

Abstract
Nontuberculous mycobacteria (NTM) have recently been recognized as important species that cause disease even in immunocompetent individuals. The mechanisms that these species use to infect and persist inside macrophages are not well characterised. To gain insight concerning this process we used THP-1 macrophages infected with M. abscessus, M. fortuitum, M. celatum, and M. tuberculosis. Our results showed that slow-growing mycobacteria gained entrance into these cells with more efficiency than fast-growing mycobacteria. We have also demonstrated that viable slow-growing M. celatum persisted inside macrophages without causing cell damage and without inducing reactive oxygen species (ROS), as M. tuberculosis caused. In contrast, fast-growing mycobacteria destroyed the cells and induced high levels of ROS. Additionally, the macrophage cytokine pattern induced by M. celatum was different from the one induced by either M. tuberculosis or fast-growing mycobacteria. Our results also suggest that, in some cases, the intracellular survival of mycobacteria and the immune response that they induce in macrophages could be related to their growth rate. In addition, the modulation of macrophage cytokine production, caused by M. celatum, might be a novel immune-evasion strategy used to survive inside macrophages that is different from the one reported for M. tuberculosis.
1. Introduction
Tuberculosis remains a global public health problem of enormous scope and proportion, with an estimated one-third of the world's population harbouring a latent infection and approximately 1.5 million deaths occurring annually from the active disease [1]. The unfortunate intersection of the tuberculosis and HIV pandemics in many developing nations has led to enormous morbidity and mortality [2], with the additional recognition of some nontuberculous mycobacteria (NTM) as important etiological agents of disease, aside from that caused by Mycobacterium tuberculosis (representative species of the M. tuberculosis complex) [3]. The three most common NTM that cause skin and soft tissue infections in humans are Mycobacterium abscessus, Mycobacterium chelonae, and Mycobacterium fortuitum [4], and the most common causes of pulmonary disease are M. abscessus and M. avium [5]. Some other species have been described to be uncommon causes of human diseases, such as Mycobacterium branderi, Mycobacterium celatum, and Mycobacterium bohemicum. Despite this, M. celatum was previously shown to be an underestimated cause of pulmonary and disseminated infections which may even be fatal [6] and has sometimes been misled with M. tuberculosis or M. terrae-like organisms [7]. We now know that NTM not only cause disease in immunocompromised individuals [3, 8–11], but they also affect the immunocompetent individuals [3, 6, 7, 12].
NTM are widely distributed in the environment with high isolation rates worldwide. These microorganisms can be found in soil and in both natural and treated water sources. Since there is no evidence of animal-to-human or human-to-human transmission of NTM, human disease is suspected to be acquired from environmental exposures. The most common clinical manifestation of NTM is lung disease, but lymphatic, skin/soft tissue, and disseminated diseases are also important [3].
Little is known about the immune response against NTM, but it has been extensively studied for M. tuberculosis. Infection with this pathogen is produced by the inhalation of aerosols containing a small number of bacilli [13]. Alveolar macrophages (MΦ) not only represent the first line of defence with their intracellular killing of bacteria and antigen presentation to lymphocytes but also provide the habitat where mycobacteria can reside [14, 15]. By arresting phagosome maturation and phagolysosomal fusion [13], M. tuberculosis causes the destruction of the MΦ, which allows the bacilli to infect new MΦ and thus perpetuate the infection [16, 17]. The outcome depends on the resistance of the host and the virulence of the infecting strain [18, 19]. If mycobacteria are not killed in this initial stage, they survive and proliferate inside the macrophages. This intracellular survival is well documented for M. tuberculosis [14, 20–24], but it has also been described for a few NTM [14], including M. marinum [25] and M. fortuitum [26].
Infection of human acute monocytic leukemia THP-1 cells and infection of bone marrow-derived macrophages (BMDM) are widely used models mimicking MΦ-Mtb interactions [15, 27–29]. Through these models, it has been demonstrated that some M. tuberculosis subcellular components, such as the cell wall lipoarabinomannan, trigger a cascade of proinflammatory cytokines and chemokines which, in turn, upregulate key components of the host defence against M. tuberculosis [15, 18, 28, 30, 31]. Since further analysis is needed to better understand this immune phenomenon between NTM and MΦ, we conducted the present study using different NTM species to elucidate (1) their ability to infect, persist, and proliferate inside their main host cell, the macrophage; (2) their effect on the initial innate immune response of macrophages. We studied two slow-growing mycobacteria, M. celatum and M. tuberculosis, and two fast-growing mycobacteria, M. abscessus and M. fortuitum. We report here that intracellular survival of mycobacteria and initial immune response elicited from macrophages might be related to their growth rate. To the best of our knowledge, this is the first evidence of persistence and survival of M. celatum inside human macrophages inducing a cell response which is different from that of M. tuberculosis, the typical pathogenic slow-growing bacillus.
2. Material and Methods
2.1. Mycobacterial Culture
M. tuberculosis H37Rv, M. celatum ATCC 51130 (strain 1908), M. fortuitum ATCC 6841, and a clinical isolate of M. abscessus (previously identified by rRNA 16S sequencing) were cultured in Dubos medium with 10% of albumin-dextrose-catalase supplement (bovine albumin fraction V 50 g/L, dextrose 20 g/L, and catalase 0.04 g/L) (Becton Dickinson, Franklin Lakes, NJ, USA) and incubated at 37°C. The bacteria were then pelleted by centrifugation and resuspended in RPMI 1640 medium (Gibco, Life Technologies, Grand Island, NY, USA) [17]. Viability was evaluated by counting the colony-forming units (CFUs), and the bacterial suspension was aliquoted and frozen at −70°C.
2.2. Cell Line Culture and Infection
The human monocyte THP-1 cell line was used for this study (ATCC TIB 202). The cells were cultured in RPMI 1640 (Gibco) supplemented with 10% heat-inactivated foetal calf serum and 0.45% dextrose (Sigma Aldrich, St. Louis, MO, USA) and incubated at 37°C in 5% CO2 incubator to a density of 1 × 106 cells/mL. Cells were counted in a Neubauer chamber after trypan blue staining. THP-1 cells were treated with phorbol-12-myristate-13-acetate (PMA, Sigma Aldrich) at a concentration of 20 nM for 72 h to induce differentiation into macrophage. Mycobacteria were added to the macrophage culture at a multiplicity of infection (MOI) of 5. After 6 h at 37°C and 5% CO2, the infected macrophages were washed with Hanks solution 1x (Gibco) to remove extracellular mycobacteria and incubated with fresh medium.
