

Abstract
In eukaryotes, the recognition of the DNA postreplication errors and initiation of the mismatch repair is carried out by two MutS homologs: MutSα and MutSβ. MutSα recognizes base mismatches and 1 to 2 unpaired nucleotides whereas MutSβ recognizes longer insertion-deletion loops (IDLs) with 1 to 15 unpaired nucleotides as well as certain mismatches. Results from molecular dynamics simulations of native MutSβ:IDL-containing DNA and MutSα:mismatch DNA complexes as well as complexes with swapped DNA substrates provide mechanistic insight into how the differential substrate specificities are achieved by MutSα and MutSβ, respectively. Our simulations results suggest more extensive interactions between MutSβ and IDL-DNA and between MutSα and mismatch-containing DNA that suggest corresponding differences in stability. Furthermore, our simulations suggest more expanded mechanistic details involving a different degree of bending when DNA is bound to either MutSα or MutSβ and a more likely opening of the clamp domains when noncognate substrates are bound. The simulation results also provide detailed information on key residues in MutSβ and MutSα that are likely involved in recognizing IDL-DNA and mismatch-containing DNA, respectively.
Introduction
The DNA mismatch repair (MMR) process is crucial for maintaining the stability of both prokaryotic and eukaryotic genomes by correcting the replication errors that might have escaped proof-reading by the replication complex (1–4). A replication error can result either from the misincorporation of a base or from strand slippage at repetitive sequences during the DNA replication process. If left undetected or unrepaired, such errors can lead to higher mutation rates (5), microsatellite instabilities (6), or genetic defects (1,2,4). The crucial first step for the initiation of the mismatch repair process is the recognition of a mismatch site. The enzyme responsible for recognizing both mispaired and unpaired bases in prokaryotic E. coli is the homodimeric MutS protein. For eukaryotes, there are a number of heterodimeric MutS homologs, with MutSα (MSH2/MSH6) and MutSβ (MSH2/MSH3) being the most important ones. Like MutS, MutSα recognizes a mismatch site or one or two unpaired bases. MutSβ, on the other hand, preferentially recognizes longer insertion-deletion loops (IDLs) with up to 15 nucleotides, as well as selected base-base mispairs (7). The two complexes are only partially redundant, owing to their different substrate specificities (1,2).
Available crystal structures of prokaryotic MutS (8,9) and its eukaryotic homologs (10,11), complexed with mismatched DNA heteroduplexes, indicate a high degree of similarity in the overall structure and domain organization of these mismatch recognition complexes. However, the exact mechanism by which different substrate specificities are realized is still unclear. The sequence similarities between the three MutS homologs: MSH2, MSH3, and MSH6 range from 27% to 39%, and the five structural domains observed are conserved within each MutS homolog. Domains I and IV interact directly with the bound DNA duplex as mismatch binding domain (MBD) and clamp, respectively. Domains II and III connect domains I and IV to domain V, an ATP-Binding Cassette-family ATPase domain (Fig. 1). Although mismatch DNA-bound MutSα was crystallized with ADP bound to both MSH2 and MSH6 domains (10), IDL-bound MutSβ structures have an ADP molecule bound to only the MSH2 site (11). For both, MutSα and MutSβ heterodimers, the bound DNA heteroduplexes are severely bent at the mismatch or IDL site. The extent of bending is greater for IDLs owing to the presence of unpaired nucleotides at the IDL site. Similar to the prokaryotic MutS, MutSα interacts with the mismatch site via a conserved “Phe-Xaa-Glu” motif present in the MBD (domain 1 of MSH6). MutSβ, on the other hand, lacks this motif and instead uses a group of residues to recognize IDLs. Another structural difference between the crystal structures of eukaryotic MutS homologs is the presence of dimerization domains (DMD) at the C termini of both MSH2 and MSH3 in MutSβ. The DMDs are believed to strengthen the heterodimer (11).
Figure 1.
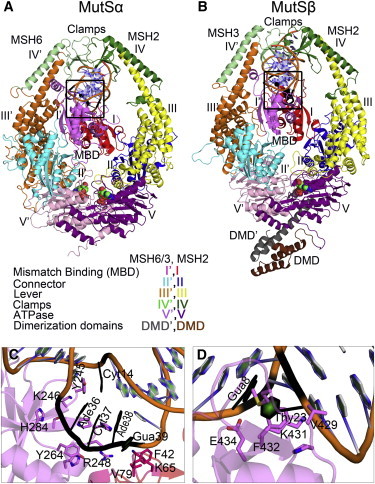
(A) Crystal structure of human MutSα bound to a G:T mismatch containing DNA (PDB ID: 2O8B). (B) human MutSβ bound to unpaired IDL-containing DNA (PDB ID: 3THX). Structural domains of the two subunits are shown in different colors and the mismatch/insertion sites in the DNA heteroduplexes are shown in black. ADP molecules bound at the ATPase domain are shown with a spherical representation. The black boxes indicate the interactions of protein with the mismatch site. These crystal structure contacts are shown in inset panels between (C) MutSα and G:T mismatch site and (D) MutSβ and IDL site. To see this figure in color, go online.
