

Abstract
Troponin I (TnI) is the molecular switch of the sarcomere. Cardiac myocytes express two isoforms of TnI during development. The fetal heart expresses the slow skeletal TnI (ssTnI) isoform and shortly after birth ssTnI is completely and irreversibly replaced by the adult cardiac TnI (cTnI) isoform. These two isoforms have important functional differences; broadly, ssTnI is a positive inotrope, especially under acidic/hypoxic conditions, whereas cTnI facilitates faster relaxation performance. Evolutionary directed changes in cTnI sequence suggest cTnI evolved to favor relaxation performance in the mammalian heart. To investigate the mechanism, we focused on several notable TnI isoform and trans-species-specific residues located in TnI’s helix 4 using structure/function and molecular dynamics analyses. Gene transduction of adult cardiac myocytes by cTnIs with specific helix 4 ssTnI substitutions, Q157R/A164H/E166V/H173N (QAEH), and A164H/H173N (AH), were investigated. cTnI QAEH is similar in these four residues to ssTnI and nonmammalian chordate cTnIs, whereas cTnI AH is similar to fish cTnI in these four residues. In comparison to mammalian cTnI, cTnI QAEH and cTnI AH showed increased contractility and slowed relaxation, which functionally mimicked ssTnI expressing myocytes. cTnI QAEH molecular dynamics simulations demonstrated altered intermolecular interactions between TnI helix 4 and cTnC helix A, specifically revealing a new, to our knowledge, electrostatic interaction between R171of cTnI and E15 of cTnC, which structurally phenocopied the ssTnI conformation. Free energy perturbation calculation of cTnC Ca2+ binding for these conformations showed relative increased calcium binding for cTnI QAEH compared to cTnI. Taken together, to our knowledge, these new findings provide evidence that the evolutionary-directed coordinated acquisition of residues Q157, A164, E166, H173 facilitate enhanced relaxation performance in mammalian adult cardiac myocytes.
Introduction
Cardiac contractility is a fine-tuned process of allosteric regulation by Ca2+ activation of thin and thick myofilament interaction and force production. The heterotrimeric troponin complex is the key regulator in contractility, sensing Ca2+ and subsequently regulating myosin interaction with actin (1,2). Alterations in troponin affinity for Ca2+ due to isoform switching, biochemical milieu, or human mutations dramatically alter troponin and heart performance (3). Understanding the mechanism of troponin function is therefore critical to ultimately developing strategies that redress pathogenic alterations in contractility.
Cardiac troponin is composed of troponin C (cTnC), the Ca2+ sensitive subunit, troponin I (cTnI), the inhibitory subunit, and troponin T (cTnT), the tropomyosin binding subunit. Upon excitation of cardiac myocytes, Ca2+ is released from the sarcoplasmic reticulum, resulting in a rapid and marked increase in intracellular Ca2+. Cardiac TnC binds Ca2+ to partially open the N-terminal hydrophobic pocket and allow the switch region of cTnI (∼residues 147–163) to then interact and fully open this hydrophobic pocket of cTnC (4). Interaction of the cTnI switch peptide region with cTnC is modeled to pull the cTnI inhibitory region (∼residues 128–146) and mobile domain (∼residues 172–210) away from actin, resulting in movement of tropomyosin and revealing myosin strong binding sites on actin (5). Release of Ca2+ from cTnC and the subsequent disengagement of cTnI allows the inhibitory region and mobile domain of cTnI to again strongly interact with actin to inhibit myosin strong binding and thus initiate relaxation. The dynamic Ca2+-dependent interchange between cTnI and cTnC/actin makes cTnI the molecular switch of the sarcomere.
In mammalian cardiac development, two TnI isoforms are sequentially expressed, ssTnI (TNNI1), the neonatal isoform, and cTnI (TNNI3), the adult isoform. In the first 20 days after birth in rodents and in newborn humans, a complete and irreversible stoichiometric shift from ssTnI to the cTnI isoform occurs in the cardiac sarcomere (6,7); experimentally blocking this transition by the transgenic expression of ssTnI in adult mice results in diastolic dysfunction (8). In terms of cardiac function, ssTnI and cTnI show three main biophysical differences: 1), ssTnI shows increased Ca2+ sensitivity under physiologic conditions; 2), ssTnI shows marked resiliency to ischemia/acidosis-mediated Ca2+ desensitization of the thin filament; and 3), cTnI has a unique 32 amino acid N-terminal extension necessary for PKA-regulated Ca2+ desensitization of the thin filament required to hasten relaxation performance (9,10). These functional differences along with the highly regulated and nonreversible developmental TnI isoform switch suggest that mammalian cTnI has evolved to favor enhanced relaxation performance over preserved inotropy in ischemia/acidosis.
Phylogenetic analysis of TnI through chordate evolution reveals that the amphibian and avian cTnI isoform is more similar to mammalian ssTnI than to mammalian cTnI (11). This difference is highly notable in the switch/helix 4 region of cTnI, the region that interacts with cTnC during Ca2+ binding. Four amino acid charge changes in mammalian cTnI compared to other chordate TnIs (either cTnI or ssTnI) are evident in this analysis: Q157R, A164H, E166V, and H173N (mammalian cTnI to ssTnI residues, respectively). Based on this analysis, we proposed a model whereby a key histidine to alanine transition at position 164 was evolutionarily selected for enhanced relaxation performance in the endothermic adult heart (11). A single histidine residue in ssTnI (H132) is responsible for the marked resistance to pH-dependent desensitization (12,13). Substitution of cognate residue A164 with histidine in cTnI results in marked protection from ischemia/acidosis-mediated Ca2+ desensitization in vitro and in vivo (13–16). Mechanistically, this histidine is hypothesized to be protonated under acidic conditions to then interact with E19 of cTnC increasing affinity of ssTnI for cTnC (11,17). Further evidence from molecular dynamics (MD) simulations and NMR suggest that this electrostatic interaction also occurs in cTnI A164H (18,19). Taken together, these studies support the necessity of histidine 132(ssTnI)/164(cTnI) in conferring resistance to Ca2+ desensitization in acidic conditions through its interaction with E19 of cTnC (11,17–19).
