

Abstract
Background
During osteoclastogenesis, the maturation of osteoclast (OC) progenitors is stimulated by the receptor activator of nuclear factor-κB ligand (RANKL). Excess OC production plays a critical role in the pathogenesis of inflammatory bone disorders. Conversely, the inhibition of abnormal OC proliferation reduces inflammation-induced bone loss. Low concentrations of carbon monoxide (CO) are known to decrease inflammation and OC-mediated bone erosion but the molecular mechanism is unknown.
Results
To obtain insight into the biological function of CO, cultured RANKL-treated RAW 264.7 cells were used in an in vitro experimental model of osteoclastogenesis. The results showed that CO inhibited: 1) tartrate-resistant acid phosphatase (TRAP)-positive cell formation; 2) F-actin ring production; 3) c-fos pathway activation; 4) the expression of cathepsin K, TRAP, calcitonin receptor, and matrix metalloproteinase-9 mRNAs; 5) the expression of nuclear factor of activated T cells, cytoplasmic, calcineurin-dependent 1 in translation. Protein-protein interaction analysis predicted mitogen-activated protein kinase kinase kinase 4 as the controlling hub.
Conclusions
Low-concentrations of CO (250 ppm) may inhibit osteoclastogenesis. Data from STRING- and IPA-based interactome analyses suggested that the expression of proteins with the functions of signal transduction, enzymes, and epigenetic regulation are significantly altered by CO during RANKL-induced osteoclastogenesis. Our study provides the first interactome analysis of osteoclastogenesis, the results of which supported the negative regulation of OC differentiation by CO.
Keywords: Carbon monoxide, Osteoclastogenesis, Interactome, RANKL, RAW 264.7
Background
Bone homeostasis is strictly regulated through a dynamic balance between osteoblastogenesis (bone formation) and osteoclastogenesis (bone resorption) [1-5]. In the former, osteoblasts (OBs) control bone formation through the synthesis of bone matrix proteins. In osteoclastogenesis, large multinucleated osteoclasts (OCs) remove the mineralized matrix of bone tissue, resulting in bone resorption. OCs are derived from hematopoietic precursors of the monocyte-macrophage lineage, and their differentiation is regulated by macrophage colony-stimulating factor (M-CSF) [6]. Mutations in the M-CSF gene induce defects in the formation of macrophages and OCs, which suggests that immune cells and bone cells are derived from the same progenitors [7].
The receptor activator of nuclear factor (NF)-κB ligand (RANKL), a member of the tumor necrosis factor (TNF) family, regulates OC maturation and differentiation [8,9]. Bone-forming OBs express RANKL as do activated T cells which indicates a role for the immune system in osteoclastic bone resorption [9,10]. In addition, many inflammatory cytokines are known to modulate RANKL expression, including TNF-α [9,11], and the RANKL dependence of several inflammatory bone diseases has been reported [10]. Bone dysfunction may also arise following the production of RANKL by activated T cells, by directly triggering osteoclastogenesis [12].
Osteoclasts express the receptor activator of NF-κB (RANK), a type I membrane protein. Thus, the RANKL/RANK signaling cascade regulates not only the maturation of OC progenitors, but also the activity of OCs in normal bone remodeling [13,14]. Given the critical functions of OCs, an understanding of the process that control their differentiation is required for the successful treatment of many bone pathologies.
Carbon monoxide (CO) is an invisible and odorless gas with a binding affinity for heme that is 240 times higher than that of oxygen. Concentrations of CO in the air exceeding 3% (30000 ppm) are usually lethal [15,16]. CO is endogenously produced by mammals, by the degradation of heme, albeit in very low concentrations [17,18]. Interestingly, however, in small amounts (250 ppm) CO exhibits anti-apoptosis, anti-proliferation, anti-inflammatory, and many other biological activities [19-22]. For example, CO-releasing molecule-2 dose-dependently inhibits RANKL-induced osteoclastogenesis [23].