2.3. Phagocytosis Rate Evaluation
200,000 macrophages were plated in each well of an 8-well chamber slide (Nunc, Thermo Scientific, Rockford, IL, USA) and infected with the indicated mycobacterial strain (MOI = 5). After 6, 12, 24, 48, and 72 h, cells were fixed with 4% p-formaldehyde, stained with the Kinyoun method (Difco, Becton Dickinson), and analysed by optical microscopy (1000x). The infection/ingestion percentage was determined by counting macrophages with more than one intracellular mycobacterium and macrophages without intracellular mycobacteria. The integrity of the THP-1 macrophage monolayer was evaluated by optical microscopy (1000x; Carl Zeiss Axiostar Plus microscope, Germany). For this purpose, the percentage of integrity was determined by counting macrophages in infected and noninfected cultures (control) at the same time. 100% of integrity corresponded to the macrophage total number in noninfected cultures.
2.4. Quantification of Viable Intracellular Mycobacteria
500,000 macrophages were plated in each well of a 24-well flat-bottom plate (Costar, Sigma Aldrich) and infected with the indicated mycobacterial strain (MOI = 5). After 6, 24, and 48 h, the cells were lysed with 0.05% Tween 20 (Sigma Aldrich) and the supernatants were cultured to determine mycobacterial CFUs. In order to investigate if macrophages were responsible for intracellular multiplication control of the mycobacteria tested, CFUs counts obtained from the infected cells were compared to those of the same mycobacteria cultured under the same conditions, but without macrophages. In this latter condition, mycobacteria were grown in RPMI medium because it has been reported previously [32] that this microorganism can grow optimally in this kind of medium.
2.5. Production of Reactive Oxygen Species by Macrophages
150,000 macrophages were plated in each well of a 96-well flat-bottom plate (Costar, Sigma Aldrich) and infected with the indicated mycobacterial strain (MOI = 5). 1% Nitro blue tetrazolium chloride (Sigma Aldrich) was added at t = 0 after 6 h or 24 h and then incubated for 15 min and the reaction was stopped by adding 10% sodium dodecyl sulphate/0.08 N NaOH; the colour intensity was quantified with a microplate reader (Thermo Multiscan EX, USA) at 600 nm. Cells restimulated with 20 nM PMA were used as our positive control for ROS production (PMA group). Nonstimulated cells (noninfected PMA treated cells) were used as a negative control and considered as our baseline level for ROS.
2.6. Production of Cytokines by Macrophages
1 × 106 macrophages were plated in each well of a 24-well flat-bottom plate (Costar, Sigma Aldrich) and infected with the indicated mycobacterial strain (MOI = 5). After 6 and 24 h, the supernatants were collected and analysed by ELISA (TiterZyme, Assay Designs, Ann Arbor, MI, USA) to determine the concentrations of TNF-α, IL-1β, IL-8, and TGF-β, according to manufacturer's protocol.
2.7. Statistical Analysis
All the experiments were performed in triplicate and repeated on three independent occasions. The results were analysed by one-way ANOVA with Tukey's post-test, using SigmaStat v.3.1 (Systat Software, San Jose, CA, USA). A P < 0.05 was considered statistically significant.
3. Results
3.1. Slow-Growing Mycobacteria Gain Entrance to THP-1 Macrophages with More Efficiency than Fast-Growing Mycobacteria
Quantification of intracellular mycobacteria in infected THP-1 macrophages showed that, 6 h after infection, the percentage of infected macrophages was higher in the presence of M. tuberculosis (100%) and M. celatum (90%), as compared to M. abscessus (70%) and M. fortuitum (80%) (Figure 1). Therefore, we suggest that macrophages phagocyted the slow growers mycobacteria more quickly than the fast growers. Although significant, we propose that the small differences observed between phagocyted M. celatum and fast growers (10% and 20% difference) were also relevant. This is because, although macrophages ingested quite similar quantities of M. celatum, M. fortuitum, and M. abscessus, the cell monolayer was severely compromised throughout the infection of the two fast growers, a fact that was not observed in macrophages infected with M. celatum (see next paragraph of results regarding monolayer integrity). Independently of the species of mycobacteria, we observed a 100% rate of infected macrophages at 24 h postinfection (data not shown).
Figure 1.
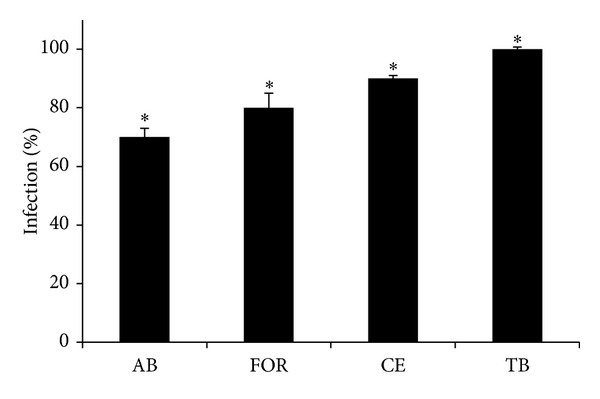
Infection rate of THP-1 cells at 6 h of mycobacterial infections. 2 × 105 THP-1 macrophages were infected with fast-growing (M. abscessus and M. fortuitum) or slow-growing mycobacteria (M. celatum and M. tuberculosis) using a MOI of 5. After 6 h of infection, cells were washed and the percentage of macrophages with more than one intracellular mycobacterium was determined. ν: M. abscessus; σ: M. fortuitum; μ: M. celatum; ο: M. tuberculosis. The experiment was performed in triplicate and repeated on three independent occasions. ANOVA with Tukey's post-test was carried out using a P ≤ 0.05. *Significant difference among mycobacteria.