Although cTnI A164H mimics ssTnI resistance to Ca2+ desensitization in acidic conditions, it differs from ssTnI in two ways. First, cTnI A164H has the N-terminal extension that contains the two critical PKA sites for increased relaxation performance during adrenergic stimulation (20). Second, cTnI A164H shows little to no alteration in Ca2+ sensitivity compared to wild-type (WT) cTnI at neutral pH (13). Earlier work showed that ssTnI H132A functionally mimicked cTnI, showing that ssTnI H132 imparts pH resistance while retaining ssTnI baseline pH Ca2+ sensitivity (12). Because cTnI A164H does not have significant increased Ca2+ sensitivity (like ssTnI) at baseline, other TnI isoform-specific residues must impart heightened baseline Ca2+ sensitivity to ssTnI. Stated differently, it is unclear why cTnI A164H confers selected Ca2+ sensitivity enhancement at acidic pH and not at neutral pH distinguishing it from ssTnI function. Dargis and colleagues (16) showed in thin filament regulated myosin ATPase activity assays that the ssTnI isoform substitutions Q157R/A164H/E166V in cTnI cause apparent increased Ca2+ sensitivity, whereas Q157R/E166V cTnI showed no increased Ca2+ sensitivity compared to cTnI. This suggests that a specific histidine (cTnI A164H) is necessary for increased Ca2+ sensitivity but only in the context of other, yet to be defined TnI isoform-specific residues (16).
A central aim of this study is to dissect TnI residues key for enabling cardiac contractile performance in health and disease. To gain mechanistic insight into Ca2+ desensitization of cTnI compared to ssTnI, we determine here the importance of the evolutionary conserved isoform-specific residues Q157, A164, E166, and H173 of cTnI. Functional analysis in adult cardiac myocytes paired with MD simulations and free energy perturbation analysis of cTnI Q157R/A164H/E166V/H173N (QAEH) establishes a basis for increased TnI-TnC interaction resulting in increased contractility. Based on these new, to our knowledge, findings we propose that two salt bridges between cTnI QAEH and cTnC alter Ca2+ binding to cTnC and therefore increase contractility and slow relaxation. These findings will be useful in informing future gene, cell, and small molecule-based strategies to enhance cardiac contractility in disease.
Methods
Generation of cTnI isoform substitutions and viral vector construction
We used a pGEM3Z vector containing cTnI-Flag, A164H cTnI-flag, A164H/E166V cTnI-flag, and Q157R/A164H/E166V cTnI-flag, and the QuikChange mutagenesis kit (Stratagene, LaJolla, CA) to generate site-directed mutants according to the manufacturer’s protocol. Primers used for mutagenesis for Q157R: sense 5′- GCA GAT GCC ATG ATG CGG GCA CTA CTG GGG ACC -3′, antisense 5′- GGT CCC CAG TAG TGC CCG CAT CAT GGC ATC TGC -3′ for H173N: sense 5′- CCT TGG ACC TGA GGG CCA ACC TCA AGC AGG TGA AG -3′, antisense 5′- CTT CAC CTG CTT GAG GTT GGC CCT CAG GTC CAA GG -3′. Mutated DNA was sequenced before subcloning of mutant cTnI cDNA into pDC315 adenovirus shuttle vector. All DNA sequences were verified by overlapping sequence runs. Recombinant Ad5 adenoviruses were produced and purified as described previously (21). Adenoviruses for ssTnI, cTnI-flag, and A164H cTnI-flag were generated previously (9,13) and used as controls here.
Ventricular myocyte isolation, gene transfer, primary culture, immunoblot, indirect immunofluorescence, and contractility were performed as previously described (22,23) and detailed in the Supporting Material.
MD simulations
The simulations were conducted as previously described (19). Briefly, the starting structures were obtained from Protein Data Bank (PDB) 1J1E chains D and F for cTnC (residues 1–90) and cTnI (residues 148–174), respectively. For sTnI (residues115–140) PDB 1YTZ was used and sTnI was modeled onto cTnC. Missing amino acids G50 and Q51 of cTnC and missing hydrogen atoms were added and energy minimized using OPLS 2005 force field (24). The imidazole groups of all histidines in the structure were ionized according to the calculated pKas. All simulations were carried out using NAMD version 2.6 with CHARMM27 protein force field and TIP3P water model (25). Each protein complex was solvated in a rectangular box with a 15 Å water buffer from the protein. Na+ and Cl− counter ions were added at 5 Å from the box boundary to neutralize the total charge of the system. Each system was initialized by a 5000 steps conjugate gradient energy minimization with protein heavy atoms restrained at 50 kcal/(mol•Å2). The restraint system was gradually heated from 25 to 300 K at 25 K increments at every 10 ps interval for 100 ps followed by a 100 ps equilibration with gradual removal of the heavy atoms restraint at 10 ps interval under a NVT condition. The final unrestrained equilibration was carried out for 100 ps followed by 40 ns of production simulation at 1 atm and 300 K NPT condition. The simulations were carried out under a periodic boundary condition using particle mesh Ewald (26). The SHAKE (27) method was employed to restrain all hydrogen bonds. A 2 fs time step was used with coordinates saved at 1 ps time intervals, resulting in a total of 40,000 configurations for simulation. Three to five 40 ns simulations were carried out for each of the modeled complexes by random initialization of the starting velocities. These simulations were conducted using computational resources from the University of Minnesota Supercomputing Institute.
Interatomic distance calculations
All distances were evaluated in angstrom units over the entire 40 ns of the simulation. For the cTnI QAEH HSD and cTnI QAEH HSP simulations of the cTnI:cTnC complex and the sTnI of the sTnI:cTnC complex, the average distance was evaluated between cTnI H164 (NE2) or sTnI H132 (NE2) and cTnC E19 (OE2). Distance measurements for the WT structure were taken between cTnI A164 (CB) or sTnI A132 (CB), respectively, and cTnC E19 (OE2).Other distance analysis was done for all the simulations between cTnI R171 (NH2) and cTnC E15 (OE2), between cTnC E40 (OE2) and cTnC K39 (NZ) and cTnC K43 (NZ).
Theoretical pKa calculations
The pKa was carried out as previously described (19,28). Briefly, the protein pKa of ionizable residue, , is evaluated by
| (1) |
Here, is the model pKa of the amino acid side chain in water. The electrostatic contribution toward the self-free energy, was obtained by evaluating the average electrostatic potential based on the numerical solution to the linearized Poisson-Boltzmann equation
| (2) |
where ε(r) is the distance dependent dielectric constant, ϕ(r) is the electrostatic potential at point r, is the permanent charge density of the solute and κ is inverse Debye-Huckle salt screening length. The electrostatic contribution toward the changes in the change in the self-free energy, , of a given residue, i, can be described by
| (3) |
where is the interaction between the ionized group with the protein permanent dipoles and is free energy of desolvation of moving the group from water to the protein.