Mouse leukemic monocyte macrophage RAW 264.7 cells are derived from Abelson-virus-treated mice ascites and recognized as pre-OCs [24]. Following their stimulation with RANKL, these cells produce OCs in a pathway involving mitogen-activated protein kinases (MAPKs), extracellular-regulated kinase (ERK), p38, jun N-terminal kinase (JNK), and c-fos [25-27]. In this study, we used RANKL-treated RAW264.7 cells as an vitro model to investigate the effect of CO on the signaling pathway of RANKL-induced osteoclastogenesis. In addition, in an approach using interactomics to obtain a protein-protein interaction (PPI) network, we derived a genome-scale PPI map to deduce the potential signaling pathways and member proteins involved in the differentiation and activation of OCs, and the effects of CO on these pathways [28].
Results
Inhibition of RANKL-induced osteoclastogenesis in RAW 264.7 cells by low-dose CO without preventing growth or inducing apoptosis
RAW 264.7 cells incubated with 10, 15, and 20 ng RANKL/mL for 96 h gave rise to TRAP(+) multinucleated cells in a dose-dependent manner. When these RANKL-treated progenitor cells were exposed to 250 ppm CO, the formation of TRAP(+) multinucleated cells was inhibited by 73 + 5% (10 ng/mL), 70+ 5% (15 ng/mL), and 41+ 10% (20 ng/mL) (Figure 1C,D), We also check the effects of CO on osteoclastogenesis in bone marrow macrophages and the results are the same as RAW cells (Additional file 1). Moreover, after incubation with 250 ppm CO for 72 or 96 h, RANKL-treated RAW 264.7 cells retained their ability to proliferate and the expression of activated caspase-3, a marker of apoptosis, was not induced (Figure 1B).
Figure 1.
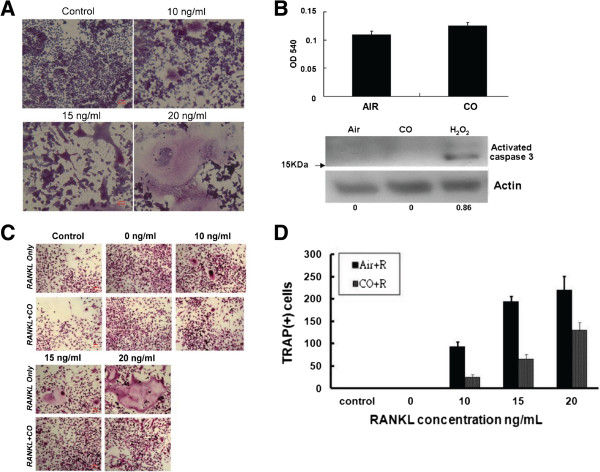
CO reduces osteoclastogenesis by RANKL-treated RAW 264.7 cells in vitro. (A) RAW 264.7 cells treated with 10, 15, and 20 ng RANKL/mL and examined by TRAP staining after 96 h. (B) Upper panel, RAW 264.7 cells exposed to air or 250 ppm CO for 96 h and then stained with 0.05% methylene blue, as described in Methods. The data are the mean from three experiments with triplicate samples; bars, +SD. Lower panel, Western blot analysis of activated caspase 3 expression by RAW 264.7 cells. (C) TRAP staining of RAW 264.7 cells treated with 20 ng RANKL/mL and then exposed to 250 ppm CO. (D) Bar chart showing the inhibitory effect of CO (250 ppm) on RAW 264.7 cells treated as in (C).
CO-induced inhibition of F-actin ring formation by osteoclasts
Formation of the F-actin ring by OCs is a necessary step in bone resorption. As shown in Figure 2A and Figure 2B, in RAW 264.7 cells treated with 20 ng RANKL/mL and 250 ppm CO for 96 h, F-actin ring formation was reduced (Additional file 2). A similar inhibitory effect of osteoclast pit formation, an indicator of bone resorption, was observed on dentin discs in the CO but not the Air group.
Figure 2.