3.2. Viable Slow-Growing Mycobacteria Persist inside THP-1 Macrophages
In order to demonstrate that intracellular mycobacteria were alive, we performed a viability test (CFUs/ml determination). Our results showed that at least during 48 h, THP-1 macrophages were unable to eliminate any of the mycobacterial species. Regarding fast-growing mycobacteria, there was a 2-log decrease in the CFUs of M. abscessus that were present inside the THP-1 macrophages, compared to the CFUs of M. abscessus cultured in medium alone (24 h after infection, Figure 2(a)). However, this decrease was no longer evident at 48 h of infection (Figure 2(a)). Although the number of M. fortuitum remained the same between those two different conditions (inside macrophages or in a macrophage-free culture), it increased by 2-log from 6 h to 48 h postinfection (Figure 2(b)). In contrast, the numbers of M. celatum and M. tuberculosis inside the macrophages were similar, and we did not observe any changes in the numbers of these two mycobacteria with the passage of time (Figures 2(c) and 2(d)). The CFUs of these two mycobacteria that were present inside the macrophages were no different from the CFUs of mycobacteria cultured in medium alone, which may indicate that at this MOI (1 : 5) M. celatum and M. tuberculosis survived but did not proliferate inside macrophages during the first 48 h of infection.
Figure 2.

Quantification of viable mycobacteria inside THP-1 macrophages. 5 × 105 THP-1 macrophages were infected with fast-growing (M. abscessus and M. fortuitum) or slow-growing mycobacteria (M. celatum and M. tuberculosis) using a MOI of 5. After 6 h of infection, cells were washed and incubated with fresh RPMI medium. Infected cells were incubated for 6 h, 24 h, and 48 h and washed before CFUs determinations (solid lines). As control, each mycobacterium was cultured in medium alone (without macrophages), and the CFUs were determined at the same times (dashed lines). (a) M. abscessus; (b) M. fortuitum; (c) M. celatum; (d) M. tuberculosis. The experiment was performed in triplicate and repeated on three independent occasions. ANOVA with Tukey's post-test was carried out using a P ≤ 0.05. *Significant differences between the CFUs of mycobacteria that were present inside the macrophages, compared to the CFUs of mycobacteria cultured in medium alone.
3.3. Fast-Growing Mycobacteria Damaged the Monolayer Integrity of THP-1 Macrophages Starting at 48 h of Infection
The integrity of the THP-1 macrophage monolayer decreased in the presence of fast-growing mycobacteria. Seventy-two hours after infection, just 10% of the monolayer remained intact in the presence of M. abscessus and M. fortuitum, while 80–90% of the monolayer was intact in the presence of slow-growing mycobacteria (Figure 3(a)). The characteristic cellular morphology of THP-1 macrophages was not altered at 48 h of infection with slow-growing mycobacteria (Figure 3(b)). According to these results, while fast-growing mycobacteria can produce 60 to 80% damage of the macrophage monolayer integrity at 48 h of infection, slow-growing mycobacteria only produced 0 to 20%. Since macrophage monolayer damage at 48 h postinfection produced by fast-growing mycobacteria was too extensive, all of the following experiments were performed within the first 24 h after infection.
Figure 3.
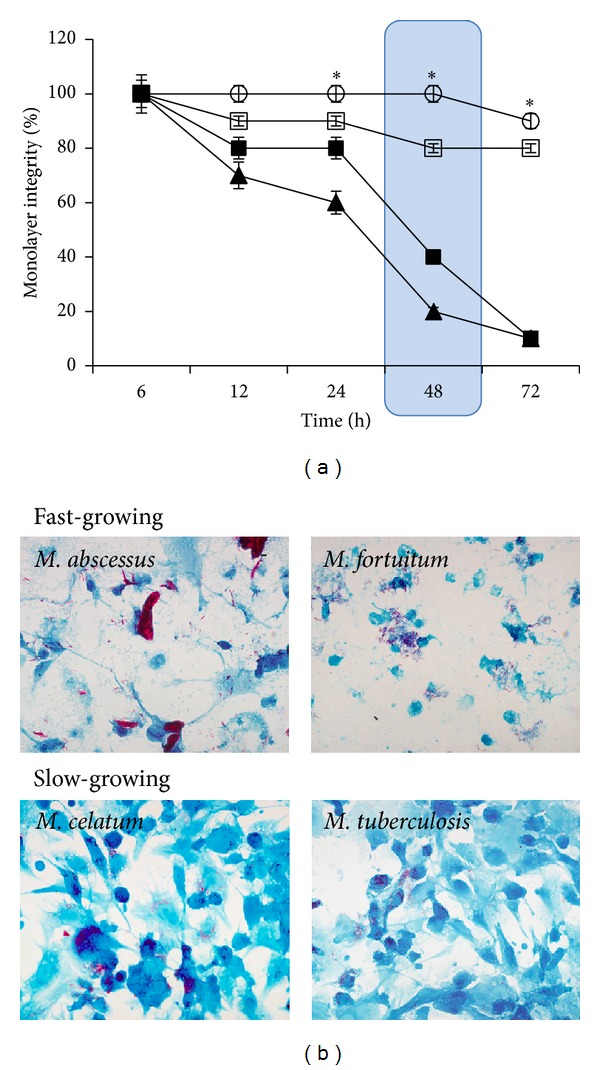
Integrity of THP-1 macrophages monolayer after mycobacterial infection. 2 × 105 THP-1 macrophages were infected with fast-growing (M. abscessus and M. fortuitum) or slow-growing mycobacteria (M. celatum and M. tuberculosis) using a MOI of 5. After 6 h of infection, cells were washed and incubated with fresh RPMI medium. Infection kinetics were stopped at 6 h, 12 h, 24 h, 48 h, and 72 h, fixed using 4% p-formaldehyde, and visualized by microscopy. Monolayer integrity of infected cells was compared with monolayer of noninfected macrophages. (a) Monolayer integrity percentage of infected macrophages. Large cell damage is shown within a blue box. ν: M. abscessus; σ: M. fortuitum; μ: M. celatum; ο: M. tuberculosis. The experiment was performed in triplicate and repeated on three independent occasions. (b) Microscope photographs of THP-1 macrophages at 48 h of infection with the indicated mycobacteria (600x, Kinyoun stain).
3.4. Slow-Growing Mycobacteria Do Not Induce the Production of Reactive Oxygen Species (ROS) by THP-1 Macrophages
Fast-growing mycobacteria induced the production of ROS by THP-1 macrophages at 6 and 24 h postinfection. In contrast, no ROS could be detected with the slow-growing mycobacteria, M. celatum and M. tuberculosis, even after 24 h of infection (Figure 4). It is probable that the high levels of ROS induced by fast-growing mycobacteria could be associated with the extensive cellular damage that we reported above.
Figure 4.