These first two terms account for the free energies associated with moving the indicated ionizable group in its neutral (AH) and ionized (A−) state from water to the protein when all other groups are in their uncharged state and the latter term, , account for the charge-charge interaction among all ionizable groups inside the protein. The protein pKa, therefore, is given by
| (4) |
where is the intrinsic pKa of the ionizable residue inside the protein when all other ionizable groups are in their neutral states and represents the pKa shifts due to the charge-charge interaction between the indicated titratable group with all other ionized groups in their average ionization states in the protein. This methodology allows for a straightforward decomposition of the total computed pKa value as the sum of the effects due to the change in moving the ionized group from water to the polar environment of the protein when all ionizable groups are set to their neutral state and its interaction with all other ionizable groups . Thus, the calculated pKa values can be used to characterize the strength of electrostatic interactions between neighboring groups in protein.
In this work the calculations were carried out based on the web-based implementation of the linearized Poisson-Boltzmann implicit solvent model available via H++ server (29). A protein dielectric constant of 4 and 40 was used for the intrinsic pKa calculation and the , respectively. Three independent frames from three simulations were used to calculate the average pKa measures shown in Table S1.These frames were stable conformations and spaced equally throughout the simulations at 20, 30, and 40 ns. H++ does not allow pKa values to exceed 12, therefore, arginine are not a true representation of electrostatic interaction strength.
Free energy perturbation calculation
The relative binding free energy of the Ca2+ ion between the WT and its mutant was evaluated using the free energy perturbation (FEP) method that has been used previously for the studies of charges in solution (30,31) and proteins (32). The binding free energy, ΔG, associated for ionizing a charge within its water or protein-water environment involves the adiabatic charging process where the charge (Q) of the Ca2+ ion is gradually changed from = 0 → = where = +2 for Ca2+ ion. The free energy is given by
| (5) |
where is the mapping potential and is the mapping parameter. The complete charging process involves 11 mapping steps with each mapping step performed at 300 K for 20 ps with a step size of 1 fs. To prevent the diffusion of the Ca2+ ion from the binding pocket when its charge is gradually changed to zero, a harmonic restraint of 200 kcal/mol Å2 was applied to restrain the nearby chelating residues (Asp-65, Asp-67, Asp-73 and Glu-76, and Ser-69) to crystallography observed Ca2+ ion position and distances. Four independent frames from 10 to 40 ns were analyzed for each simulation on the 3–5 simulations run per variant. The relative association binding free energy was then evaluated by the difference between the WT and its mutant. The FEP calculations were conducted within the Molaris 9.09 software modeling package (33).
Statistics
All results are expressed as mean ± SE. Multigroup comparisons were assessed using one-way analysis of variance with the Bonnferoni post-hoc test with P < 0.05 considered statistically different.
Results
Replacement and incorporation of cTnI substitutions by gene delivery
To determine the structure-function relationship of the isoform-specific residue substitutions between cTnI and ssTnI, we used in vitro cellular methods. Recombinant adenoviruses were generated to express the cTnI to ssTnI substitutions as shown in Fig. 1. The four isoform-specific residues are highlighted and denote residues that change significantly during chordate evolution (11,34). This study is aimed at identifying the impact of these isoform-specific residues through substitution of ssTnI residues, Q157R, A164H, E166V, and H173N in the context of the mammalian cTnI template.
Figure 1.
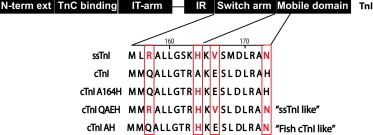
Troponin I isoform sequence alignment highlighting the substitutions used in this study. Top bar depicts the domains of cTnI showing the sequence region of the switch arm and a portion of the mobile domain. Differences between cTnI and ssTnI boxed in red. Substitutions from cTnI to ssTnI are marked in red for the ssTnI amino acid. To see this figure in color, go online.
To study the effects of the cTnI substitutions in vitro, adult rat cardiac myocytes were transduced with recombinant adenoviral vectors encoding the cTnI to ssTnI substitutions Q157R/A164H/E166V/H173N (QAEH) or A164H/H173N (AH). This system approaches 100% efficiency for myocyte transduction and takes advantage of the well-documented stoichiometric replacement of endogenous TnI (9,23,35,36). All cTnI substitutions were Flag-tagged to track the modified TnIs through differential migration in SDS-PAGE compared to the endogenous and through immunofluorescence for analysis of incorporation into the sarcomere. We showed previously that this epitope does not alter function (12,19,23,36). Three days after transduction cardiac myocytes were harvested for expression and replacement studies via Western blot and indirect immunofluorescence for detection of incorporation into the thin filament. As has been well documented (9,19,23,37), cTnI gene transfer showed stoichiometric replacement of the endogenous protein with the cTnI-Flag substitutions or the ssTnI. Fig. 2 A shows that the endogenous cTnI band decreases as the cTnI-Flag band incorporates into the sarcomere. The Western blots were quantified using Odyssey analysis to determine the extent of replacement. All the TnI substitutions showed 70–80% stoichiometric replacement as shown in Fig. 2 B. Sarcomeric incorporation of these cTnI substitutions was determined via indirect immunofluorescence through the Flag epitope. Fig. 2 C shows that the cTnI substitutions have a sarcomeric staining pattern consistent with proper incorporation into the thin filament.
Figure 2.
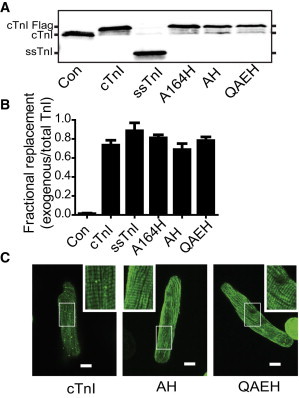
Replacement and incorporation of the adenoviral transduced adult cardiac myocytes. (A) Representative Western blot of adult rat ventricular myocytes day 3 after transduction probed with a pan-TnI antibody. Flag-cTnI, endogenous cTnI and ssTnI labels denote expected sizes for each isoform. All cTnI isoforms are Flag tagged. (B) Quantification of TnI replacement based on Western blot comparing exogenous (Flag band) to total TnI (n = 9–12 for each group from 3 to 4 independent experiments). (C) Indirect immunofluorescence of FLAG-tagged substitutions showing normal sarcomeric staining. Measurement bar = 10 μm. Inset shows normal thin filament staining for the two substitutions cTnI AH and cTnI QAEH compared to WT cTnI. To see this figure in color, go online.
Sarcomere dynamics in living cells under physiologic and acidic pH conditions
Sarcomere length dynamics in single adult rat cardiac myocytes transduced with cTnI substitutions were conducted to ascertain the functional impact of these substitutions at pH 7.4 and pH 6.2. All studies were conducted on day 3 after gene transfer. The cTnI substitutions QAEH and AH were compared to cTnI, cTnI A164H, and ssTnI (Fig. 3). Fractional shortening %, is a measure of contractility and shows that ssTnI, cTnI QAEH, and cTnI AH have heightened contractility, whereas cTnI A164H shows normal contractility under physiologic conditions (Fig. 3 C). As a measure of relaxation, time to baseline 50% shows that cTnI QAEH and cTnI AH have slowed relaxation (Fig. 3 D). Taken together these data are consistent with cTnI QAEH and cTnI AH as Ca2+ sensitive substitutions, much like ssTnI, and functionally resemble ssTnI more than either cTnI or cTnI A164H.