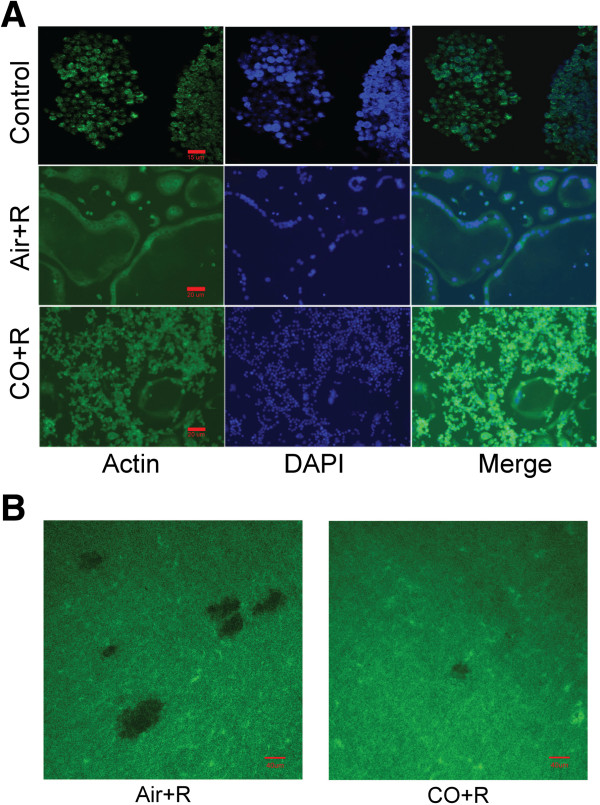
CO inhibits F-actin ring formation by RANKL-treated RAW 264.7 cells. (A) Effect of 250 ppm CO on the distribution of clustered F-actin filaments in RANKL-treated RAW 264.7 cells. Arrows show the position of the F-actin ring. (B) Pit formation assay of RANKL-treated RAW 264.7 cells cultured on dentin discs and incubated with air or CO.
CO-induced suppression of RANKL-induced JNK and c-jun phosphorylation, and c-fos but not IκB-α expression
The activation of NF-κB plays an important role in the differentiation of pre-OCs. As shown in Figure 3A, in RAW 264.7 cells exposed to 20 ng RANKL/mL and 250 ppm CO there was no change in the expression of IκB-α, which is normally induced by NF-κB and serves as its inhibitor. Furthermore, this same dose of CO inhibited the phosphorylation of RANKL-induced JNK and c-jun but not that of either ERK or mitogen-activated protein kinase p38 (p38) within 120 min (Figure 3B). The same concentration of CO down-regulated the expression of c-fos in similarly treated RAW 264.7 cells (Figure 3C).
Figure 3.
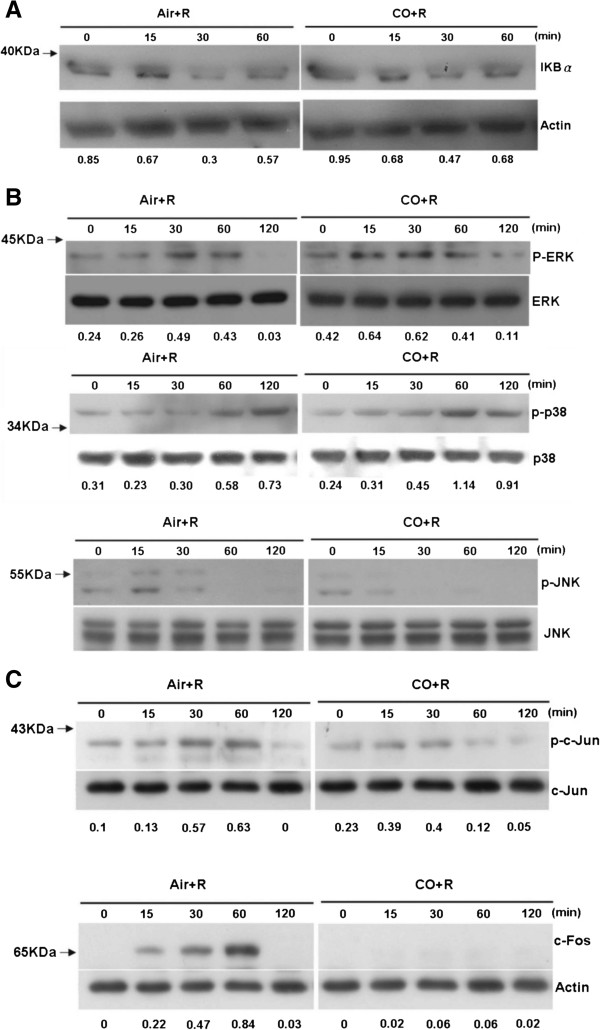
CO suppresses JNK and c-jun phosphorylation and c-fos expression in RANKL-treated RAW 264.7 cells. Western blots showing (A) the expression of IκB-α, as an indicator of NF-κB activity, (B) the phosphorylation of ERK, p38, and JNK, and (C) c-Jun and c-Fos expression.
