox Res. 2003;5:521–528. doi: 10.1007/BF03033162. [DOI] [PubMed] [Google Scholar]
- Gao Z, Todorov B, Barrett CF, van Dorp S, Ferrari MD, van den Maagdenberg AM, De Zeeuw CI, Hoebeek FE. Cerebellar ataxia by enhanced Ca(V)2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1a(S218L) mutant mice. J Neurosci. 2012;32:15533–15546. doi: 10.1523/JNEUROSCI.2454-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauck V, Jaeger D. The control of rate and timing of spikes in the deep cerebellar nuclei by inhibition. J Neurosci. 2000;20:3006–3016. doi: 10.1523/JNEUROSCI.20-08-03006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebre SA, Reeber SL, Sillitoe RV. Parasagittal compartmentation of cerebellar mossy fibers as revealed by the patterned expression of vesicular glutamate transporters VGLUT1 and VGLUT2. Brain Struct Funct. 2012;217:165–180. doi: 10.1007/s00429-011-0339-4. [DOI] [PubMed] [Google Scholar]
- Gerlai R, Millen KJ, Herrup K, Fabien K, Joyner AL, Roder J. Impaired motor learning performance in cerebellar En-2 mutant mice. Behav Neurosci. 1996;110:126–133. doi: 10.1037/0735-7044.110.1.126. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Patel N, Slager RE, Tokarz ME, Bucan M, Wiley JL, Elsea SH. How much is too much? Phenotypic consequences of Rai1 overexpression in mice. Eur J Hum Genet. 2008;16:941–954. doi: 10.1038/ejhg.2008.21. [DOI] [PubMed] [Google Scholar]
- Gold DA, Gent PM, Hamilton BA. ROR alpha in genetic control of cerebellum development: 50 staggering years. Brain Res. 2007;1140:19–25. doi: 10.1016/j.brainres.2005.11.080. [DOI] [PubMed] [Google Scholar]
- Graham DJ, Wylie DR. Zebrin-immunopositive and –immunonegative stripe pairs represent functional units in the pigeon vestibulocerebellum. J Neurosci. 2012;32:12769–12779. doi: 10.1523/JNEUROSCI.0197-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel C, Hawkes R. Parasagittal organization of the rat cerebellar cortex: direct comparison of Purkinje cell compartments and the organization of the spinocerebellar projection. J Comp Neurol. 1990;291:79–102. doi: 10.1002/cne.902910107. [DOI] [PubMed] [Google Scholar]
- Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22:271–283. doi: 10.1093/hmg/dds427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess EJ, Wilson MC. Tottering and leaner mutations perturb transient developmental expression of tyrosine hydroxylase in embryologically distinct Purkinje cells. Neuron. 1991;6:123–132. doi: 10.1016/0896-6273(91)90127-L. [DOI] [PubMed] [Google Scholar]
- Hirota J, Ando H, Hamada K, Mikoshiba K. Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem J. 2003;372:435–441. doi: 10.1042/BJ20030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano S, Sawada K, Kawano M, Kanemoto M, Xiong G, Mogi K, Sakata-Haga H, Takeda J, Fukui Y, Nogami H. Expression of inorganic phosphate/vesicular glutamate transporters (BNPI/VGLUT1 and DNPI/VGLUT2) in the cerebellum and precerebellar nuclei of the rat. Brain Res Mol Brain Res. 2002;107:23–31. doi: 10.1016/S0169-328X(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Hoebeek FE, Stahl JS, van Alphen AM, Schonewille M, Luo C, Rutteman M, van den Maagdenberg AM, Molenaar PC, Goossens HH, Frens MA, De Zeeuw CI. Increased noise level of purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron. 2005;45:953–965. doi: 10.1016/j.neuron.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Holt GR, Softky WR, Koch C, Douglas RJ. Comparison of discharge variability in vitro and in vivo in cat visual cortex neurons. J Neurophysiol. 1996;75:1806–1814. doi: 10.1152/jn.1996.75.5.1806. [DOI] [PubMed] [Google Scholar]
- Horn KM, Pong M, Gibson AR. Functional relations of cerebellar modules of the cat. J Neurosci. 2010;30:9411–9423. doi: 10.1523/JNEUROSCI.0440-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. The cerebellum and neural control. New York: Raven; 1984. [Google Scholar]