Figure 3.
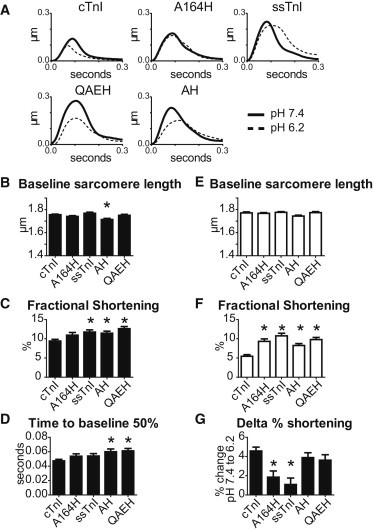
Sarcomere length dynamics at physiologic pH and in acidosis. (A) Representative traces of sarcomere length dynamics normalized to baseline of the five groups (Black = pH 7.4, White = pH 6.2). (B–D) pH 7.4 sarcomere length dynamics showing baseline sarcomere length, fractional shortening, and the relaxation parameter time from peak amplitude to 50% baseline. (E and F) pH 6.2 sarcomere length dynamics showing baseline sarcomere length and fractional shortening. (G) The change in fractional shortening from pH 6.2 to pH 7.4. n = 45–60 myocytes from six independent experiments for each group. ∗P < 0.05 from one-way analysis of variance with the Bonferroni post hoc test.
We previously showed that cTnI A164H and ssTnI preserve contractile function under acidic conditions compared to cTnI (9,13,19). To determine the impact of cTnI substitutions on contractile function in acidosis, we tested sarcomere length dynamics at pH 6.2. Consistent with previous publications (9,13,19), cTnI A164H and ssTnI maintain fractional shortening at pH 6.2 compared to cTnI (Fig. 3 F). cTnI QAEH and cTnI AH showed similar fractional shortening in acidic conditions to cTnI A164H and ssTnI. The change in fractional shortening between pH 7.4 and pH 6.2 was calculated to determine if each substitution preserved contractile function in acidosis. Fig. 3 G shows that only cTnI A164H and ssTnI have delta fractional shortening that is not statistically different from 0% indicating that these TnI constructs preserve contractile function in acidosis. Although cTnI QAEH and cTnI AH have similar pH 6.2 fractional shortening to cTnI A164H and ssTnI, they show delta fractional shortening values that are similar to cTnI. This suggests that they are baseline Ca2+ sensitizers and are not pH titratable molecular inotropes as found in cTnI A164H.
MD simulations
To gain structural insight into the mechanism of modified cTnI function we performed MD simulations. Simulations were carried out with sTnI-cTnC, cTnI-cTnC, and cTnI QAEH-cTnC in both the histidine protonated (HSP) and deprotonated state (HSD). Starting structures were truncated cTnC residues 1–90 and cTnI residues 148–174 from the Ca2+ bound human cTn crystal structure (PDB, 1J1E, chains D and F) (38), and sTnI 115–140 from the Ca2+ bound chicken fast-skeletal troponin crystal structure (PDB, 1YTZ) (39) and mutated computationally. We used chicken fast-skeletal TnI as a surrogate for ssTnI, as these two proteins do not differ in the sequence region of interest (11). Three simulations were run with different starting velocities for each molecule over 40 ns to probe conformational space. Representative structures at 40 ns show major conformational differences between sTnI and cTnI (Fig. 4). The switch region in all simulations is generally in the same position and making the same contacts. The C-terminal half in sTnI, helix 4, is unstructured and bends to interact with cTnC helix A (Fig. 4 B), whereas in cTnI helix 4 is not interacting with cTnC (Fig. 4 B). Substitutions that make cTnI more ssTnI-like in function, cTnI QAEH, show a similar overall conformation as sTnI with a bend of helix 4 and closer proximity to cTnC helix A, only when the histidine is protonated to mimic acidosis (HSP) (Fig. 4 B).
Figure 4.
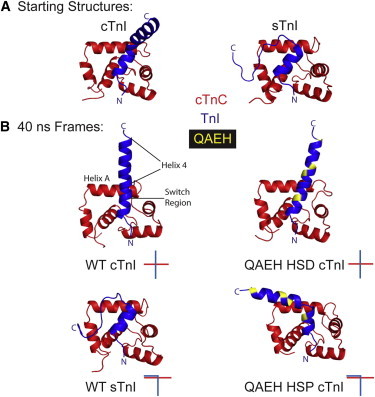
(A) Atomistic ribbon structure representations of the starting structures used for simulation. cTnI starting structure was the same for all cTnI variants. (B) Atomistic ribbon structure representations of the 40 ns frame from TnC-TnI MD simulations. Red is cTnC 1–90, Blue is cTnI 148–174 or sTnI 115–140, yellow shows the position of the QAEH residues that have been substituted with the ssTnI residue. HSP (histidine protonated), HSD (histidine deprotonated). Atomistic stick figure represents the cTnC helix A (red) position in relation to the TnI switch peptide-helix 4 position (blue). To see this figure in color, go online.
cTnI QAEH HSP helix 4 interactions
Previous work provided evidence that in the protonated state cTnI A164H and residue H132 of sTnI interact with E19 of cTnC (17,19). In this study, MD analysis of cTnI QAEH HSP showed H164 in close proximity to cTnC E19 throughout the 40 ns simulation for two of the three simulations (Fig. 5 A). Average as a measure of shift in pKa due to electrostatic interactions showed cTnC E19 - cTnI QAEH HSP has strong electrostatic interactions (−1.22) comparable to sTnI (−1.41). The electrostatic interaction for cTnI H164 was less (0.30) but was considerably different than cTnI QAEH HSD (−0.35) where this interaction did not occur (Table S1). This histidine to glutamic acid electrostatic interaction has been suggested to be the mechanism of maintained contractile function under acidic conditions when histidine would be protonated. Further analysis of the simulations revealed that cTnI QAEH HSP has additional interactions of cTnI helix 4 with cTnC. A key new interaction found here was R171 of cTnI as it is within 3 Å of E15 of cTnC in two out of three simulations (Fig. 5 D). These are the same two simulations that showed H164-E19 interaction. pKa analysis showed strong electrostatic interactions with cTnC E15 (−1.26) with cTnI QAEH HSP, although this residue has no apparent electrostatic interactions in cTnI WT or cTnI QAEH HSD (Table S1). H++ does not allow values to be >12, therefore, arginines do not show any and are not included in this analysis. This further interaction of helix 4 is similar to sTnI, which has many side chains within 3 Å of cTnC side chains in this region of the protein. In contrast, cTnI A164H simulations published by Palpant et al. (19) showed the cTnI H164-cTnC E19 interaction but no further interactions in helix 4.