In MutS and MutS homologs, the specificity for mismatch- and IDL-containing heterodimers is mediated by subunits that interact specifically with the mismatch sites. As observed from the crystal structures for IDL-bound MutSβ, the MBDs of both MSH2 and MSH3 monomers interact with the IDL-DNA heteroduplexes. On the other hand, for mismatch-DNA bound MutSα, only the MBD of MSH6 interacts with the mismatch site whereas MSH2 forms only nonspecific interactions with the backbone. Comparing the crystal structures of both MutSα and MutSβ, it can be seen that the MSH2 subunit is oriented differently with the MBD. It is shifted by 5.3Å and rotated by 21° in MutSβ with respect to the MutSα structure (11). Different roles of MSH2 in MutSα and MutSβ are also evident from experimental studies that have the MBD removed from MSH2. MutSα is almost fully functional when the MBD is removed from MSH2, whereas such a mutation in MutSβ renders the enzyme completely nonfunctional (12).
The crystal structures suggest that the different substrate specificities of MutSα and MutSβ are related to different protein-DNA contacts, but the exact mechanism remains unclear. To examine this question in more detail and follow up on previous computational studies of MutS and MutS homologs from our lab (13–15), we carried out molecular dynamics simulations of MutSβ (MSH2/MSH3) and MutSα (MSH2/MSH6) complexed with DNA. In particular, we simulated MutSβ and MutSα in complex with both, the native DNA substrates and swapped substrates, i.e., MutSβ bound with DNA containing a Gua:Thy mismatch and MutSα bound with DNA containing an IDL loop, to gain insight into how the different substrate specificities are realized. Our simulations suggest that MutSβ prefers highly bent heteroduplexes over MutSα. Although the stronger bending appears to be necessary to form stable interactions with IDL-containing DNA, the reduced bending of DNA when bound to MutSα is presumed to be energetically more favorable for recognizing mismatch-containing DNA. In the following, the simulation methodology is briefly described before the results are presented and discussed.
Methods
Simulated systems and simulation protocol
Molecular dynamics (MD) simulations of MutSα in complex with DNA containing a Gua:Thy mismatch and MutSβ in complex with DNA containing a 3-nucleotide (nt) IDL loop were carried out with explicit solvent. The initial conformations of the MutSα-DNA and MutSβ-DNA complexes were taken from the crystal structures 2O8B (10) and 3THX (11), respectively. In the crystal structure of DNA-bound MutSβ (PDB ID: 3THX), the unpaired bases of the 3-nt loop (Ade36, Cyt37, and Ade38) show isomerization. There are two alternate conformations observed with equal probability. In one conformation, the Cyt14:Gua39 pair downstream of the IDL is intact resulting in a 3-nt loop (designated as 3L). In a second, alternate conformation, the Cyt14:Gua39 pair is broken with the Gua39 base flipped out and interacting with F42 of MSH2. This latter conformation results in a 4-nt loop conformation (designated as 4L) with unpaired bases Ade36, Cyt37, Ade38, and Gua39. We have simulated the MutSβ structures bound to both of these alternate conformations of DNA heteroduplexes. Missing protein residues were completed using the loop modeling function (16) in MODELLER9v10 (17). More specifically, in the structure of MutSβ, fragments involving residues 1045-1046, 1070-1078, 1172-1185, 1634-1643, and 1730-1746 in MSH3 and 97-100, 126-133, 304-312, 507-508, 535-536, 635-636, 703-711, and 846-860 in MSH2 were built. For the MutSα structure, the fragments involving residues 550-552, 651-653, 718-728, 933-935, 991-993, 1098-1105, 1122-1126, 1178-1188, and 1270-1284 in MSH6 as well as fragments involving residues 108-112, 139-145, 243-247, 316-321, and 713-723 in MSH2 were built. Swapped structures were created by aligning MSH2/MSH6 to the MSH2/MSH3 proteins with their natively bound DNA and then replacing the mismatch DNA in MSH2-MSH6 with the IDL-containing DNA (in the 3L conformation) from MSH2-MSH3 and the IDL-containing DNA in MSH2-MSH3 with the mismatch DNA from MSH2-MSH6. Table 1 summarizes all the systems that were simulated in this study.
Table 1.
Simulated systems and abbreviations used in this paper
| Protein | Nucleic acid | |
|---|---|---|
| WT_MutSα | MSH2/MSH6 | Gua:Thy mismatch DNA |
| WT_MutSβ-3L | MSH2/MSH3 | 3-nt insertion/deletion loop DNA |
| WT_MutSβ-4L | MSH2/MSH3 | 4-nt insertion/deletion loop DNA |
| Swap_MutSα-4L | MSH2/MSH6 | 4-nt insertion/deletion loop DNA |
| Swap_MutSβ-MM | MSH2/MSH3 | Gua:Thy mismatch DNA |
The Chemistry at Harvard Molecular Modeling (CHARMM)27 (18,19) force field with the CMAP correction (20) was used to describe molecular interactions. All simulations were run with NAMD2.9 (21). MutSα and MutSβ complexes were solvated in a rectangular TIP3P solvent box and neutralized with Na+ ions. Additional Na+ and Cl- ions were added at a concentration of 0.15 M NaCl added salt. The total simulation systems consisted of ∼ 600,000 atoms. Long-range electrostatic interactions were treated using the Particle Mesh Ewald technique (using a spacing of 1 Å and an Ewald coefficient (κ) of 0.312341). A nonbonded cutoff of 10 Å (switched at 8.5 Å) was applied to the Lennard-Jones potential. The direct part of the Ewald sum was also truncated at 10 Å. A 2 fs time step was used for all MD simulations in combination with SHAKE (22) to holonomically constrain bonds involving hydrogens. Rigid water molecules were constrained using the SETTLE algorithm (23).The systems were minimized under harmonic restraints of 3 kcal/mol/Å2 in three steps. Initially, the backbone atoms of the protein and DNA were restrained while allowing only the water molecules to reorient for 50,000 steps. Then, only restraints on the Cα atoms of the protein were applied to allow the protein and DNA to relax during an additional 50,000 steps of minimization. Finally, the restraints were reduced to 1 kcal/mol/Å2, and the structures were minimized further for another 25,000 steps. The minimized structures were heated gradually from 0 to 300 K at a rate of 30 K/60 ps while still maintaining the restraints. The simulation at 300 K was then continued for 100 ps before gradually releasing the restraints from 1 kcal/mol/Å2 to 0 during another 200 ps. The final structures were then subjected to three independent production runs in the NPT ensembles reaching a total simulation time of 220 ns for each system.