CO-induced reduction of Acp5, Ctsk, Calcr, Mmp9, and NFATc1 expression
RANK is expressed on the cell surfaces of pre-OCs and OCs. As shown in Figure 4A, RANK mRNA expression in RAW 264.7 cells was not affected by 250 ppm CO within 96 h. Tartrate-resistant acid phosphatase (Acp5), the calcitonin receptor (Calcr), cathepsin K (Cstk), and matrix metalloproteinase-9 (mmp9) are unique markers of mature OCs. In RAW 264.7 cells treated with 20 ng RANKL/mL for 96 h, 250-ppm CO reduced the mRNA expression levels of all four genes (Figure 4B). Moreover, the expression of the protein nuclear factor of activated T cells, cytoplasmic, calcineurin-dependent 1 (NFATc1), which is the master regulator of osteoclastogenesis, was likewise significantly reduced (Figure 4C).
Figure 4.
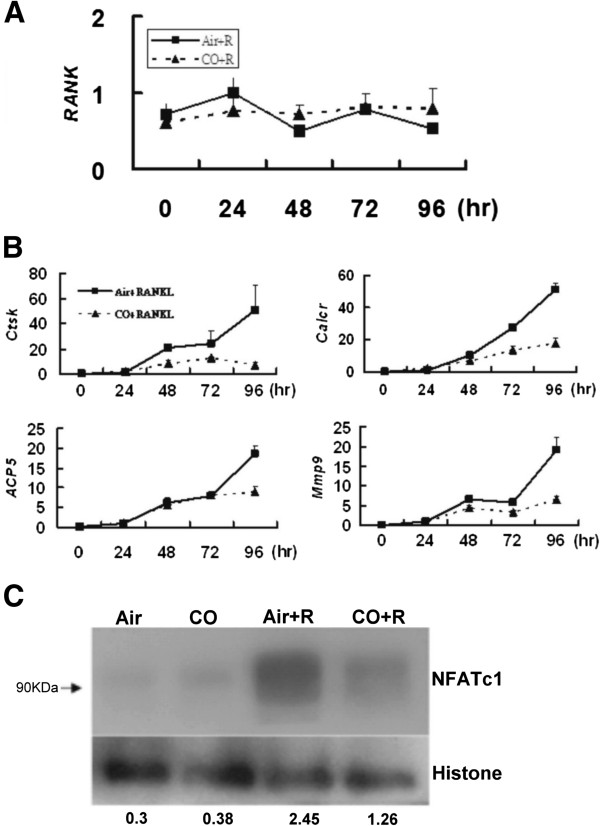
CO reduces the expression levels of the genes Acp5, Ctsk, Calcr, and Mmp9 and of the protein NFATc1 but not that of RANK in RANKL-treated RAW 264.7 cells. (A) Western blot analysis of RANK expression. (B) qRT-PCR analysis of Acp5, Calcr, Cstk, and Mmp9 expression. (C) Western blot analysis of NFATc1 expression.
Critical roles of MAP3K4 and the JNK signaling pathway in RANKL-induced osteoclastogenesis
The STRING database was used to construct a PPI map of RANKL-induced osteoclastogenesis. The map showed three protein clusters joined by mitogen-activated protein kinase kinase kinase 4 (MAP3K4) as a potential hub protein (Figure 5A). Using IPA software (see Methods), we identified the JNK protein family signaling pathway as a potential participant in the maturation of RAW 264.7 cells treated with 20 ng RANK/mL and exposed to 250 ppm CO (Figure 5B).
Figure 5.