Figure 5.
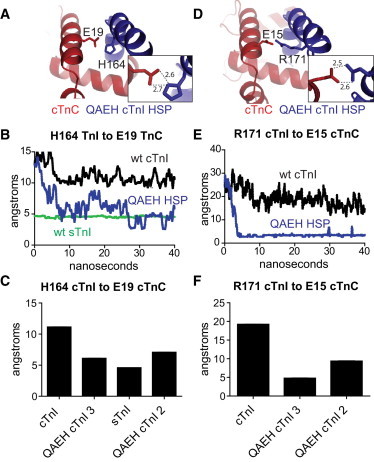
Ribbon structures of TnI Helix 4 interactions with cTnC. (A) Representative frame of QAEH cTnI 3 HSP showing H164 of TnI interaction with E19 of cTnC, cTnC Red, cTnI blue. Inset shows distances between E19 and H164. (B) Representative distance trace from H164 to E19 comparing one simulation from WT cTnI (black), sTnI (green), and QAEH cTnI (blue). (C) Average distance over the entire 40 ns error bars are +/− SE. (D) Representative frame of QAEH cTnI 3 HSP showing R171of cTnI interaction with E15 of cTnC, cTnC Red, cTnI blue. Inset shows distance between E15 and R171. (E) Representative distance trace from R171 to E15 comparing one simulation from WT cTnI (black) and QAEH cTnI (blue). (F) Average distance over the entire 40 ns, error bars are +/− SE. To see this figure in color, go online.
QAEH HSD cTnI alters cTnC intramolecular interactions
The deprotonated cTnI QAEH (HSD) showed an overall structure similar to cTnI. The switch region is tilted slightly but no overall gross conformational changes were observed. Analysis of pKa measurements revealed an electrostatic interaction in cTnC E40-K39 in the cTnI WT, simulations that shift to E40-K43 in the sTnI, cTnI QAEH HSP, and cTnI QAEH HSD (Table S1). Distance measurements across the entire 40 ns simulations showed this shift in TnC intramolecular interaction is stable throughout the simulations (Fig. 6). We speculate this shift may alter further interactions through helix C and site I of cTnC and therefore affect site II Ca2+ binding (40,41).
Figure 6.
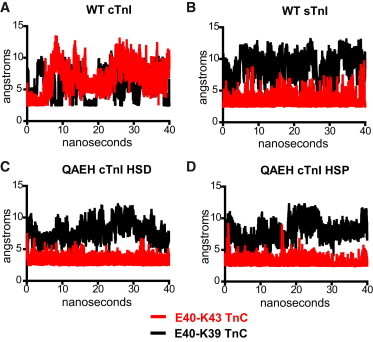
MD-derived intramolecular analysis of E40 cTnC distance measurements to cTnC residues K39 and K43. Black trace is E40-K39, Red trace is E40-K43. HSD (deprotonated histidines), HSP (protonated histidines). To see this figure in color, go online.
Free energy of cTnC Ca2+ binding
To investigate if the conformations obtained throughout the simulations affect Ca2+ binding affinity, we performed FEP calculations on sample frames from 10 to 40 ns for all isoforms. Distance constraints were put on the cTnC site II Ca2+ chelating residues Asp-65, Asp-67, Asp-73, Glu-76, and Ser-69, whereas the charge of the Ca2+ ion was gradually changed from 0 to +2. The binding free energy was averaged over the multiple simulations snapshots for the given isoform. The relative free energy of Ca2+ binding is shown in Fig. 7. This analysis showed decreased Ca2+ binding free energy for both cTnI QAEH HSD and HSP relative to cTnI, consistent with the proposition that these conformations seen throughout the simulation could facilitate increased Ca2+ binding affinity. We further performed FEP on the previously published simulations for cTnI A164H HSD and HSP (19). Here, cTnI A164H HSD showed no difference to WT cTnI, whereas cTnI A164H HSP showed marked decreased free energy of Ca2+ binding. These findings are consistent with the cellular data shown here where A164H cTnI has no neutral pH effect while preserving contractility in acidosis (Fig. 3).
Figure 7.
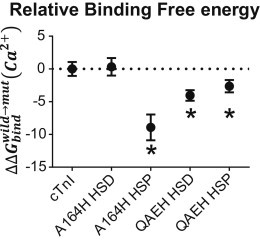
Relative Ca2+ binding free energy. The change in Ca2+ binding free energy normalized to the average of WT cTnI plotted as, . Four representative frames from all simulations for a given isoform were calculated and averaged, error bars are +/− SE. ∗p < 0.05 from a Student’s t-test to cTnI value.
Discussion
This study provides new, to our knowledge, biophysical insights into the TnI isoform-dependence of cardiac muscle contractility. The combination of sarcomere length dynamics in living adult cardiac myocytes together with state-of-the-art MD simulations reveals an emerging model of Ca2+ sensitivity in the mammalian adult cardiac myocyte. The main new findings presented here are that four evolutionary conserved mammalian-specific cTnI residues, Q157, A164, E166, and H173, in concert, facilitate normal contractility and relaxation properties in the mammalian adult heart. These four evolutionarily coordinated changes in TnI residues establish an important biophysical trade-off: gaining relaxation performance at the expense of preserving contractile vigor in ischemia.
In support of this model, free energy perturbation analysis of stable TnI-TnC conformations in MD simulations indicates that TnI’s Helix 4 conformation and interaction with cTnC is a key determinate of contractility and the basis for the difference in cTnI function compared to ssTnI. We propose salt bridge formation between H164 of cTnI and E19 of cTnC and R171 of cTnI and E15 of cTnC initiates an intramolecular conformation change in cTnC to cause increased Ca2+ binding affinity as revealed by cTnI QAEH. This leads to a model prediction of increased TnI-cTnC interaction leading to increased contractility and slowed relaxation because of ssTnI substitutions Q157R/A164H/E166V/H173N in cTnI. By this deeper understanding of TnI-TnC interactions a strong foundation is established to instruct potential gene, protein, and small molecule therapeutics in heart disease.