Analysis of the trajectories
Trajectory analysis, in particular, structure analyses involving the variations in DNA bending angles and maintenance of native protein-DNA contacts, were carried out using visual molecular dynamics (24). The bending angle for the mismatch containing DNA heteroduplex was defined, following previous work (25), as the angle between the centers of mass of heavy atoms of 3-nt blocks of the pentadecamer bases 2-5/26-29, 6-10/21-25, and 11-14/17-20. For the IDL-containing DNA heteroduplex, the bending angle was defined as the angle between the centers of mass of heavy atoms of the 3-nt blocks: 4-10/43-49, 11-15/35-42, and 16-21/29-34.
Cluster analysis of the sampled conformations (at 5 ps intervals) was performed using the kclust program in the Molecular Modeling Toolset for Structural Biology Tool Set (26). Simulated DNA conformations for both the mismatch- and IDL-containing heteroduplexes were grouped based on mutual root mean square deviations (RMSD) of the nucleotide heavy atoms using a 3.0 Å radius cutoff. The two terminal base pairs were ignored in this analysis.
Results and Discussion
We simulated eukaryotic MutS homologs (MutSα, MutSβ) in the presence of cognate and noncognate bound DNA (see Table 1). For brevity’s sake, we will subsequently refer to the crystal structure DNA-bound states as WT states (WT_MutSα, WT_MutSβ-3L, WT_MutSβ-4L); and the complexes with noncognate DNA substrates as DNA-swapped states (Swap_MutSα-4L, Swap_MutSβ-MM).
DNA-induced changes on conformations and dynamics of MutS homologs
To compare DNA-binding induced changes in the structure and dynamics of the enzymes, we monitored the time series of the RMSD for the protein Cα atoms and DNA backbone atoms relative to the respective x-ray crystal structures (Fig. 2). In the WT DNA-bound simulations, we observe RMSD values of 3 to 5 Å for each monomer that is typical for stable large multidomain protein complexes with intrasubunit and intersubunit domain motions. In contrast, the RMSDs of the DNA-swapped systems were consistently higher than the WT systems. The DNA heteroduplexes, when bound to the noncognate complexes, also exhibited higher deviations than the deviations observed when bound to the cognate complexes. When MutSα was simulated with the noncognate IDL-DNA heteroduplex, the deviations were especially high in the MSH2 moiety. We further analyzed the deviations within each structural domain of both of the monomers after aligning the rest of the molecule with respect to the crystal structure (Figs. S1 and S2 in the Supporting Material). We found that the large deviations in the MSH2 domain of the swapped MutSα were primarily because of motions of the MBD domain (domain I) of MSH2 around the noncognate IDL-DNA.
Figure 2.
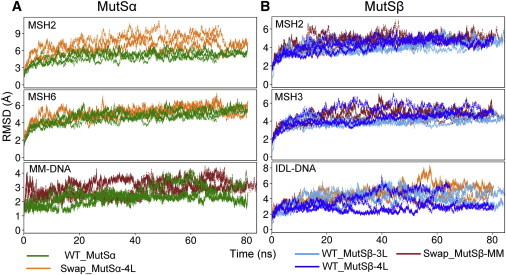
RMSD of protein and DNA backbone atoms with respect to the crystal structure for (A) MutSβ and (B) MutSα systems. The top two panels show results for the protein subunits whereas the bottom panels show the structural deviations of the DNA-heteroduplexes. To see this figure in color, go online.
We observed reorientation of MSH2 subunits for systems swapped with both mismatch- and IDL-containing DNA, although the reorientation was more prominent when IDL-containing DNA was added to the MutSα environment (Fig. 2) than when mismatch-containing DNA was added to the MutSβ environment. This may be attributable to the presence of the stabilizing dimerization domains at the C termini of MutSβ, which are thought to strengthen the heterodimer (11). We note, however, that the changes in MSH2 upon swapping the DNA substrates did not fully recover the different crystallographically observed structures of MSH2 in the presence of IDL- or mismatch-containing DNA. Presumably, this is attributable to the different second moieties (MSH3 and MSH6) that prevent a full relaxation of MSH2 to the respective crystallographic structures with either IDL- or mismatch-containing DNA. However, it is also possible, that our simulation timescales were too short to fully relax the complex structures in the presence of the swapped substrates.