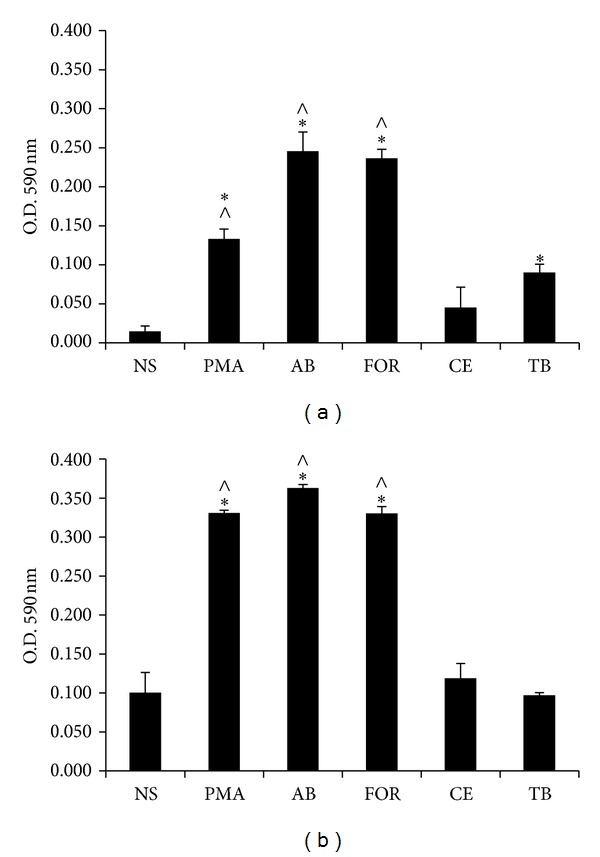
Production of reactive oxygen species by THP-1 macrophages after mycobacterial infection. 1.5 × 105 THP-1 macrophages were infected with fast-growing (AB, M. abscessus and FOR, M. fortuitum) or slow-growing mycobacteria (CE, M. celatum and TB, M. tuberculosis), using a MOI of 5. After 6 h of infection, cells were washed and incubated with fresh RPMI medium and 0.1% NBT. Reaction was stopped using 10% SDS in 0.8 N NaOH, and the reduction of NBT was measured at 6 h (a) and 24 h (b). The experiment was performed in triplicate and repeated on three independent occasions. ANOVA with Tukey's post-test was carried out using a P ≤ 0.05; NS: nonstimulated cells; PMA: phorbol-12-myristate-13-acetate. *Significant difference compared to nonstimulated cells; ∧significant difference compared to M. tuberculosis.
3.5. The Cytokine Pattern Induced by M. celatum Is Different from the Cytokine Pattern Induced Either by M. tuberculosis or by Fast-Growing Mycobacteria
Cytokines have an essential role in the modulation of the immune response and, to a large extent, determine the course of a disease [31, 33]. THP-1 macrophages produced no detectable amounts of TNF-α at 6 h postinfection in the four tested mycobacteria, but they produced similar amounts of the aforementioned cytokine after 24 h of infection in all studied mycobacteria (approximately 2800 pg/μl; data not shown). IL-1β was detected 6 and 24 h after infection; after 6 h no significant differences were observed in any of the mycobacteria, but after 24 h the levels of this cytokine were significantly higher in the presence of fast-growing mycobacteria even though monolayer was compromised (80% integrity at 24 h for M. abscessus infections and 60% for M. fortuitum). The lowest levels of IL-1β were produced in response to M. celatum (Figures 5(a) and 5(b)). The fast-growing mycobacteria and M. celatum induced the production of high levels of IL-8 at 6 and 24 h postinfection, while no detectable levels of this cytokine were found in the presence of M. tuberculosis (Figures 5(a) and 5(b)). At 6 h and 24 h postinfection, the four tested mycobacteria induced the production of the anti-inflammatory cytokine TGF-β, with M. celatum inducing the highest levels (Figures 5(a) and 5(b)). According to our results we propose that M. celatum is a slow-growing mycobacterium with a particular induced cytokine pattern that does not correspond to that produced by a typical slow- (such as M. tuberculosis) or fast-growing mycobacterium. We therefore suggest that the immune response caused by M. celatum should be further characterized.
Figure 5.
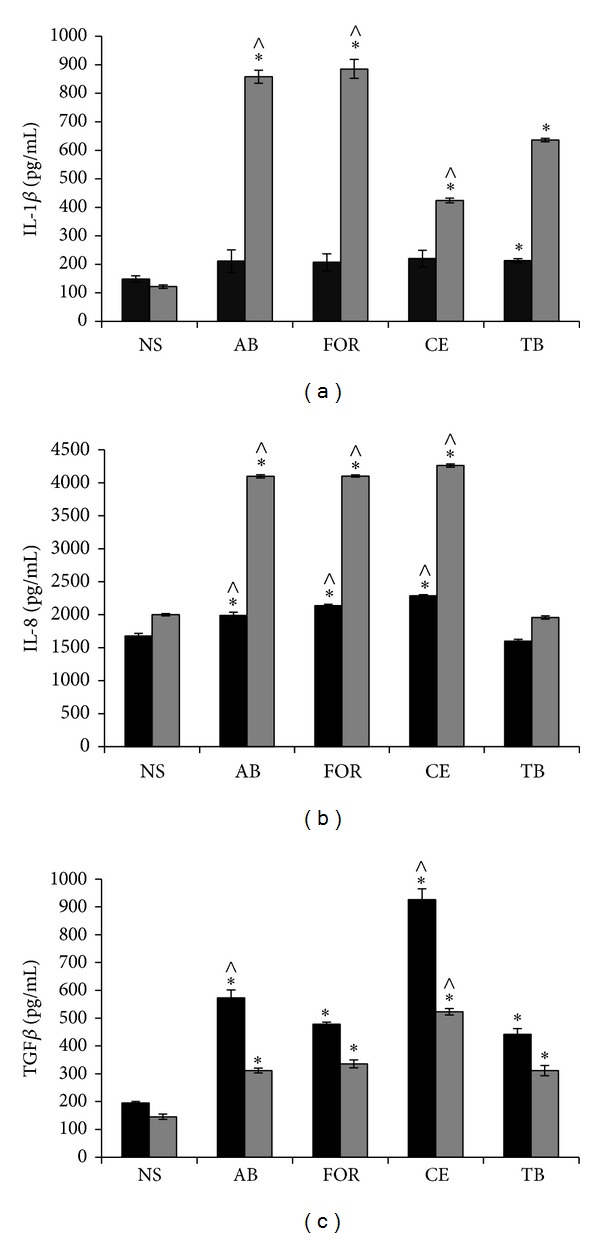
Production of cytokines by THP-1 macrophages after mycobacterial infection. 1 × 106 THP-1 macrophages were infected with fast-growing (AB, M. abscessus and FOR, M. fortuitum) or slow-growing mycobacteria (CE, M. celatum and TB, M. tuberculosis), using a MOI of 5. After 6 h of infection, cells were washed and incubated with fresh RPMI medium. Cytokines were quantified at 6 h (black bars) and 24 h (gray bars) after infection. The proinflammatory cytokines IL-1β (a) and IL-8 (b) and the anti-inflammatory cytokine TGF-β (c) were analysed. The experiment was performed in triplicate and repeated on three independent occasions. ANOVA with Tukey's post-test was carried out using a P ≤ 0.05. *Significant differences compared to nonstimulated cells; ∧significant differences compared to M. tuberculosis. NS: nonstimulated; PMA: phorbol 12-myristate 13-acetate.