- Jeong YG, Kim MK, Hawkes R. Ectopic expression of tyrosine hydroxylase in Zebrin II immunoreactive Purkinje cells in the cerebellum of the ataxic mutant mouse, pogo. Brain Res Dev Brain Res. 2001;129:201–209. doi: 10.1016/S0165-3806(01)00212-7. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Yan J, Zhao Y, Donahue LR, Beamer WG, Li X, Roe BA, Ledoux MS, Gu W. Carbonic anhydrase-related protein VIII deficiency is associated with a distinctive lifelong gait disorder in waddles mice. Genetics. 2005;171:1239–1246. doi: 10.1534/genetics.105.044487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinovsky A, Boukhtouche F, Blazeski R, Bornmann C, Suzuki N, Mason CA, Scheiffele P. Development of axon-target specificity of ponto-cerebellar afferents. PLoS Biol. 2011;9:e1001013. doi: 10.1371/journal.pbio.1001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon GT, Medina JF, Mauk MD. A mathematical model of the cerebellar-olivary system I: self-regulating equilibrium of climbing fiber activity. J Comput Neurosci. 1998;5:17–33. doi: 10.1023/A:1008874209991. [DOI] [PubMed] [Google Scholar]
- Kim CH, Oh SH, Lee JH, Chang SO, Kim J, Kim SJ. Lobule-specific membrane excitability of cerebellar Purkinje cells. J Physiol. 2012;590:273–288. doi: 10.1113/jphysiol.2011.221846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemerle B, Zanjani H, Joyner A, Herrup K. Pattern deformities and cell loss in Engrailed-2 mutant mice suggest two separate patterning events during cerebellar development. J Neurosci. 1997;17:7881–7889. doi: 10.1523/JNEUROSCI.17-20-07881.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, Strazielle C. Spontaneous and induced mouse mutations with cerebellar dysfunctions: behavior and neurochemistry. Brain Res. 2007;1140:51–74. doi: 10.1016/j.brainres.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Lang EJ, Sugihara I, Llinás R. GABAergic modulation of complex spike activity by the cerebellar nucleoolivary pathway in rat. J Neurophysiol. 1996;76:255–275. doi: 10.1152/jn.1996.76.1.255. [DOI] [PubMed] [Google Scholar]
- Larouche M, Che PM, Hawkes R. Neurogranin expression identifies a novel array of Purkinje cell parasagittal stripes during mouse cerebellar development. J Comp Neurol. 2006;494:215–227. doi: 10.1002/cne.20791. [DOI] [PubMed] [Google Scholar]
- Larsell O. The morphogenesis and adult pattern of the lobules and fissures of the cerebellum of the white rat. J Comp Neurol. 1952;97:281–356. doi: 10.1002/cne.900970204. [DOI] [PubMed] [Google Scholar]
- Larsell O. The comparative anatomy and histology of the cerebellum from monotremes through apes. Minneapolis, MN: University of Minnesota; 1970. [Google Scholar]
- Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Libster AM, Yarom Y. In and out of the loop: external and internal modulation of the olivo-cerebellar loop. Front Neural Circuits. 2013;7:73. doi: 10.3389/fncir.2013.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzini MC, Ward MS, Zhang Q, Lieberman MD, Mason CA. The stop signal revised: immature cerebellar granule neurons in the external germinal layer arrest pontine mossy fiber growth. J Neurosci. 2006;26:6040–6051. doi: 10.1523/JNEUROSCI.4815-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason CA, Gregory E. Postnatal maturation of cerebellar mossy and climbing fibers: transient expression of dual features on single axons. J Neurosci. 1984;4:1715–1735. doi: 10.1523/JNEUROSCI.04-07-01715.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt CJ, Ebner TJ, Bloedel JR. Relationships between simultaneously recorded Purkinje cells and nuclear neurons. Brain Res. 1987;425:1–13. doi: 10.1016/0006-8993(87)90477-X. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Identification and characterization of the vesicular GABA transporter. Nature. 1997;389:870–876. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- Miale IL, Sidman RL. An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp Neurol. 1961;4:277–296. doi: 10.1016/0014-4886(61)90055-3. [DOI] [PubMed] [Google Scholar]