The opening and closing behavior of the clamp domains during the simulations was analyzed by tracking the distances between the centers of mass of the two clamps (Fig. 3). For the WT MutS homologs, the clamps generally remained closed during the simulation near the crystal structure values although there was a slight transient opening in two out of three simulations for WT_MutSβ-4L. In contrast, the clamps opened significantly more in the simulations with swapped DNA as comparatively larger distances between the centers of mass of the two clamps are observed for longer times throughout the swapped trajectories. This suggests that the presence of cognate DNA stabilizes the bound states with the clamps closed, which is likely to be essential for initiating mismatch repair. Opening of the clamps more likely allows the MutS homologs to continue scanning DNA and/or dissociate entirely. We note that previous simulations of MutSα in the absence of bound DNA showed considerable opening of the clamps (15) not very different from what we observed in the swapped simulations in this study.
Figure 3.
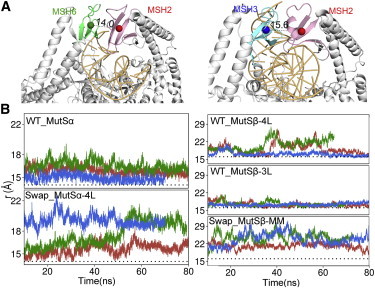
DNA-binding induced movement of clamp domains. (A) Distances between the centers of masses (spheres) for the clamps (colored regions) of each subunit as observed for the crystal structure of MutSα and MutSβ. (B) Time variations of these distances, r (Å), are plotted for multiple runs for WT, Swapped, and APO states of MutSα and MutSβ. The crystal structure values are shown as black dotted lines. To see this figure in color, go online.
Figs. 2 and 3 also provide evidence for the degree of convergence achieved in our simulations. The initial structural responses of MutS to different DNA substrates appear to take place within the first 20 to 60 ns and similar key features are observed in multiple trajectories such as the significant opening of the clamps in two out of three trajectories of MutSα-4L. We believe therefore that the simulations presented in this study, capture the structural response and dynamics on the 100-ns time scale. Different dynamics, possibly linked to repair initiation, may occur on much longer time scales of μs-ms, but these are not the target of the present study.
Conformations of the DNA heteroduplexes
Higher RMSD values were observed for the DNA backbone atoms in the swapped systems (Fig. 2). This indicates that not just the protein but also the DNA adjusted depending on the MutS homolog. The DNA heteroduplexes that contain mismatches are highly bent structures, as observed from the crystal structures. To characterize the conformational changes when present in the native or nonnative environments, we analyzed the variations in the bending angle for the heteroduplexes (Fig. 4). We found that when the mismatch-containing DNA was bound to MutSβ, the kink angle varied over a broad range of angles, from 110° to 140° with two peaks near 120° and 135°, with a preference for smaller bending angles. In contrast, mismatch-containing DNA bound to WT_MutSα exhibited a narrower distribution with a small peak at around 120° and a large well-defined peak at 130°. In a previous study from our lab (25), we found that DNA bending comes at a significant cost that increases with the extent of bending. At the same time, differences in the free energies associated with bending of DNA heteroduplexes vs. homoduplexes became pronounced for bending angles below 130° (25). Therefore, bending DNA at angles around 130° would allow discrimination of mismatch-containing DNA with the least energetic cost. The peak of the distribution for mismatch-containing DNA bound to MutSα lies just at that angle. On the other hand, mismatch-containing DNA is bent more strongly when bound to MutSβ. This should still allow discrimination, but it would incur a higher energetic cost that could be too high for bending some sequences. Consequently, MutSβ may miss some mismatch sites altogether by continuing to scan DNA instead of incurring the energetic cost of strongly bending DNA. This hypothesis is further supported by the experiments that have shown that yeast MutSβ recognizes DNA heteroduplexes with C:C mismatches with greater efficiency than G:G, A:C, and G:T mismatches. According to our previous simulation study, DNA with C:C mismatches is more easily bent than the other three mismatches (25). In contrast, MutSα does not significantly bind C:C heteroduplexes (7), possibly because the discrimination from homoduplex DNA at shallower bending angles is not sufficient.
Figure 4.
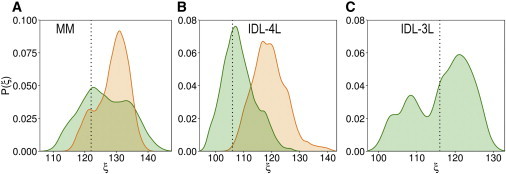
Probability distributions of DNA kink angles, ξ, (as defined in Methods) observed during the simulations (ignoring the first 10 ns). (A) Mismatch containing heteroduplexes when in the native environment bound to MutSα (WT_MutSα, green) and in the swapped environment bound to MutSβ (Swap_MutSβ-MM, orange). (B) 4L in native environment bound to MutSβ (WT_MutSβ-4L, green) and in swapped environment bound to MutSα (Swap_MutSα-4L, orange). (C) 3L in native environment bound to MutSβ (WT_MutSβ-3L, green). To see this figure in color, go online.
The IDL-DNA with 4L conformation is highly bent with a peak near the crystal structure value of 107° when bound to MutSβ. When the same IDL-DNA is simulated in complex with MutSα, the kink angle increases significantly to 120° corresponding to a partial straightening of the IDL-DNA. The IDL-DNA with 3L conformation also samples relatively small bending angles with two peaks, one near ∼ 110° and one near ∼ 122° compared with the crystal structure value of 117°. This observation, together with the conformations seen in the crystal structures, suggests that stronger bending (at smaller bending angle values) may be required to successfully recognize IDL-DNA and therefore MutSβ is better suited to this task than MutSα. This is consistent with previous studies that suggest that DNA with extra bases more likely prefers bent conformations (27).