4. Discussion
The interaction of mycobacteria with their host cell is a complex event, which has not yet been fully characterised, but it has been reported that macrophage receptors bind to molecules on the surface of mycobacteria and trigger phagocytosis. Phagocytosis, as the main process that mediates the entry of mycobacteria into these cells, has been suggested previously [34, 35]. Therefore, we investigated the effect of two fast-growing (M. abscessus and M. fortuitum) and two slow-growing mycobacteria (M. celatum and M. tuberculosis) on the response of human macrophages, in order to evaluate whether the growth rate and other particular characteristics of these microorganisms affect their host-pathogen interaction.
Our results showed that M. tuberculosis was the species that reached 100% entrance into THP-1 cells at an earlier time (compared to NTM infections), reflecting the high recognition of its surface components by host cells. It has been reported that several mycobacterial envelope lipids are involved in this recognition. These lipids include the family of lipoarabinomannans (LAMs), whose effects on the host innate immune response depend on the chemical modifications of their distal arabinose residues. While M. tuberculosis has mannosylated LAMs (ManLAMs), M. smegmatis, M. fortuitum, and other fast-growing mycobacteria have LAMs with phosphatidylinositol (PiLAMs) [36, 37] and M. chelonae has unmodified LAMs [36–38]. This fact might help explain the differences in the phagocytosis process between NTM and M. tuberculosis. Even when M. celatum entered the host cell more efficiently than M. fortuitum and M. abscessus at 6 h postinfection, it reached the 100% level of cells infected at the same time as M. fortuitum. Hence, we can assume that these two mycobacteria (M. celatum and M. fortuitum) might have similar LAMs. M. celatum surface lipids should be further analysed in order to confirm our hypothesis.
At the same time, we observed a delayed entry of M. abscessus into macrophages that might be explained by the fact that this species produces a glycopeptidic biofilm [38], which could mask its surface lipids, resulting in a less phagocytable microorganism. We simultaneously analysed the mycobacterial intracellular viability, the THP-1 monolayer integrity, and the ROS production of those cells. Our results demonstrated that during the first 48 h the slow-growing mycobacteria, M. celatum and M. tuberculosis, were able to persist inside THP-1 macrophages without causing cellular damage, albeit without proliferating (as has been demonstrated previously for the tubercle bacilli; see [39]), and this could be related to the lack of ROS production in response to these mycobacteria. M. tuberculosis actively blocks reactive oxygen species production [16, 29, 40–43], which is related to its immune-evasion strategy ability to persist inside macrophages [44]. It remains to be determined if M. celatum blocks reactive oxygen species production by similar mechanisms.
Although both fast-growing mycobacteria were able to proliferate at 48 h postinfection, in an environment almost free of macrophages (10–20%), our results suggest that this phenomenon was due to monolayer destruction. For M. abscessus, it has been reported that infection of THP-1 macrophages induces an oxidative environment inside the cell [45] that could explain the decrease in the number of viable intracellular mycobacteria observed at 24 h after infection (ROS related, see Figure 4), when monolayer integrity was still sufficient (80%) to control mycobacterial growth. Although M. fortuitum induced a strong ROS response, cells were not able to control the bacterial growth even at 24 h postinfection, at which time we observed significant monolayer reduction (Figure 3(a)). This early macrophage destruction might be due to a known cellular process, such as apoptosis, which has been reported to occur in THP-1 infections with the fast growers M. smegmatis and M. fortuitum. Macrophage destruction can also be a consequence of a robust production of reactive oxygen species (ROS) in THP-1 cells infected with M. abscessus and M. fortuitum (Figure 4), as has been reported previously with different host cell/pathogen systems [46–48]. These results confirm the hypothesis that fast-growing mycobacteria induce a very potent immune response when compared to typical pathogenic mycobacteria (such as M. tuberculosis) [49].
Macrophages represent the first line of host defense against most bacterial pathogens. Following interaction with the bacteria, macrophages initiate inflammatory responses by secreting cytokines and chemokines [50–52]. Control of M. tuberculosis infection is associated with a proinflammatory class, Th1-type cytokine profile [53, 54]. Among the cytokines, two key proinflammatory cytokines for antimicrobial responses are TNF-α and IL-1β [18, 52, 55]. In our model, TNF-α was induced at the same levels by the four tested mycobacteria (≈2800 pg/μl), independently of their growth rate and virulence, which reflects the importance of this cytokine in the control of any mycobacterial infection (data not shown).
IL-1β is an important cytokine for host immune defense against M. tuberculosis, since several studies have demonstrated that IL-1β- and IL-1-receptor-knockout mice are more susceptible to M. tuberculosis infections than the wild-type animals [56–59]. Our results suggest that (at 6 h postinfection) the secretion of IL-1β must be important to control the infection regardless of the mycobacterial species present in the media as this cytokine facilitates the recruitment of other innate immune cells to the site of infection and as it induces the production of IL-8 and mediates proliferation, cellular differentiation, and apoptosis [60, 61]. Cytokine IL-1β also seems to be crucial for the inflammasome formation of pathogenic and nonpathogenic mycobacteria [52, 59]. At 24 h postinfection we observed a differential secretion of IL-1β among the mycobacteria studied: fast growers induced the highest level. In consequence, the important induction of IL-1β secretion by fast-growing mycobacteria is consistent with their ROS production, since both mechanisms contribute to inflammasome activation. This has been suggested for the fast grower M. abscessus [62] and has been previously demonstrated only for the slow grower M. kansasii [52]. In contrast, M. celatum induced the lowest level of IL-1β (even lower than that of M. tuberculosis), which suggests that this may be another immune-evasion strategy.