- Miki T, Zwingman TA, Wakamori M, Lutz CM, Cook SA, Hosford DA, Herrup K, Fletcher CF, Mori Y, Frankel WN, Letts VA. Two novel alleles of tottering with distinct Ca(v)2.1 calcium channel neuropathologies. Neuroscience. 2008;155:31–44. doi: 10.1016/j.neuroscience.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Yamasaki M, Hashimoto K, Yamazaki M, Abe M, Usui H, Kano M, Sakimura K, Watanabe M. Cav2.1 in cerebellar Purkinje cells regulates competitive excitatory synaptic wiring, cell survival, and cerebellar biochemical compartmentalization. J Neurosci. 2012;32:1311–1328. doi: 10.1523/JNEUROSCI.2755-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostofi A, Holtzman T, Grout AS, Yeo CH, Edgley SA. Electrophysiological localization of eyeblink-related microzones in rabbit cerebellar cortex. J Neurosci. 2010;30:8920–8934. doi: 10.1523/JNEUROSCI.6117-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdick J, Schilling K, Smeyne RJ, Corbin JG, Bocchiaro C, Morgan JI. Control of segment-like patterns of gene expression in the mouse cerebellum. Neuron. 1993;10:1007–1018. doi: 10.1016/0896-6273(93)90050-2. [DOI] [PubMed] [Google Scholar]
- Oh WJ, Noggle SA, Maddox DM, Condie BG. The mouse vesicular inhibitory amino acid transporter gene: expression during embryogenesis, analysis of its core promoter in neural stem cells and a reconsideration of its alternate splicing. Gene. 2005;351:39–49. doi: 10.1016/j.gene.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Orr HT. Cell biology of spinocerebellar ataxia. J Cell Biol. 2012;197:167–177. doi: 10.1083/jcb.201105092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paukert M, Huang YH, Tanaka K, Rothstein JD, Bergles DE. Zones of enhanced glutamate release from climbing fibers in the mammalian cerebellum. J Neurosci. 2010;30:7290–7299. doi: 10.1523/JNEUROSCI.5118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The mouse brain in stereotaxic coordinates. Amsterdam: Elsevier; 2004. [Google Scholar]
- Person AL, Raman IM. Synchrony and neural coding in cerebellar circuits. Front Neural Circuits. 2012;6:97. doi: 10.3389/fncir.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeber SL, Sillitoe RV. Patterned expression of a cocaine- and amphetamine-regulated transcript peptide reveals complex circuit topography in the rodent cerebellar cortex. J Comp Neurol. 2011;519:1781–1796. doi: 10.1002/cne.22601. [DOI] [PubMed] [Google Scholar]
- Reeber SL, Gebre SA, Sillitoe RV. Fluorescence mapping of afferent topography in three dimensions. Brain Struct Funct. 2011;216:159–169. doi: 10.1007/s00429-011-0304-2. [DOI] [PubMed] [Google Scholar]
- Reeber SL, White JJ, George-Jones NA, Sillitoe RV. Architecture and development of olivocerebellar circuit topography. Front Neural Circuits. 2012;6:115. doi: 10.3389/fncir.2012.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith RM, McKenna J, Wu H, Hashmi SS, Cho SH, Dash PK, Gambello MJ. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2013;51:93–103. doi: 10.1016/j.nbd.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Rong Y, Wang T, Morgan JI. Identification of candidate Purkinje cell-specific markers by gene expression profiling in wild-type and pcd(3J) mice. Brain Res Mol Brain Res. 2004;132:128–145. doi: 10.1016/j.molbrainres.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Ruigrok TJ. Ins and outs of cerebellar modules. Cerebellum. 2011;10:464–474. doi: 10.1007/s12311-010-0164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Kakizaki T, Hayashi R, Nishimaru H, Furukawa T, Nakazato Y, Takamori S, Ebihara S, Uematsu M, Mishina M, Miyazaki J, Yokoyama M, Konishi S, Inoue K, Fukuda A, Fukumoto M, Nakamura K, Obata K, Yanagawa Y. The physiological roles of vesicular GABA transporter during embryonic development: a study using knockout mice. Mol Brain. 2010;3:40. doi: 10.1186/1756-6606-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarna JR, Hawkes R. Patterned Purkinje cell death in the cerebellum. Prog Neurobiol. 2003;70:473–507. doi: 10.1016/S0301-0082(03)00114-X. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Marzban H, Watanabe M, Hawkes R. Complementary stripes of phospholipase Cbeta3 and Cbeta4 expression by Purkinje cell subsets in the mouse cerebellum. J Comp Neurol. 2006;496:303–313. doi: 10.1002/cne.20912. [DOI] [PubMed] [Google Scholar]
- Sawada K, Fukui Y. Expression of tyrosine hydroxylase in cerebellar Purkinje cells of ataxic mutant mice: its relation to the onset and/or development of ataxia. J Med Invest. 2001;48:5–10. [PubMed] [Google Scholar]