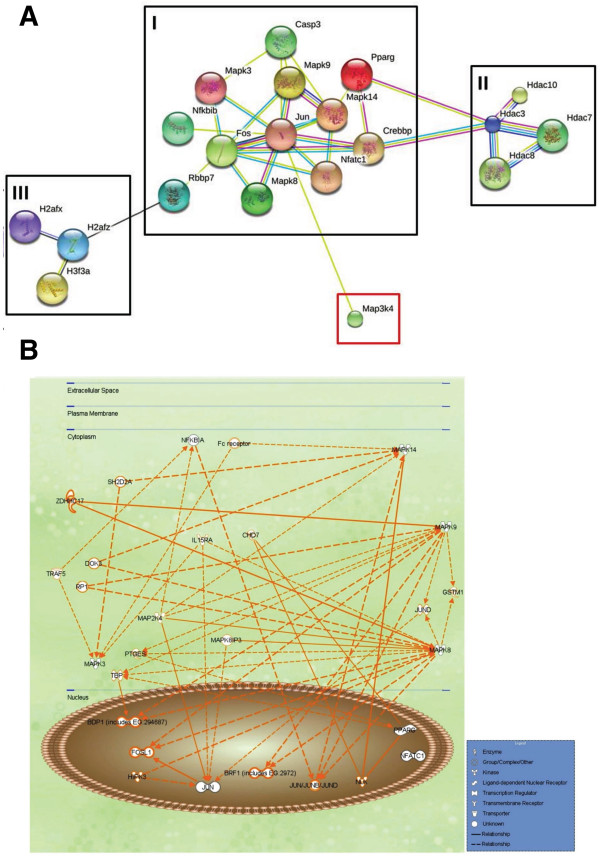
The PPI network and signaling pathway analysis. (A) The PPI network, as shown in the interaction view, generated by the STRING database. Black squares indicate the protein clusters, and the red square the controlling hub, MAP3K4. (B) The computed predicted signaling pathways that are activated in RANKL-treated RAW 264.7 cells.
Discussion
The potential benefits of CO have led to many studies of its possible therapeutic applications, which have been examined in different disease models. Sato et al. showed that CO prolonged the survival of mouse-to-rat cardiac grafts [29]. In mice with ischemic lung injury, the inhalation of CO was reported to prevent their death [30]. The protective effects of CO have also been demonstrated in animal models of autoimmune diseases [31-33].
Bone remodeling is a tightly regulated cycle involving the formation of bone by OBs and its resorption by OCs. The latter are monocyte-macrophage lineage-derived cells whose differentiation and maturation is regulated by OBs via RANKL. Excessive bone resorption, initiated by proinflammatory cytokines, also proceeds through RANKL and leads to abnormal bone destruction. Given the involvement of OCs in these processes, an understanding of their formation and function is crucial to prevent bone loss in inflammatory diseases such as arthritis.
In our study, small doses of exogenous CO not only reduced the degree of inflammation and the expression of RANKL. Consistent with these results, CO was shown to reduce joint destruction in a murine model of collagen-induced arthritis [34]. However, the effect of CO on RANKL-induced osteoclastogenesis has not been investigated. In this study, the differentiation of pre-OC murine macrophage-like RAW 264.7 cells into multinucleated OCs was stimulated using recombinant RANKL protein. This system was then used to examine the potential role of CO in osteoclastogenesis. In these experiments, the expression of several OC markers was examined, including the glycosylated monomeric metalloenzyme TRAP, which is expressed at high levels by. differentiated OCs. In the presence of 250 ppm CO, TRAP expression by RANKL-stimulated RAW 264.7 cells that had differentiated into TRAP (+) multinucleated OCs was blocked (Figure 1). Cytoskeletal reorganization in mature functioning OCs, including the formation of dot-like F-actin rings, was likewise inhibited in RANKL-treated RAW 264.7 cells, whereas their growth was not, nor was apoptosis induced (Figure 2). Thus, collectively, our data show that one of the effects of CO is to inhibit the maturation of pre-OCs induced by RANKL.