The fast-growing mycobacteria and M. celatum induced the production of similar levels of IL-8 at both times analysed (6 h and 24 h), while M. tuberculosis induced a statistically significantly lower level. IL-8 not only is involved in attracting PMNs (Polymorphonuclears) to the site of infection [63] but also facilitates the elimination of microorganisms by increasing the efficiency of the bactericidal activity of granulocytes [64]. It is also important in the production of an inflammatory response because it is involved in maintaining balance in the granuloma-active infection. In higher concentrations, however, it can help eliminate the mycobacteria by nonoxidative pathways, as has been demonstrated in M. fortuitum [64] and M. smegmatis [65] infections. The high amounts of IL-8 in the supernatants of fast-growing mycobacteria infections are in concordance with the production of IL-1β. However, in the case of M. celatum, cytokines other than IL-1β might be responsible for inducing the production of IL-8 at 24 h postinfection.
Anti-inflammatory cytokines prevent the cellular damage that can be caused by an excessive Th1-like response. The fast-growing mycobacteria, M. abscessus and M. fortuitum, induced the production of TGF-β at the same levels as in M. tuberculosis; but for infections with the fast growers, TGF-β production was not enough to prevent cell damage induced by a strong proinflammatory response (as has been demonstrated to occur in M. tuberculosis infections). M. celatum induced the highest levels of TGF-β, which could reduce the innate immune response and favour the persistence of this mycobacterium in the host, despite the proinflammatory response mediated by IL-8 secretion. To the best of our knowledge, this is the first report that suggests the modulation of macrophages cytokine production by M. celatum, particularly of IL-1β and TGF-β, and of the absence of reactive oxygen species during its infection.
5. Conclusions
This study provides evidence that growth rate might be related, in some cases, to the intracellular survival of mycobacteria and the immune response that they induce in THP-1 macrophages. Growth rate, however, is not the only determinant of the outcome of the interaction of mycobacteria-macrophages; other factors such as envelope cell lipids and the particular virulence factors of each mycobacterium should be further considered. We suggest that the ability to block reactive oxygen species production by slow-growing mycobacteria is an immune-evasion strategy that putatively promotes their survival and cytokine production in the host, even in NTM species. Finally, our data provides insight into the novel mechanisms that M. celatum uses to persist inside its host cell, which should further be characterized in order to gain knowledge about the pathogenic NTM species that cause disease in immunocompetent patients.
Acknowledgments
The authors thank Dr. T. Horn-Copeland, M.D., for helpful English language review of the paper, and Dr. I. Wong-Baeza, for the critical review of the paper. The study was supported by CONACyT, Mexico, Grant CB 2010-156347-M (JAG-y-M) and Grant CB 2011-169063 (ACH-R) and The European Community, Grant HEALTH-F3-2008-200999 (JAG-y-M). A. Cecilia Helguera-Repetto was supported by a CONACYT Postdoctoral Scholarship in CINVESTAV (BM Department), IPN, México. Sandra Rivera-Gutierrez, Jorge F. Cerna-Cortes, and Jorge A. Gonzalez-y-Merchand are fellows of COFAA and EDI, IPN.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Global tuberculosis report. 2013, http://www.who.int/tb/publications/global_report/en/
- 2.Baena A, Porcelli SA. Evasion and subversion of antigen presentation by Mycobacterium tuberculosis . Tissue Antigens. 2009;74(3):189–204. doi: 10.1111/j.1399-0039.2009.01301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. The American Journal of Respiratory and Critical Care Medicine. 2007;175(4):367–416. doi: 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 4.Yu JR, Heo ST, Lee KH, et al. Skin and soft tissue infections due to rapidly growing mycobacteria: case series and literature review. Infection and Chemotherapy. 2013;45(1):85–93. doi: 10.3947/ic.2013.45.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koh W-J, Kim YH, Kwon OJ, et al. Surgical treatment of pulmonary diseases due to nontuberculous mycobacteria. Journal of Korean Medical Science. 2008;23(3):397–401. doi: 10.3346/jkms.2008.23.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bux-Gewehr I, Hagen HP, Rusch-Gerdes S, Feurle GE. Fatal pulmonary infection with Mycobacterium celatum in an apparently immunocompetent patient. Journal of Clinical Microbiology. 1998;36(2):587–588. doi: 10.1128/jcm.36.2.587-588.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tjhie JHT, Van Belle AF, Dessens-Kroon M, Van Soolingen D. Misidentification and diagnostic delay caused by a false-positive amplified Mycobacterium tuberculosis direct test in an immunocompetent patient with a Mycobacterium celatum infection. Journal of Clinical Microbiology. 2001;39(6):2311–2312. doi: 10.1128/JCM.39.6.2311-2312.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bull TJ, Shanson DC, Archard LC, Yates MD, Hamid ME, Minnikin DE. A new group (type 3) of Mycobacterium celatum isolated from AIDS patients in the London area. International Journal of Systematic Bacteriology. 1995;45(4):861–862. doi: 10.1099/00207713-45-4-861. [DOI] [PubMed] [Google Scholar]
- 9.Tortoli E, Piersimoni C, Bacosi D, et al. Isolation of the newly described species Mycobacterium celatum from AIDS patients. Journal of Clinical Microbiology. 1995;33(1):137–140. doi: 10.1128/jcm.33.1.137-140.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gholizadeh Y, Varnerot A, Maslo C, et al. Mycobacterium celatum infection in two HIV-infected patients treated prophylactically with rifabutin. European Journal of Clinical Microbiology and Infectious Diseases. 1998;17(4):278–281. doi: 10.1007/BF01699987. [DOI] [PubMed] [Google Scholar]
- 11.Piersimoni C, Tortoli E, de Lalla F, et al. Isolation of Mycobacterium celatum from patients infected with human immunodeficiency virus. Clinical Infectious Diseases. 1997;24(2):144–147. doi: 10.1093/clinids/24.2.144. [DOI] [PubMed] [Google Scholar]
- 12.Piersimoni C, Zitti PG, Nista D, Bornigia S. Mycobacterium celatum pulmonary infection in the immunocompetent: case report and review. Emerging Infectious Diseases. 2003;9(3):399–402. doi: 10.3201/eid0903.020342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaufmann SHE. How can immunology contribute to the control of tuberculosis? Nature Reviews Immunology. 2001;1(1):20–30. doi: 10.1038/35095558. [DOI] [PubMed] [Google Scholar]