Major conformations of the nucleic acids sampled during the simulations were obtained through clustering. The representative structures closest to the cluster centroids are shown in Fig. 5. Only one cluster was found for mismatch-containing DNA when bound to MutSα suggesting a very stable complex. However, when mismatch DNA is bound to MutSβ, there was greater conformational variability. In two clusters (Cluster Nos. 1 and 4), the mismatch G:T pair opened up and type II kinks were formed as the two bases unpaired and stacked onto the 5ʹ neighboring bases of the respective strands. The other two clusters (Cluster Nos. 2 and 3) consist of conformations that showed bending at the mismatch site, with different reorientations of the mismatch pairing. However, no type I kink similar to the crystal structure was observed. The structural changes around the mismatch site when bound to MutSβ may be in part attributable to the missing phenylalanine that inserts at the mismatch site and stacks with the mismatched base in MutSα. Our previous simulations of mismatch-containing DNA in the absence of protein suggested that type II kinks are found at larger bending angles (25). Since MutSβ bent the mismatch-containing DNA more than MutSα, the emergence of type II kinks at the mismatch site is therefore not surprising.
Figure 5.
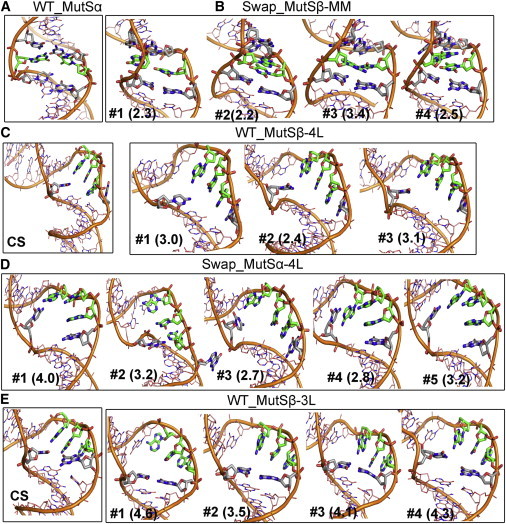
Representative structures of nucleic acid conformations from clustering. The mismatch and IDL parts are shown in green with neighboring residues in gray. The CS panel represents the crystal structures and clusters are ordered with respect to their cluster size. Values given in parentheses correspond to heavy atom RMSDs with respect to the experimental structure. To see this figure in color, go online.
For the 4L-containing DNA heteroduplexes, the conformations of the unpaired nucleotides forming IDL bound to MutSβ were similar to the crystal structure. However, in complex with MutSα, the structural ensemble became more diverse. In most of the conformations, Gua39 which is flipped out in the crystal structure to stack with a nonconserved phenylalanine of MSH2, flipped in and stacked with Ade38, one of the unpaired nucleotides (Clusters Nos. 1, 3 to 5). However, Cyt14 retained a single hydrogen bond with Ade36 as seen in the crystal structure. Furthermore, the second-most probable conformation (Cluster No. 2) involved a reorientation of the whole IDL loop whereas Cyt14 moved down to pair with Ade38 and Gua39 completely flipped out into the major groove.
For the 3L-containing DNA heteroduplexes, the conformations were also largely similar to the crystal structure when bound to MutSβ (Clusters Nos. 1 to 3). However, we noticed a slight reorientation of the unpaired nucleotides while retaining the Cyt14:Gua39 base pair. These structures correspond to the major population of structures with less DNA bending (larger bending angles; Fig. 4 C). In addition, in a minor population with increased DNA bending (Cluster No. 4), the Cyt14:Gua39 base pair opened up whereas Cyt14 formed a single hydrogen pair with Ade36.
Interactions of DNA heteroduplexes with the protein subunits
To further understand the role of the enzymes in differentially binding to different DNA substrates, we analyzed the interactions of DNA with different protein subunits during the simulations. Any protein residue with a heavy atom located within 5 Å from a heavy atom of the DNA is considered as protein-DNA contact. According to this definition, mismatch-containing DNA formed more contacts with MSH6 of MutSα than with MSH3 of MutSβ in the swapped simulation, both upstream and downstream of the mismatch site (Figs. 6 and 7 ). In the native MutSα environment, the mismatch pair Gua8:Thy23 formed persistent specific interactions with the conserved “F432-X-E434” motif of the MSH6 subunit. F432 of MSH6 formed stacking interactions with Thy23 of the Gua8:Thy23 pair, and E434 formed hydrogen bond interactions with Thy23. In addition to these specific interactions, Gua8 formed nonspecific interactions with V429, G430, and K431 of the MBD domain of MSH6, and Thy23 interacted with V450, M452, S459, and G460 (Fig. 8 A) of MSH6. Interestingly, we observed that some of these specific or nonspecific interactions were partially compensated by similar residues in the MBD domain of MSH3. The F432 and E434 residues of MSH6 were replaced by K246 and R248 in MSH3, respectively, although K246 cannot form the same stacking interactions as phenylalanine (Fig. 8 C). However, the hydrogen bonding interactions of E434 of MSH6 with N7 of mismatch base Thy23 was substituted by the hydrogen bonding interaction between O2 of Thy23 and Nζ of K246 or with Nε of R248 in MSH2 subunit.