The regulatory mechanisms by which RANKL and its intracellular signaling pathways enable pre-OCs to differentiate have been extensively studied [8,35,36]. RANKL binds to RANK expressed on the plasma membrane of OC precursors [25,27,37-41], which then activates an intricate signaling cascade that includes NF-κB, c-fos, c-jun, MAPKs, and NFATc1 [36,42]. Support for the role of this pathway in osteoclastogenesis comes from a previous study in which inhibition of c-fos expression in mice blocked OC differentiation and caused osteopetrosis [39]. IκB-α binds to NF-κB in the cytoplasm, thus maintaining the protein in an inactive form and tightly regulating its transcriptional activity. In our study, CO had no effect on the NF-κB (IκB-α) pathway, as shown in a western blot analysis of IκB-α expression and measurements of RANK mRNA levels (Figure 3A, 4A). However, CO did inhibit the maturation of RANKL-treated RAW 264.7 cells. So what is the nature of the signaling pathway involved in CO-inhibited osteoclastogenesis? Given the CO-mediated reductions in c-jun and JNK phosphorylation and c-fos expression in RANKL-treated RAW 264.7 cells (Figure 3B, Figure 3C), CO could inhibit RANKL-induced osteoclastogenesis by deactivating AP-1, thus down-regulating c-fos, a component of this complex. The activity of AP-1 (containing c-fos) is crucial for the auto-amplification of NFATc1 [43], which is induced and activated by RANKL signaling during the terminal differentiation of OCs [36,42]. Among the OC-specific genes regulated by NFATc1 are Acp5, Calcr, Cstk, and mmp9[43-45]. In the presence of CO, the mRNA levels of all four genes was significantly reduced (Figure 4B). In addition, CO inhibited the translation of NFATc1 (Figure 4C). Together, these findings suggest that CO acts on the MAPK pathway to inhibit osteoclastogenesis by RAW 264.7 cells induced to differentiate by stimulation with RANKL.
Interactomics yields deep insights into the molecular mechanism of diseases and their intracellular signaling pathways [28] but, to the best of our knowledge, it has not been used to profile osteoclastogenesis. In this study, a STRING database analysis of RANKL-induced osteoclastogenesis established a PPI map with three clusters (Figure 5A). Cluster I is a highly symmetric, connected protein cluster with strong interactions. It contains Fos, Jun, retinoblastoma binding protein 7 (RBBP7), nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, beta (NFKBIB), CREB binding protein (CREBBP, CBP), peroxisome proliferator-activated receptor gamma (PPARG), NFATc1, and MAPKs. RBBBP7, also called RbAp46, is a histone H4 binding protein that binds to the c-fos transcriptional activation site to inhibit cell mitosis [46]. We found that the prevention of differentiation by RANKL-treated RAW264.7 cells was accompanied by a decrease in c-fos expression, which suggests that CO alters the action of RBBBP7 on c-fos during osteoclastogenesis. Another cluster I protein, the nuclear receptor PPARG, is pivotal for adipogenesis and interacts with histone deacetylase 3 (HDAC3) [47], contained in cluster II along with HDAC7. HDACs catalyze the removal of acetyl groups from an ϵ-N-acetyl lysine amino acid on histones, which are contained in cluster III. In a previous report the use of shRNA to inhibit HDAC3 expression also inhibited OC formation whereas similar inhibition of HDAC7 accelerated OC differentiation [48]. These findings suggest that the balance between HDAC3 and HDAC7 expression decides the fate of pre-OCs exposed to CO. Among the cluster III histones are the histone H2A family (H2AF) members X and Z (H2AFX, H2AFZ), and histone H3 family member 3A (H3F3A). Histones are highly alkylated and comprise the major protein component of chromatin. The epigenetic regulation of histones by methylation and acetylation may provide regulatory control of OC differentiation. For example, the expression of NFATc1 induced by RANKL is associated with the demethylation of trimethylated histone H3 lysine 4 and lysine 27 (H3K4me3, H3K27me3) [49].
Proteins identified as controlling hubs are major, central proteins in PPI networks. As shown in Figure 5A, c-Jun strongly interacted with other proteins in cluster I, such as jnk1 and jnk2. Furthermore, this cluster interacts with MAP3K4 (also called MTK1), a protein in the MAPK pathway, through c-Jun. Abell et al. [50] showed that MP3K4 regulates jnk1 and jnk2 to control the activity of histone acetyltransferase. It also controls CBP activity in trophoblast stem cells during the epithelial-mesenchymal transition. In our interactomics analysis, MAP3K4 was designated as a hub protein that interacts with c-Jun, thereby controlling the interaction between CBP and HDAC3 during OC differentiation. PPI maps derived using the IPA software have been widely used to gain insight into molecular interactions, signaling pathways, and pathogenesis [51]. One of the advantages of this approach is data from a limited number of experiments can be analyzed. We therefore took advantage of this method to obtain a global understanding of the signaling pathways that are activated during osteoclastogenesis in the presence of CO (Figure 5B). Our results showed that CO significantly inhibited the expression of the transcriptional factors c-JUN and c-FOS, the protein partners of AP-1 (Figure 3C), which suggests their involvement in the CO-mediated blockade of OC differentiation. JNK1 and JNK2, two proteins controlled by MAP2K4, were shown to interact with JUND and thereby alter the transcriptional activity of JUN. The IPA PPI showed that in RANKL-treated RAW264.7 cells exposed to CO, MAP2K4, a downstream regulator of MAP3K4 [52], interacts with IκB-α and P38.