In Fig. 8, we present a synthesis of key isoform-specific TnI residues that impact structure and function based on our data presented here as well as previously published results (11,19). At neutral pH, cTnI and cTnI A164H have similar contractility. whereas ssTnI, cTnI QAEH, and cTnI AH have increased contractility. In acidosis, cTnI has decreased contractility, whereas all the other constructs maintain normal contractility. The change in contractility from neutral pH to acidic pH (delta pH change) shows that cTnI, cTnI QAEH, and cTnI AH have marked contractility changes, whereas ssTnI and cTnI A164H have comparatively small contractility changes. Relaxation time shows cTnI and cTnI A164H have normal relaxation at neutral pH, whereas ssTnI, cTnI QAEH, and cTnI AH have increased relaxation times. The atomistic structure depicting cTnC helix A in relation to cTnI switch peptide and helix 4 models a 90° bend of TnI for ssTnI and cTnI QAEH due to H164-E19 interaction and further interactions of helix 4 with cTnC helix A, specifically R171-E15 for cTnI QAEH. Furthermore, cTnI A164H has a slight bend in TnI, due to H164-E19 interaction but no further interactions of helix 4 with cTnC helix A. MD simulations were not performed for cTnI AH (N.D. for structure). The model suggests that the bent conformation with H164-E19 interaction seen in ssTnI, cTnI A164H, and cTnI QAEH maintains contractility in acidosis, although further interactions of TnI helix 4 with cTnC Helix A as for ssTnI and cTnI QAEH results in increased contractility and relaxation time at neutral pH. Taken as a whole, cTnIs QAEH and AH structurally and functionally mimic ssTnI, suggesting that these evolutionary conserved residues are necessary for normal mammalian heart function.
Figure 8.
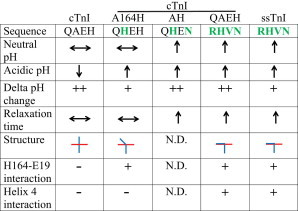
Summary of isoform-specific residues as molecular determinants of TnI function. Compilation of functional data depicting neutral pH and acidic pH contractility, the change in contractility from neutral pH to acidic pH (Delta pH change) and relaxation time at neutral pH. ↔ indicates no change in contractility/relaxation time compared to cTnI neutral pH, ↑ indicates increased contractility/relaxation time compared to cTnI, ↓ indicates decreased contractility with acidosis, + indicates small change in contractility from neutral pH to acidic pH, ++ indicates marked change in contractility from neutral pH to acidic pH. Sequence of the isoform-specific residues with black residues from cTnI and green residues from ssTnI. Atomistic stick figure showing cTnC helix A (red) in relation to cTnI switch peptide and helix 4 (blue) from MD simulations. N.D. (not determined). To see this figure in color, go online.
Studies comparing cTnI protein sequences across species reveal that in chordate evolution at the mammalian divergence there was a key histidine to alanine transition at position 164 (numbering corresponding to rodent cTnI) (11). All non-mammalian chordate cTnIs have a histidine at this position and resemble ssTnI in sequence. At this same evolutionary divergence point, three other amino acids in this same region of cTnI also transitioned in concert: R157Q, V166E, and N173H. Thus, all mammalian cTnIs studied to date have residues Q157, A164, E166, and H173. These changes result in an overall potential charge difference of negative two, if you protonate the histidines. We previously speculated (19) that this could alter the conformation and/or interaction of cTnI with cTnC to alter function of mammalian cTnI. Indeed, Dargis et al. (16) showed that substitutions of Q157R/A164H/E166V in cTnI increased Ca2+ sensitivity in thin filament ATPase assays. They also showed that cTnI H164 is necessary for this effect. Our findings in intact myocytes show that cTnI Q157R/A164H/E166V/H173N (QAEH) increases contractility and slows relaxation under physiologic pH conditions, functionally mimicking ssTnI. MD simulations show that cTnI QAEH HSP helix 4 bends and has multiple contacts with helix A of cTnC that structurally mimic sTnI. Taken together, these data are evidence that cTnI isoform-specific residues Q157, A164, E166, and H173 are required to maintain normal contractile function in the adult cardiac myocyte. Furthermore, cTnI A164H/H173N (AH) functionally mimics ssTnI at pH 7.4, suggesting that this second histidine (H173) is sufficient for maintaining normal contractile function under neutral pH conditions in A164H cTnI. Previous investigation of ssTnI N141H (the cTnI substitution in ssTnI) showed this improves relaxation of adult cardiac myocytes compared to ssTnI while still maintaining Ca2+ sensitivity in acidosis (12). Together these two pieces of evidence indicate that two histidines (H164 and H173) are necessary for maintaining normal contractility at neutral pH while preserving function at acidic pH. Structurally, H173 does not interact directly with cTnC, but when present, as in A164H MD simulations from Palpant et al. (19), the R171-E15 salt bridge is not observed. This second histidine may alter helix 4 conformation when protonated, disrupting the R171-E15 salt bridge. Further investigation of the role of H173 structurally is necessary to clarify this point. Construction of a fish-like cTnI (AH) in these four residues suggests that fish cTnI is relatively Ca2+ sensitive and pH resistant. Previously, trout cTnI was shown to increase Ca2+ binding affinity in the context of mammalian cTnC compared to mammalian cTnI (42). Our data suggest that cTnI helix 4 residue differences may contribute to the increased Ca2+ sensitivity of trout. The requirement for decreased Ca2+ sensitivity in the heart of endotherms has been proposed (11) and is supported by our results that sequence divergence at the mammalian lineage results in negative inotropy with enhanced relaxation performance.
MD simulations shown here support a mechanism of increased TnI-cTnC interaction following switch peptide binding to cTnC, thus increasing affinity of TnI to TnC. Specifically, MD simulations reveal a new potential TnI-TnC interaction, cTnI R171-cTnC E15, that is critical to further enhance function. R171 is highly conserved throughout TnI evolution and a human mutation, R170Q (human numbering) has been implicated as a cause of preadolescent HCM suggesting its position is susceptible to alter troponin function (11,43). Increasing or decreasing affinity of TnI to cTnC has been proposed to be part of the mechanism of small molecule Ca2+ sensitizers and desensitizers respectively (44). Recent NMR data suggest that increased affinity is how sTnI maintains contractile function under acidic conditions through H132-E19 interaction (17). The increased interaction in cTnI QAEH HSP R171 with cTnC E15 correlates with a more negative free energy of Ca2+ binding supporting that this conformation increases Ca2+ sensitivity relative to WT cTnI. These simulations and FEP calculations further corroborate the cTnI H164-cTnC E19 salt bridge as being the mechanism of increased Ca2+ sensitivity in acidosis of natural and genetically engineered histidine containing TnIs. The FEP analysis of cTnI A164H from Palpant et al. (19) shows a strong correlation to the cellular physiologic data presented here. There is no change in free energy of Ca2+ binding in the deprotonated cTnI A164H mimicking the pH 7.4 cellular data showing no statistical change in contractility measures. Furthermore, the protonated form shows a more negative free energy of Ca2+ binding mimicking the pH 6.2 cellular data of maintained contractility. Interestingly, the FEP data show that the deprotonated form of cTnI QAEH has lower free energy of Ca2+ binding than cTnI. Again, this correlates with the cellular dynamics data where this substitution has increased contractility and slowed relaxation at physiologic pH 7.4. These correlations between cellular contractility and FEP analysis of cTnC simulations are evidence that FEP analysis is a valuable tool to understand structural implications to Ca2+ binding in cTnC.