Figure 6.
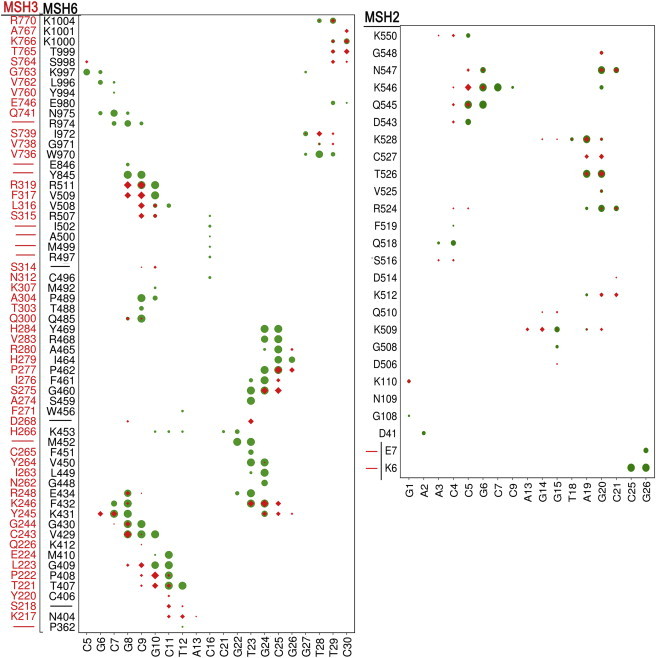
Contacts present for at least 20% of the simulation time (ignoring the first 10 ns) between protein subunits (residues on the y axis) and mismatch containing DNA (nucleotides on the x axis). The mismatch pair Gua8:Thy23 is indicated with a black dotted line. Green circles denote interactions between mismatch DNA and cognate MSH6 (left panel) or MSH2 (right panel) in WT_MutSα; red diamonds denote the interaction of mismatch DNA with noncognate MSH3 (left panel) or MSH2 (right panel) in Swap MutSβ-MM. The extent of the interaction is indicated by the size of the symbol, with largest size corresponding to the interaction present persistently and smaller size corresponding to the intermittent interaction. Residue correspondence between MSH3 and MSH6 was obtained by structurally aligning MSH3 subunit to the MSH6 subunit using STAMP (29). A dash in the first two columns indicates a lack of a corresponding residue. The MSH2 subunit of MutSα has the first N-terminal 12 nucleotides resolved, which are absent in MutSβ. To see this figure in color, go online.
Figure 7.
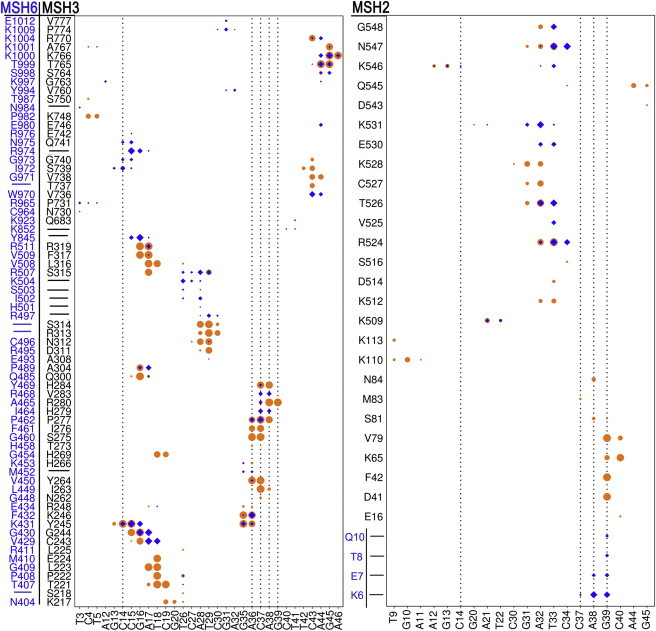
Protein-DNA contacts as in Fig. 6 but for IDL-containing DNA. Unpaired residues Cyt14, Ade36-Ade38, and Gua39 are indicated by black dotted lines. Orange circles denote the interaction of IDL DNA with cognate MSH3 (left panel) or MSH2 (right panel) in WT_MutSβ and blue diamonds denote the interaction of IDL DNA with noncognate MSH6 (left panel) or MSH2 (right panel) in Swap_MutSα-4L. To see this figure in color, go online.
Figure 8.
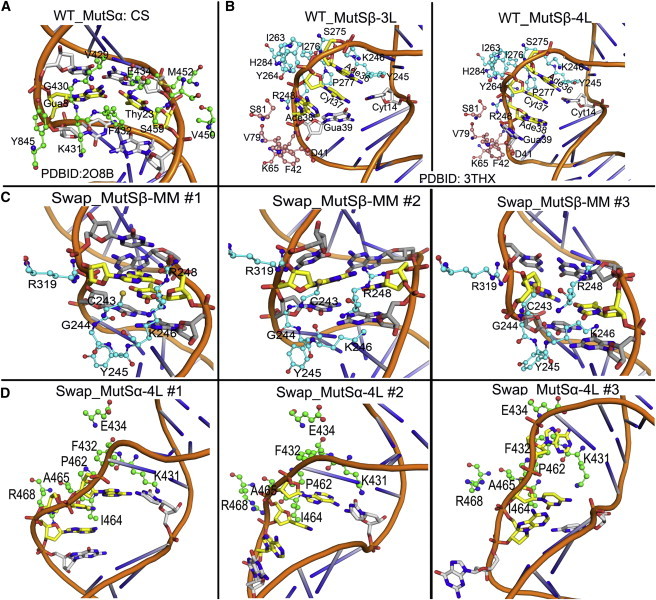
Interactions at the mismatch site for DNA heteroduplexes observed in the crystal structure of (A) MutSα and (B) MutSβ compared with the simulated structures for (C) Swap_MutSβ-MM and (D) Swap_MutSα-4L. Three average structures are shown for each of the three simulations. The mismatch site is shown with yellow sticks, and the adjacent base pairs are shown in gray. The interacting protein residues are shown in stick-and-ball representation (green for MSH6, cyan for MSH3, and pink for MSH2). To see this figure in color, go online.