- 14.Tufariello JM, Chan J, Flynn JL. Latent tuberculosis: mechanisms of host and bacillus that contribute to persistent infection. The Lancet Infectious Diseases. 2003;3(9):578–590. doi: 10.1016/s1473-3099(03)00741-2. [DOI] [PubMed] [Google Scholar]
- 15.Iona E, Pardini M, Gagliardi MC, et al. Infection of human THP-1 cells with dormant Mycobacterium tuberculosis . Microbes and Infection. 2012;14(11):959–967. doi: 10.1016/j.micinf.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion vesicular proton-ATPase. Science. 1994;263(5147):678–681. doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- 17.Sanjurjo L, Amezaga N, Vilaplana C, et al. The scavenger protein apoptosis inhibitor of macrophages (AIM) potentiates the antimicrobial response against Mycobacterium tuberculosis by enhancing autophagy. PLoS ONE. 2013;8(11) doi: 10.1371/journal.pone.0079670.e79670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pieters J. Mycobacterium tuberculosis and the Macrophage: maintaining a balance. Cell Host and Microbe. 2008;3(6):399–407. doi: 10.1016/j.chom.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Marques da Silva TR, De Freitas JR, Chagas Silva Q, et al. Virulent Mycobacterium fortuitum restricts NO production by a gamma interferon-activated J774 cell line and phagosome-lysosome fusion. Infection and Immunity. 2002;70(10):5628–5634. doi: 10.1128/IAI.70.10.5628-5634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. Journal of the American Medical Association. 1999;282(7):677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 21.Stewart GR, Robertson BD, Young DB. Tuberculosis: a problem with persistence. Nature Reviews: Microbiology. 2003;1(2):97–105. doi: 10.1038/nrmicro749. [DOI] [PubMed] [Google Scholar]
- 22.Rivero-Lezcano OM, González-Cortés C, Reyes-Ruvalcaba D, Diez-Tascón C. CCL20 is overexpressed in Mycobacterium tuberculosis-infected monocytes and inhibits the production of reactive oxygen species (ROS) Clinical and Experimental Immunology. 2010;162(2):289–297. doi: 10.1111/j.1365-2249.2010.04168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vandal OH, Pierini LM, Schnappinger D, Nathan CF, Ehrt S. A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis . Nature Medicine. 2008;14(8):849–854. doi: 10.1038/nmXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh A, Crossman DK, Mai D, et al. Mycobacterium tuberculosis WhiB3 Maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathogens. 2009;5(8) doi: 10.1371/journal.ppat.1000545.e1000545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouley DM, Ghori N, Mercer KL, Falkow S, Ramakrishnan L. Dynamic nature of host-pathogen interactions in Mycobacterium marinum granulomas. Infection and Immunity. 2001;69(12):7820–7831. doi: 10.1128/IAI.69.12.7820-7831.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menendez MC, Garcia MJ, Navarro MC, et al. Characterization of an rRNA operon (rrnB) of Mycobacterium fortuitum and other mycobacterial species: implications for the classification of mycobacteria. Journal of Bacteriology. 2002;184(4):1078–1088. doi: 10.1128/jb.184.4.1078-1088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Theus SA, Cave MD, Eisenach KD. Activated THP-1 cells: an attractive model for the assessment of intracellular growth rates of Mycobacterium tuberculosis isolates. Infection and Immunity. 2004;72(2):1169–1173. doi: 10.1128/IAI.72.2.1169-1173.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chacón-Salinas R, Serafín-López J, Ramos-Payán R, et al. Differential pattern of cytokine expression by macrophages infected in vitro with different Mycobacterium tuberculosis genotypes. Clinical and Experimental Immunology. 2005;140(3):443–449. doi: 10.1111/j.1365-2249.2005.02797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simeone R, Bobard A, Lippmann J, et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathogens. 2012;8(2) doi: 10.1371/journal.ppat.1002507.e1002507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Doerfler M, Lee TC, Guillemin B, Rom WN. Mechanisms of stimulation of interleukin-1β and tumor necrosis factor-α by Mycobacterium tuberculosis components. Journal of Clinical Investigation. 1993;91(5):2076–2083. doi: 10.1172/JCI116430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Broser M, Cohen H, et al. Enhanced interleukin-8 release and gene expression in macrophages after exposure to Mycobacterium tuberculosis and its components. Journal of Clinical Investigation. 1995;95(2):586–592. doi: 10.1172/JCI117702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bange FC, Brown AM, Jacobs WR., Jr. Leucine auxotrophy restricts growth of Mycobacterium bovis BCG in macrophages. Infection and Immunity. 1996;64(5):1794–1799. doi: 10.1128/iai.64.5.1794-1799.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arriaga AK, Orozco EH, Aguilar LD, Rook GAW, Hernández Pando R. Immunological and pathological comparative analysis between experimental latent tuberculous infection and progressive pulmonary tuberculosis. Clinical and Experimental Immunology. 2002;128(2):229–237. doi: 10.1046/j.1365-2249.2002.01832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vieira OV, Jordao L. Tuberculosis: new aspects of an old disease. International Journal of Cell Biology. 2011;2011:13 pages. doi: 10.1155/2011/403623.403623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 2000;288(5471):1647–1650. doi: 10.1126/science.288.5471.1647. [DOI] [PubMed] [Google Scholar]
- 36.Dao DN, Kremer L, Guérardel Y, et al. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infection and Immunity. 2004;72(4):2067–2074. doi: 10.1128/IAI.72.4.2067-2074.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torrelles JB, Schlesinger LS. Diversity in Mycobacterium tuberculosis mannosylated cell wall determinants impacts adaptation to the host. Tuberculosis. 2010;90(2):84–93. doi: 10.1016/j.tube.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guérardel Y, Maes E, Elass E, et al. Structural study of lipomannan and lipoarabinomannan from Mycobacterium chelonae: presence of unusual components with α1,3-mannopyranose side chains. Journal of Biological Chemistry. 2002;277(34):30635–30648. doi: 10.1074/jbc.M204398200. [DOI] [PubMed] [Google Scholar]