- Sawada K, Sakata-Haga H, Fukui Y. Alternating array of tyrosine hydroxylase and heat shock protein 25 immunopositive Purkinje cell stripes in zebrin II-defined transverse zone of the cerebellum of rolling mouse Nagoya. Brain Res. 2010;1343:46–53. doi: 10.1016/j.brainres.2010.04.062. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seil FJ, Johnson ML, Hawkes R. Molecular compartmentation expressed in cerebellar cultures in the absence of neuronal activity and neuron-glia interactions. J Comp Neurol. 1995;356:398–407. doi: 10.1002/cne.903560307. [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, Chou CH, Oddo S, Sailer CA, Knaus HG, Gutman GA, Barish ME, LaFerla FM, Chandy KG. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J Clin Invest. 2004;113:582–590. doi: 10.1172/JCI200420216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda Y, Sugiuchi Y, Futami T, Izawa R. Axon collaterals of mossy fibers from the pontine nucleus in the cerebellar dentate nucleus. J Neurophysiol. 1992;67:547–560. doi: 10.1152/jn.1992.67.3.547. [DOI] [PubMed] [Google Scholar]
- Sillitoe RV, Hawkes R. Handbook of cerebellar disorders. The Netherlands: Springer; 2013. Zones and stripes: development of cerebellar topography; pp. 43–59. [Google Scholar]
- Sillitoe RV, Benson MA, Blake DJ, Hawkes R. Abnormal dysbindin expression in cerebellar mossy fiber synapses in the mdx mouse model of Duchenne muscular dystrophy. J Neurosci. 2003;23:6576–6585. doi: 10.1523/JNEUROSCI.23-16-06576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe RV, Stephen D, Lao Z, Joyner AL. Engrailed homeobox genes determine the organization of Purkinje cell sagittal stripe gene expression in the adult cerebellum. J Neurosci. 2008;28:12150–12162. doi: 10.1523/JNEUROSCI.2059-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe RV, Vogel MW, Joyner AL. Engrailed homeobox genes regulate establishment of the cerebellar afferent circuit map. J Neurosci. 2010;30:10015–10024. doi: 10.1523/JNEUROSCI.0653-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe RV, Fu Y, Watson C. The cerebellum. In: Watson C, Paxinos G, Puelles L, editors. The mouse nervous system. Amsterdam: Elsevier; 2011. [Google Scholar]
- Singec I, Knoth R, Ditter M, Frotscher M, Volk B. Neurogranin expression by cerebellar neurons in rodents and non-human primates. J Comp Neurol. 2003;459:278–289. doi: 10.1002/cne.10600. [DOI] [PubMed] [Google Scholar]
- Snow WM, Anderson JE, Fry M. Regional and genotypic differences in intrinsic electrophysiological properties of cerebellar Purkinje neurons from wild-type and dystrophin-deficient mdx mice. Neurobiol Learn Mem. 2014;107:19–31. doi: 10.1016/j.nlm.2013.10.017. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Cellular and genetic regulation of the development of the cerebellar system. Prog Neurobiol. 2004;72:295–339. doi: 10.1016/j.pneurobio.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Development of “Pinceaux” formations and dendritic translocation of climbing fibers during the acquisition of the balance between glutamatergic and gamma-aminobutyric acidergic inputs in developing Purkinje cells. J Comp Neurol. 2008;506:240–262. doi: 10.1002/cne.21501. [DOI] [PubMed] [Google Scholar]
- Strømme P, Dobrenis K, Sillitoe RV, Gulinello M, Ali NF, Davidson C, Micsenyi MC, Stephney G, Ellevog L, Klungland A, Walkley SU. X-linked Angelman-like syndrome caused by Slc9a6 knockout in mice exhibits evidence of endosomal-lysosomal dysfunction. Brain. 2011;134:3369–3383. doi: 10.1093/brain/awr250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarov A, Joyner AL. Cerebellum morphogenesis: the foliation pattern is orchestrated by multi-cellular anchoring centers. Neural Dev. 2007;2:26. doi: 10.1186/1749-8104-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarov A, Turnbull RK, Kim EJ, Lebel-Potter M, Guillemot F, Joyner AL. Ascl1 genetics reveals insights into cerebellum local circuit assembly. J Neurosci. 2011;31:11055–11069. doi: 10.1523/JNEUROSCI.0479-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara I. Microzonal projection and climbing fiber remodeling in single olivocerebellar axons of newborn rats at postnatal days 4–7. J Comp Neurol. 2005;487:93–106. doi: 10.1002/cne.20531. [DOI] [PubMed] [Google Scholar]
- Sugihara I. Compartmentalization of the deep cerebellar nuclei based on afferent projections and aldolase C expression. Cerebellum. 2011;10:449–463. doi: 10.1007/s12311-010-0226-1. [DOI] [PubMed] [Google Scholar]