Conclusions
In this study, a low concentration of CO (250 ppm) was shown to inhibit osteoclastogenesis in RANKL-treated RAW264.7 cells. An interactome identifying the PPI network involved in the observed effects allows the following conclusions. First, proteins that function as signal transducers, enzymes, and epigenetic regulators are significantly affected by CO during RANKL-induced osteoclastogenesis. Second, CO inhibits osteoclastogenesis through the MAPK signaling pathway. Third, STRING predicted that MAP3K4 is a major controlling hub protein. Fourth, the interactomics software IPA not only predicted a critical role for MAP2K4 but also identified MAP3K4 as a hub protein. Fifth, STRING and IPA provide overlapping, complementary information. While the data obtained with these tools are similar, the latter provided both a more complete PPI and an easier approach to understanding the behavior of each protein during CO-regulated osteoclastogenesis. Our research offers new data and thus new insights into CO-regulated osteoclastogenesis. However, further detailed investigations into the molecular mechanisms underlying this process are needed.
Methods
Methylene blue solution, sarkosyl, and the TRAP staining and leukocyte acid phosphatase assay kits were purchased from Sigma (St. Louis, MO, USA). Recombinant RANKL protein was obtained from PeproTech (Rocky Hill, NJ, USA). Anti-IκB antibody was obtained from Biolegend (San Diego, CA, USA). Antibodies for phospho-p38, phospho-JNK, phospho-ERK, phospho-c-jun, and c-fos were purchased from GeneTex (Irvine, CA, USA). Anti-RANKL and anti-actin antibodies were obtained from Abcam (Cambridge, UK), and Sigma, respectively.
Cell culture
RAW 264.7 cells were cultured in DMEM with 10% FCS, 2 mM l-glutamine, 10 units penicillin/mL, and 10 μg streptomycin/mL at 37°C in a 5% CO2 humidified incubator. RAW 264.7 cells were transferred to 100-mm dishes when they reached 80% confluence and further grown in the culture medium.
Cell proliferation assay
RAW 264.7 cells were grown in 24-well plates (104 cells/mL per well). The medium was removed at day 3 and the cells were stained with methylene blue solution (0.3 mL per well) at room temperature for 30 min, followed by three to four washes with Milli-Q water. The cells were then air dried and dissolved overnight in 1% sarkosyl in phosphate-buffered saline (PBS, 0.3 mL/well). The cell solution was transferred to a 96-well plate (0.1 mL/well), and the absorbance was read at 540 nm using an ELISA plate spectrophotometer.
In vitro OC differentiation
Cells (104 per well) were grown in 24-well plates and exposed to RANKL (5–20 ng/mL) for 120 h to induce OC formation [24]. These cells were fixed and then stained for TRAP expression using a TRAP staining kit according to the manufacturer’s protocol. The cells were observed using an Olympus BX51 microscope equipped with DP controller (ver. 3.3.1.292). Those with more than three nuclei were identified as TRAP (+) OCs.
CO exposure
The cells were incubated at 37°C in the presence or absence of 5% CO2. The gas was prepared by using an air mixer to dilute an initial concentration of 1% (10,000 ppm) in compressed air with fresh air to obtain a final concentration of 250 ppm, which was then delivered into the incubator. A CO analyzer (Siemens Ultramat 23, Germany) with a sensitivity of 10–600 ppm was used to measure CO levels.
Immunofluorescence analysis
F-actin rings were detected as described previously [53]. Briefly, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS, and incubated with an anti-actin antibody at 4°C overnight. After a PBS wash, the cells were incubated with FITC-conjugated secondary antibody for 30 min at 37°C and then analyzed using a Olympus BX51 microscope equipped with a DP controller (version 3.3.1.292).