- 39.Armitige LY, Jagannath C, Wanger AR, Norris SJ. Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infection and Immunity. 2000;68(2):767–778. doi: 10.1128/iai.68.2.767-778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar D, Rao KVS. Regulation between survival, persistence, and elimination of intracellular mycobacteria: a nested equilibrium of delicate balances. Microbes and Infection. 2011;13(2):121–133. doi: 10.1016/j.micinf.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 41.Russell DG. Phagosomes, fatty acids and tuberculosis. Nature Cell Biology. 2003;5(9):776–778. doi: 10.1038/ncb0903-776. [DOI] [PubMed] [Google Scholar]
- 42.Koul A, Herget T, Klebl B, Ullrich A. Interplay between mycobacteria and host signalling pathways. Nature Reviews Microbiology. 2004;2(3):189–202. doi: 10.1038/nrmicro840. [DOI] [PubMed] [Google Scholar]
- 43.Gengenbacher M, Kaufmann SHE. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiology Reviews. 2012;36(3):514–532. doi: 10.1111/j.1574-6976.2012.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vergne I, Chua J, Singh SB, Deretic V. Cell biology of Mycobacterium tuberculosis phagosome. Annual Review of Cell and Developmental Biology. 2004;20:367–394. doi: 10.1146/annurev.cellbio.20.010403.114015. [DOI] [PubMed] [Google Scholar]
- 45.Oberley-Deegan RE, Rebits BW, Weaver MR, et al. An oxidative environment promotes growth of Mycobacterium abscessus . Free Radical Biology and Medicine. 2010;49(11):1666–1673. doi: 10.1016/j.freeradbiomed.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okahashi N, Okinaga T, Sakurai A, et al. Streptococcus sanguinis induces foam cell formation and cell death of macrophages in association with production of reactive oxygen species. FEMS Microbiology Letters. 2011;323(2):164–170. doi: 10.1111/j.1574-6968.2011.02375.x. [DOI] [PubMed] [Google Scholar]
- 47.Kim WH, Goo SY, Lee K-H, Park S-J. Vibrio vulnificus-induced cell death of human mononuclear cells requires ROS-dependent activation of p38 and ERK 1/2 MAPKs. Immunological Investigations. 2009;38(1):31–48. doi: 10.1080/08820130802500583. [DOI] [PubMed] [Google Scholar]
- 48.Aulik NA, Hellenbrand KM, Czuprynski CJ. Mannheimia haemolytica and its leukotoxin cause macrophage extracellular trap formation by bovine macrophages. Infection and Immunity. 2012;80(5):1923–1933. doi: 10.1128/IAI.06120-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bohsali A, Abdalla H, Velmurugan K, Briken V. The non-pathogenic mycobacteria M. smegmatis and M. fortuitum induce rapid host cell apoptosis via a caspase-3 and TNF dependent pathway. BMC Microbiology. 2010;10, article 237 doi: 10.1186/1471-2180-10-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delbridge LM, O’Riordan MX. Innate recognition of intracellular bacteria. Current Opinion in Immunology. 2007;19(1):10–16. doi: 10.1016/j.coi.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 51.Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140(6):771–776. doi: 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 52.Chen C-C, Tsai S-H, Lu C-C, et al. Activation of an NLRP3 inflammasome restricts Mycobacterium kansasii infection. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0036292.e36292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abebe F, Mustafa T, Nerland AH, Bjune GA. Cytokine profile during latent and slowly progressive primary tuberculosis: a possible role for interleukin-15 in mediating clinical disease. Clinical and Experimental Immunology. 2006;143(1):180–192. doi: 10.1111/j.1365-2249.2005.02976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salgame P. Host innate and Th1 responses and the bacterial factors that control Mycobacterium tuberculosis infection. Current Opinion in Immunology. 2005;17(4):374–380. doi: 10.1016/j.coi.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 55.Flynn JL, Chan J. Immunology of tuberculosis. Annual Review of Immunology. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 56.Mayer-Barber KD, Barber DL, Shenderov K, et al. Cutting edge: caspase-1 independent IL-1β production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. The Journal of Immunology. 2010;184(7):3326–3330. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayer-Barber KD, Andrade BB, Barber DL, et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary Myeloid subsets during Mycobacterium tuberculosis infection. Immunity. 2011;35(6):1023–1034. doi: 10.1016/j.immuni.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McElvania Tekippe E, Allen IC, Hulseberg PD, et al. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS ONE. 2010;5(8) doi: 10.1371/journal.pone.0012320.e12320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Briken V, Ahlbrand SE, Shah S. Mycobacterium tuberculosis and the host cell inflammasome: a complex relationship. Frontiers in Cellular and Infection Microbiology. 2013;3(article 62) doi: 10.3389/fcimb.2013.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Volpe E, Cappelli G, Grassi M, et al. Gene expression profiling of human macrophages at late time of infection with Mycobacterium tuberculosis . Immunology. 2006;118(4):449–460. doi: 10.1111/j.1365-2567.2006.02378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu B, Huang C, Kato-Maeda M, et al. Messenger RNA expression of IL-8, FOXP3, and IL-12β differentiates latent tuberculosis infection from disease. The Journal of Immunology. 2007;178(6):3688–3694. doi: 10.4049/jimmunol.178.6.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee HM, Yuk JM, Kim KH, et al. Mycobacterium abscessus activates the NLRP3 inflammasome via Dectin-1-Syk and p62/SQSTM1. Immunology and Cell Biology. 2012;90(6):601–610. doi: 10.1038/icb.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferrero E, Biswas P, Vettoretto K, et al. Macrophages exposed to Mycobacterium tuberculosis release chemokines able to recruit selected leucocyte subpopulations: focus on γδ cells. Immunology. 2003;108(3):365–374. doi: 10.1046/j.1365-2567.2003.01600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nibbering PH, Pos O, Stevenhagen A, Van Furth R. Interleukin-8 enhances nonoxidative intracellular killing of Mycobacterium fortuitum by human granulocytes. Infection and Immunity. 1993;61(8):3111–3116. doi: 10.1128/iai.61.8.3111-3116.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mcgarvey JA, Wagner D, Bermudez LE. Differential gene expression in mononuclear phagocytes infected with pathogenic and non-pathogenic mycobacteria. Clinical and Experimental Immunology. 2004;136(3):490–500. doi: 10.1111/j.1365-2249.2004.02490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]