The cTnI QAEH HSD simulations generally look similar to cTnI. Analysis of pKa measures show a difference of cTnC salt bridges between E40-K39 in cTnC, whereas in cTnI QAEH HSD and HSP, as well as sTnI, cTnC E40 interacted more closely with cTnC K43. This was corroborated through distance measurements over the entire 40 ns simulations. E40 of cTnC is the 12th residue of the inactive site I Ca2+ binding loop. Mutation of this residue to alanine shows Ca2+ desensitization of thin filament force production and has been shown to alter the closed to open transition of the hydrophobic patch in the Ca2+ bound state through MD simulation (45,46). Other Ca2+ sensitizing/desensitizing modifications in TnC helix C result in alterations of cTnC site I and are proposed to influence Ca2+ binding to site II (40,41). Based on these findings, we speculate that the altered salt bridge in cTnI QAEH HSD, cTnC E40-K39 to E40-K43, plays a key role in the decreased free energy of Ca2+ binding shown here.
Free energy perturbation analysis of the Ca2+ bound structures presented here also suggests that the switch peptide/helix 4 interactions of cTnI QAEH with cTnC determine, at least a portion of the Ca2+ sensitivity seen in cardiac myocytes. The FEP analysis only accounts for the TnI and Ca2+ bound cTnC state, therefore cTnI QAEH increases Ca2+ affinity in this state, which would result in a slower off-rate of Ca2+. A slower Ca2+ off-rate correlates with the slow relaxation shown here in cTnI QAEH expressing cardiac myocytes. In total, the FEP analysis is evidence that cTnI A164H and cTnI QAEH alter cellular function by increasing interactions between cTnI helix 4 and cTnC helix A.
An alternate explanation of how these TnI-TnC conformations give rise to altered Ca2+ sensitivity could be through direct or indirect tropomyosin/actin interaction. Directly, these substitutions are not involved in actin interaction but they are very close to the second actin binding motif in TnI. Thus, indirectly, the bent TnI conformation may position TnI’s mobile domain and second actin binding motif in a position that favors contraction rather than relaxation. Further structure/function studies are necessary to investigate this possibility.
In conclusion, this study provides, to our knowledge, new mechanistic insights into TnI-TnC interactions that underscore molecular inotropy and relaxation. We propose that cTnI residues Q157, A164, E166, and H173, in concert, facilitate mammalian cardiac myocyte-enhanced relaxation and thus essential for normal heart function in eutherians. Our results also extend to troponin action itself, defining a model of cTnI helix 4 conformation and interaction with cTnC as being a key determinate of cardiac myocyte relaxation performance. This work enhances our knowledge of the mechanisms of Ca2+ sensitivity and further establishes a foundation to aid in the design of new therapeutics targeting TnI-TnC interaction in diseases of the sarcomere, such as hypertrophic and ischemic cardiomyopathy.
Acknowledgments
We thank the Minnesota Supercomputing Institute and Prof. Arieh Warshel from University of Southern California for providing the computational resources necessary to complete these studies. We thank Dr. Brian Sykes and Dr. Nathan Palpant for helpful discussions. We thank the Lillehei Heart Institute for support.
This work was supported by grants from the National Institutes of Health (NIH) (BRT (T32) and JMM).
Supporting Material
References
- 1.Gordon A.M., Homsher E., Regnier M. Regulation of contraction in striated muscle. Physiol. Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 2.Farah C.S., Miyamoto C.A., Reinach F.C. Structural and regulatory functions of the NH2- and COOH-terminal regions of skeletal muscle troponin I. J. Biol. Chem. 1994;269:5230–5240. [PubMed] [Google Scholar]
- 3.Willott R.H., Gomes A.V., Potter J.D. Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 4.Li M.X., Spyracopoulos L., Sykes B.D. Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 5.Dong W.J., An J., Cheung H.C. Structural transition of the inhibitory region of troponin I within the regulated cardiac thin filament. Arch. Biochem. Biophys. 2006;456:135–142. doi: 10.1016/j.abb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhavsar P.K., Dhoot G.K., Barton P.J. Developmental expression of troponin I isoforms in fetal human heart. FEBS Lett. 1991;292:5–8. doi: 10.1016/0014-5793(91)80820-s. [DOI] [PubMed] [Google Scholar]
- 7.Feng H.-Z., Hossain M.M., Jin J.P. Myofilament incorporation determines the stoichiometry of troponin I in transgenic expression and the rescue of a null mutation. Arch. Biochem. Biophys. 2009;487:36–41. doi: 10.1016/j.abb.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fentzke R.C., Buck S.H., Leiden J.M. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J. Physiol. 1999;517:143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westfall M.V., Rust E.M., Metzger J.M. Slow skeletal troponin I gene transfer, expression, and myofilament incorporation enhances adult cardiac myocyte contractile function. Proc. Natl. Acad. Sci. USA. 1997;94:5444–5449. doi: 10.1073/pnas.94.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Westfall M.V., Turner I., Metzger J.M. Troponin I chimera analysis of the cardiac myofilament tension response to protein kinase A. Am. J. Physiol. Cell Physiol. 2001;280:C324–C332. doi: 10.1152/ajpcell.2001.280.2.C324. [DOI] [PubMed] [Google Scholar]
- 11.Palpant N.J., Houang E.M., Metzger J.M. Pathogenic peptide deviations support a model of adaptive evolution of chordate cardiac performance by troponin mutations. Physiol. Genomics. 2010;42:287–299. doi: 10.1152/physiolgenomics.00033.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westfall M.V., Metzger J.M. Single amino acid substitutions define isoform-specific effects of troponin I on myofilament Ca2+ and pH sensitivity. J. Mol. Cell. Cardiol. 2007;43:107–118. doi: 10.1016/j.yjmcc.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Day S.M., Westfall M.V., Metzger J.M. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat. Med. 2006;12:181–189. doi: 10.1038/nm1346. [DOI] [PubMed] [Google Scholar]