The importance of the specific interactions identified in our simulations is further supported by mutational studies of equivalent residues in yeast MSH3 (28). Experimental data suggest that if yeast MSH3 K158 (equivalent to K246 in human MSH3) or K160 (equivalent to R248 in human MSH3) are mutated to Asp/Glu/Ala/Met or Asp, respectively, there is a defect in the repair of 1-nt frameshifts while the repair of C:C, A:A, A:C, and C:T mispairs (recognized by wild-type MSH3) is prevented (28). The nonspecific interactions of Gua8 with V249 and G430 in MSH6 seem to be replaced by interactions with C243 and G244 in MSH3, although the mode of interaction varied (Fig. 8 C). The interactions of adjacent base pairs (-1/+1) with MSH6 are partially compensated by similar MSH3 residues. For instance, interactions of Cyt7 with K431 of MSH6 and between Cyt9 with R511 of MSH6 were replaced by Y245 and R319 of MSH3, respectively. Finally, Gua24 interactions with K431, F432, and G460 of MSH6 were replaced by Y245, K246, and S275 of MSH3.
In addition to the interactions with MSH6, the mismatch containing DNA heteroduplex also interacted with MSH2 subunit in MutSα, although only nonspecifically without interactions at the mismatch site. Some of these interactions in MSH2 of MutSα (V525, T526, K528, Q545, and K546) were also seen with the MSH2 subunit of MutSβ. Interestingly, the four conserved lysines (K509, K512, K546, and K550) responsible for DNA-recognition in MutSα also formed interactions with the nucleotides of DNA heteroduplexes in the MutSβ environment, although the detailed mode of interaction was not fully conserved.
The nucleotide loop of IDL-containing DNA (Fig. 8 B) interacted with the proteins primarily via contacts between the phosphate backbone and the MBD of MSH3 and to lesser extent with the MBD of MSH2. The interactions between IDL-DNA and MSH3 residues were partially replaced by interactions with structurally equivalent MSH6 residues (Fig. 7). For instance, instead of the stacking interactions between MSH3 Y245 and Cyt14 and Ade36, as Y245 was inserted between the adjacent nucleotides on the minus strand opposite of the IDL in MutSβ, MSH6 K431 is inserted in MutSα (Fig. 8 B). Furthermore, the phosphate of the first loop nucleotide Ade36 interacted with MSH3 K246, Y264, S275, I276, and P277 in MutSβ while it interacted with F432 and P462 of MSH6 in MutSα, equivalent to K246 and P277 of MSH3, respectively. The phosphate of Cyt37 interacted with I263, Y264, S275, I276, P277, and H284 of MSH3 while it interacted only with MSH6 P462, equivalent to MSH3 P277. The phosphate of Ade38 was stabilized by interactions with P277, R280, and H284 of MSH3 but again only interacted with MSH6 P462 (Fig. 8 D). P277 (MSH3) and P462 (MSH6) are highly conserved among MutS homologs. In general, specific interactions of the IDL-DNA with MSH3 were more extensive than with MSH6. These additional interactions are likely key to the successful recognition of IDL-DNA. This is supported by experiments that found that the interaction of IDL with H284 of MSH3 was critical for IDL repair (28) whereas, in our simulations, the equivalent Y469 of MSH6 did not interact with the DNA. Based on our simulations, we suggest I263, S275, Y264, I276, and R280 as additional mutation targets in MSH3 that may diminish the ability of MutSβ to recognize IDL-DNA.
IDL-DNA interacted partially with MSH2 of MutSβ as Gua39 forms stacking interactions with F42, and interacts nonspecifically with D41, K65, V79, and S81 of MSH2 (Fig. 8 B). However, no similar MSH2 residues were observed with IDL-DNA when added to the MutSα environment (Fig. 8 D), suggesting that not just interactions with MSH3 but also indirectly with MSH2 contribute to the stability of IDL-DNA when bound to MutSβ. Again, this could be tested via mutations of the MSH2 residues involved in contacts with IDL-DNA in MutSβ.
It is evident that the crystal structure interactions between MutSα and mismatch-containing DNA and between MutSβ and IDL-DNA are quite stable. When swapping the DNA substrates only some of the stabilizing interactions were maintained. Furthermore, MSH3 appears to favor kinks at the mismatch site, which favors IDL recognition and allows the recognition of some of the more easily kinked mispairs by MutSβ but fails to stabilize other mismatches to the same extent as MutSα does. On the other hand, the interaction of MSH6 appears to be strongest with the first unpaired base of the IDL loop substrate while not binding as tightly to subsequent unpaired bases as MSH3 does. This is consistent with MutSα only being unable to efficiently recognize insertion/deletion loops with up to one to two unpaired bases.