- Sugihara I, Shinoda Y. Molecular, topographic, and functional organization of the cerebellar nuclei: analysis by three-dimensional mapping of the olivonuclear projection and aldolase C labeling. J Neurosci. 2007;27:9696–9710. doi: 10.1523/JNEUROSCI.1579-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara I, Fujita H, Na J, Quy PN, Li BY, Ikeda D. Projection of reconstructed single Purkinje cell axons in relation to the cortical and nuclear aldolase C compartments of the rat cerebellum. J Comp Neurol. 2009;512:282–304. doi: 10.1002/cne.21889. [DOI] [PubMed] [Google Scholar]
- Takeda T, Maekawa K. Transient direct connection of vestibular mossy fibers to the vestibulocerebellar Purkinje cells in early postnatal development of kittens. Neuroscience. 1989;32:99–111. doi: 10.1016/0306-4522(89)90110-3. [DOI] [PubMed] [Google Scholar]
- Todorov B, Kros L, Shyti R, Plak P, Haasdijk ED, Raike RS, Frants RR, Hess EJ, Hoebeek FE, De Zeeuw CI, van den Maagdenberg AM. Purkinje cell-specific ablation of Cav2.1 channels is sufficient to cause cerebellar ataxia in mice. Cerebellum. 2012;11:246–258. doi: 10.1007/s12311-011-0302-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert DL, Pittman T, Alisky JM, Clark BR. Chronic NMDA receptor blockade or muscimol inhibition of cerebellar cortical neuronal activity alters the development of spinocerebellar afferent topography. Brain Res Dev Brain Res. 1994;80:268–274. doi: 10.1016/0165-3806(94)90112-0. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488:647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uusisaari M, De Schutter E. The mysterious microcircuitry of the cerebellar nuclei. J Physiol. 2011;589:3441–3457. doi: 10.1113/jphysiol.2010.201582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uusisaari M, Knöpfel T. Functional classification of neurons in the mouse lateral cerebellar nuclei. Cerebellum. 2011;10:637–646. doi: 10.1007/s12311-010-0240-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Patterned expression of Purkinje cell glutamate transporters controls synaptic plasticity. Nat Neurosci. 2005;8:1329–1334. doi: 10.1038/nn1539. [DOI] [PubMed] [Google Scholar]
- Watt AJ, Cuntz H, Mori M, Nusser Z, Sjöström PJ, Häusser M. Traveling waves in developing cerebellar cortex mediated by asymmetrical Purkinje cell connectivity. Nat Neurosci. 2009;12:463–473. doi: 10.1038/nn.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel A, Schild D, Zeug A. Resolution in the ApoTome and the confocal laser scanning microscope: comparison. J Biomed Opt. 2009;14 doi: 10.1117/1.3083439. 014022. [DOI] [PubMed] [Google Scholar]
- Welsh JP, Yuen G, Placantonakis DG, Vu TQ, Haiss F, O'Hearn E, Molliver ME, Aicher SA. Why do Purkinje cells die so easily after global brain ischemia? Aldolase C, EAAT4, and the cerebellar contribution to posthypoxic myoclonus. Adv Neurol. 2002;89:331–359. [PubMed] [Google Scholar]
- White JJ, Sillitoe RV. Development of the cerebellum: from gene expression patterns to circuit maps. Wiley Interdiscip Rev Dev Biol. 2013a;2:149–164. doi: 10.1002/wdev.65. [DOI] [PubMed] [Google Scholar]
- White JJ, Sillitoe RV. Postnatal development of cerebellar zones revealed by neurofilament heavy chain protein expression. Front Neuroanat. 2013b;7:9. doi: 10.3389/fnana.2013.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BK, Hess EJ. Animal models for dystonia. Movement Disord. 2013;28:982–989. doi: 10.1002/mds.25526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter L, Canto CB, Hoogland TM, de Gruijl JR, De Zeeuw CI. Strength and timing of motor responses mediated by rebound firing in the cerebellar nuclei after Purkinje cell activation. Front Neural Circuits. 2013;7:133. doi: 10.3389/fncir.2013.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaylaoglu MB, Titmus A, Visel A, Alvarez-Bolado G, Thaller C, Eichele G. Comprehensive expression atlas of fibroblast growth factors and their receptors generated by a novel robotic in situ hybridization platform. Dev Dyn. 2005;234:371–386. doi: 10.1002/dvdy.20441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A control Vgatflox/flox mouse and an L7Cre;Vgatflox/flox mutant mouse. Note the extended limbs of the mutant when it falls to its side.
Control L7Cre;Vgatflox/+ and L7Cre mice. Neither mouse exhibits motor behavior deficits.