- 14.Palpant N.J., D’Alecy L.G., Metzger J.M. Single histidine button in cardiac troponin I sustains heart performance in response to severe hypercapnic respiratory acidosis in vivo. FASEB J. 2009;23:1529–1540. doi: 10.1096/fj.08-121996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palpant N.J., Day S.M., Metzger J.M. Single histidine-substituted cardiac troponin I confers protection from age-related systolic and diastolic dysfunction. Cardiovasc. Res. 2008;80:209–218. doi: 10.1093/cvr/cvn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dargis R., Pearlstone J.R., Smillie L.B. Single mutation (A162H) in human cardiac troponin I corrects acid pH sensitivity of Ca2+-regulated actomyosin S1 ATPase. J. Biol. Chem. 2002;277:34662–34665. doi: 10.1074/jbc.C200419200. [DOI] [PubMed] [Google Scholar]
- 17.Robertson I.M., Holmes P.C., Sykes B.D. Elucidation of isoform-dependent pH sensitivity of troponin i by NMR spectroscopy. J. Biol. Chem. 2012;287:4996–5007. doi: 10.1074/jbc.M111.301499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pineda-Sanabria S.E., Robertson I.M., Sykes B.D. Interaction between the regulatory domain of cardiac troponin C and the acidosis-resistant cardiac troponin I A162H. Cardiovasc. Res. 2013;97:481–489. doi: 10.1093/cvr/cvs348. [DOI] [PubMed] [Google Scholar]
- 19.Palpant N.J., Houang E.M., Metzger J.M. pH-responsive titratable inotropic performance of histidine-modified cardiac troponin I. Biophys. J. 2012;102:1570–1579. doi: 10.1016/j.bpj.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yasuda S.-i., Coutu P., Metzger J.M. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ. Res. 2007;101:377–386. doi: 10.1161/CIRCRESAHA.106.145557. [DOI] [PubMed] [Google Scholar]
- 21.Albayya F.P., Metzger J.M. Adenoviral vectors: production and purification. Methods Mol. Biol. 2003;219:3–17. [PubMed] [Google Scholar]
- 22.Herron T.J., Vandenboom R., Metzger J.M. Calcium-independent negative inotropy by beta-myosin heavy chain gene transfer in cardiac myocytes. Circ. Res. 2007;100:1182–1190. doi: 10.1161/01.RES.0000264102.00706.4e. [DOI] [PubMed] [Google Scholar]
- 23.Davis J., Wen H., Metzger J.M. Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ. Res. 2007;100:1494–1502. doi: 10.1161/01.RES.0000268412.34364.50. [DOI] [PubMed] [Google Scholar]
- 24.Jorgensen W.L., Maxwell D.S., Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 25.Jorgensen W. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 26.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 27.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 28.Bashford D., Karplus M. pKa’s of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 29.Gordon J.C., Meyers J.B., Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33(Web Server issue):W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sham Y.Y., Warshel A. The surface constraint all atom model provides size independent results in calculations of hydration free energies. J. Chem. Phys. 1998;109:7940–7944. [Google Scholar]
- 31.Warshel A. Dynamics of reactions in polar solvents: semi-classical trajectory studies of electron transfer and proton transfer reactions. J. Phys. Chem. 1982;86:2218–2224. [Google Scholar]
- 32.Warshel A., Sussman F., King G. Free energy of charges in solvated proteins: microscopic calculations using a reversible charging process. Biochemistry. 1986;25:8368–8372. doi: 10.1021/bi00374a006. [DOI] [PubMed] [Google Scholar]
- 33.Lee F.S., Chu Z.T., Warshel A. Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the Polaris and Enzymix program. J. Comput. Chem. 1993;14:161–185. [Google Scholar]
- 34.Shaffer J.F., Gillis T.E. Evolution of the regulatory control of vertebrate striated muscle: the roles of troponin I and myosin binding protein-C. Physiol. Genomics. 2010;42:406–419. doi: 10.1152/physiolgenomics.00055.2010. [DOI] [PubMed] [Google Scholar]
- 35.Rust E.M., Westfall M.V., Metzger J.M. Stability of the contractile assembly and Ca2+-activated tension in adenovirus infected adult cardiac myocytes. Mol. Cell. Biochem. 1998;181:143–155. doi: 10.1023/a:1006802719136. [DOI] [PubMed] [Google Scholar]
- 36.Davis J., Wen H., Metzger J.M. Allele and species dependent contractile defects by restrictive and hypertrophic cardiomyopathy-linked troponin I mutants. J. Mol. Cell. Cardiol. 2008;44:891–904. doi: 10.1016/j.yjmcc.2008.02.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westfall M.V., Albayya F.P., Metzger J.M. Functional analysis of troponin I regulatory domains in the intact myofilament of adult single cardiac myocytes. J. Biol. Chem. 1999;274:22508–22516. doi: 10.1074/jbc.274.32.22508. [DOI] [PubMed] [Google Scholar]
- 38.Takeda S., Yamashita A., Maéda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 39.Vinogradova M.V., Stone D.B., Fletterick R.J. Ca(2+)-regulated structural changes in troponin. Proc. Natl. Acad. Sci. USA. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang D., McCully M.E., Regnier M. Structural and functional consequences of cardiac troponin C L57Q and I61Q Ca(2+)-desensitizing variants. Arch. Biochem. Biophys. 2013;535:68–75. doi: 10.1016/j.abb.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang D., Robertson I.M., Regnier M. Structural and functional consequences of the cardiac troponin C L48Q Ca(2+)-sensitizing mutation. Biochemistry. 2012;51:4473–4487. doi: 10.1021/bi3003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirkpatrick K.P., Robertson A.S., Gillis T.E. The influence of trout cardiac troponin I and PKA phosphorylation on the Ca2+ affinity of the cardiac troponin complex. J. Exp. Biol. 2011;214:1981–1988. doi: 10.1242/jeb.052860. [DOI] [PubMed] [Google Scholar]
- 43.Kaski J.P., Syrris P., Elliott P.M. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:436–441. doi: 10.1161/CIRCGENETICS.108.821314. [DOI] [PubMed] [Google Scholar]
- 44.Robertson I.M., Sun Y.B., Sykes B.D. A structural and functional perspective into the mechanism of Ca2+-sensitizers that target the cardiac troponin complex. J. Mol. Cell. Cardiol. 2010;49:1031–1041. doi: 10.1016/j.yjmcc.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kekenes-Huskey P.M., Lindert S., McCammon J.A. Molecular basis of calcium-sensitizing and desensitizing mutations of the human cardiac troponin C regulatory domain: a multi-scale simulation study. PLOS Comput. Biol. 2012;8:e1002777. doi: 10.1371/journal.pcbi.1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parvatiyar M.S., Pinto J.R., Potter J.D. Predicting cardiomyopathic phenotypes by altering Ca2+ affinity of cardiac troponin C. J. Biol. Chem. 2010;285:27785–27797. doi: 10.1074/jbc.M110.112326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.