Conclusions
The results from our simulation study provide mechanistic details for how the eukaryotic MutS homologs are tuned to specific recognition of mismatch-containing DNA by MutSα and longer IDL-DNA by MutSβ. Swapping the DNA substrates allowed us to identify key interactions between the DNA and the MutS homolog protein moieties with the main findings that interactions are more extensive between mismatch-containing DNA and MutSα and between IDL-DNA and MutSβ. In general, this suggests reduced stability when noncognate substrates are bound. More specifically, we found that MutSα preferentially interacts with only the first base of an IDL, which would explain why it is limited to recognizing only short IDLs. We also observed a more likely opening of the clamps when substrates are swapped between MutSα and MutSβ that suggests more likely dissociation from noncognate substrates.
Combining the results of this study with our previous analysis of DNA bending in the absence of protein (25), it appears that MutSα is exquisitely tuned to recognize mismatch-containing DNA by inducing just enough bending to allow discrimination from canonical DNA. On the other hand, the more extensive bending of DNA when bound to MutSβ seems to be necessary for IDL-DNA recognition although interfering with the recognition of mismatch-containing DNA, especially for those mismatches that are not easily kinked.
Our study provides a list of key residues that should affect the ability of MutSβ and MutSα to recognize IDL-containing and mismatch-containing DNA, respectively. It is our hope that this work will stimulate further experimental studies to confirm the hypotheses proposed in this study. However, there is also room for additional computational studies to gain additional insight into the exact recognition mechanism of mismatch- and IDL-containing DNA.
Acknowledgments
Funding from NIH GM092949 is acknowledged. Computer resources were used at XSEDE facilities (TG-MCB090003).
Supporting Material
References
- 1.Kunkel T.A., Erie D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 2.Iyer R.R., Pluciennik A., Modrich P.L. DNA mismatch repair: functions and mechanisms. Chem. Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 3.Modrich P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 5.de Wind N., Dekker M., te Riele H. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat. Genet. 1999;23:359–362. doi: 10.1038/15544. [DOI] [PubMed] [Google Scholar]
- 6.Sia E.A., Dominska M., Petes T.D. Isolation and characterization of point mutations in mismatch repair genes that destabilize microsatellites in yeast. Mol. Cell. Biol. 2001;21:8157–8167. doi: 10.1128/MCB.21.23.8157-8167.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrington J.M., Kolodner R.D. Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Mol. Cell. Biol. 2007;27:6546–6554. doi: 10.1128/MCB.00855-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Obmolova G., Ban C., Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 9.Natrajan G., Lamers M.H., Sixma T.K. Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res. 2003;31:4814–4821. doi: 10.1093/nar/gkg677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warren J.J., Pohlhaus T.J., Beese L.S. Structure of the human MutSalpha DNA lesion recognition complex. Mol. Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 11.Gupta S., Gellert M., Yang W. Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat. Struct. Mol. Biol. 2012;19:72–78. doi: 10.1038/nsmb.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S.D., Surtees J.A., Alani E. Saccharomyces cerevisiae MSH2-MSH3 and MSH2-MSH6 complexes display distinct requirements for DNA binding domain I in mismatch recognition. J. Mol. Biol. 2007;366:53–66. doi: 10.1016/j.jmb.2006.10.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Law S.M., Feig M. Base-flipping mechanism in postmismatch recognition by MutS. Biophys. J. 2011;101:2223–2231. doi: 10.1016/j.bpj.2011.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukherjee S., Law S.M., Feig M. Deciphering the mismatch recognition cycle in MutS and MSH2-MSH6 using normal-mode analysis. Biophys. J. 2009;96:1707–1720. doi: 10.1016/j.bpj.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukherjee S., Feig M. Conformational change in MSH2-MSH6 upon binding DNA coupled to ATPase activity. Biophys. J. 2009;96:L63–L65. doi: 10.1016/j.bpj.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiser A., Feig M., Sali A. Evolution and physics in comparative protein structure modeling. Acc. Chem. Res. 2002;35:413–421. doi: 10.1021/ar010061h. [DOI] [PubMed] [Google Scholar]
- 17.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 18.Foloppe N., MacKerell A.D., Jr. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21:86–104. [Google Scholar]
- 19.MacKerell A.D., Jr., Banavali N.K. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 2000;21:105–120. [Google Scholar]
- 20.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 21.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 23.Miyamoto S., Kollman P.A. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 24.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 25.Sharma M., Predeus A.V., Feig M. DNA bending propensity in the presence of base mismatches: implications for DNA repair. J. Phys. Chem. B. 2013;117:6194–6205. doi: 10.1021/jp403127a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feig M., Karanicolas J., Brooks C.L., 3rd MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Feig M., Zacharias M., Pettitt B.M. Conformations of an adenine bulge in a DNA octamer and its influence on DNA structure from molecular dynamics simulations. Biophys. J. 2001;81:352–370. doi: 10.1016/S0006-3495(01)75705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dowen J.M., Putnam C.D., Kolodner R.D. Functional studies and homology modeling of Msh2-Msh3 predict that mispair recognition involves DNA bending and strand separation. Mol. Cell. Biol. 2010;30:3321–3328. doi: 10.1128/MCB.01558-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell R.B., Barton G.J. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins. 1992;14:309–323. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.