Pit formation assay
RAW264.7 cells (1 × 103) were co-cultured with RANKL on dentin discs (Immunodiagnostic Systems Inc., Fountain Hills, AR, USA) in a 96-well plate for 72 h in the presence (CO group) or absence (air group) of CO. Typically, three discs were prepared per group. To observe the areas containing resorption lacunae, the cells were removed the discs were incubated in 0.25 M ammonium hydroxide, washed with distilled water, and then stained with 0.5% (wt/vol) toluidine blue. The resorbed areas were imaged using a reflective optical microscope (LSM 510, Zeiss).
Western blotting analyses
The cells were washed twice with PBS and protein extracts of the nucleus and cytosol were prepared using a ProteoJET cytoplasmic and nuclear protein extraction kit (Fermentas, Glen Burnie, MD, USA). The extracts were centrifuged at 10,000 × g for 5 min after which the supernatants were collected and treated with protease inhibitors. The protein concentration was determined using the Bradford protein assay. The extracts (20 μg) were then dissolved in 6× Laemmli sample loading buffer, boiled for 10 min, and subjected to SDS-PAGE on a 10% gel. The proteins in the gel were electrotransferred onto polyvinylidene fluoride membrane (Millipore) with a semi-dry transfer unit at 20 V for 30 min. After a blocking step with 5% skim milk in Tris-buffered saline (TBS; 20 mM Tris base pH 7.6, 150 mM NaCl) containing 1% Tween-20 at room temperature for 1 h, the membrane was incubated with the primary antibody at room temperature at 4°C overnight. It was then washed three times for 10 min with TBS containing 1% Tween-20, incubated with secondary antibody at room temperature for 1 h, and again washed as before. The immunoblotted protein bands were visualized by chemiluminescence using Immobilon western chemiluminescent HRP substrate (Millipore, Billerica, MA, USA) and X-ray film.
Real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis
Trizol reagent was used to isolate total RNA, which was further eluted with 20 μL of RNase-free water. For cDNA synthesis, 5 μg of total RNA was reverse transcribed at 42°C for 60 min using RevertAid first-strand cDNA synthesis kit (Fermentas). The reaction was terminated by heating at 75°C for 5 min. The sequences of the primers were as follows:
Cathepsin K: 5′–ATGTGGGGGCTCAAGGTTCTG–3′ and 5′–CATATGGGAAAGCATCTTCAGAGT C–3′
TRAP: 5′–AGCAGCCAAGGAGGACTACGTT–3′ and 5′–TCGTTGATGTCGCACAGAGG–3′
Calcitonin receptor: 5′–AGTTGCCCTCTTATGAAGGAGAAG–3′ and 5′–GGAGTGTCGTCCCAGCACAT–3′
MMP-9: 5′–GGAACTCACACGACATCTCCA–3′ and 5′–GAAACTCACACGCCAGAAGAATTT–3′
The Maxima SYBR Green/ROX qPCR master mix kit (Fermentas) was used for all qRT-PCRs. The reactions were carried out in a total volume of 20 μL containing 10 μL of 2× Maxima SYBR Green/ROX qPCR, 1 μM of the primer pair, and 5 μL of cDNA. Thermal cycling parameters were 95°C for 10 min, 40 cycles of 95°C for 15 sec, and 60°C for 60 s. The samples for each PCR were prepared in triplicate. The GAPDH gene was used as the control. Data sets were analyzed and amplification plots were obtained automatically by the 7500 Fast System Software (Applied Biosystems). The comparative threshold cycle (2–[delta] Ct) method, which converts differences of cycle numbers to the ratio of the test gene/control gene, was used to normalize the gene expression levels to GAPDH mRNA.
PPI network and signaling pathway analysis
Proteins were analyzed for their interactions and signaling pathways using Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database version 9.0 (http://string.embl.de) and the Ingenuity Pathways Analysis (IPA) software (http://www.ingenuity.com), respectively.
Statistics
All statistical analyses were performed using the Student’s t-test. Significance was defined as p < 0.05.
Abbreviations
Acp5: Tartrate resistant acid phosphatase; Calcr: Calcitonin receptor; CBP: CREB binding protein; CO: Carbon monoxide; Cstk: Cathepsin K; ERK: Extracellular regulated kinase; H2AFX: H2A histone family, member X; H2AFZ: H2A histone family, member Z; H3F3A: H3 histone, family 3A; H3K27me3: Lysine 27; H3K4me3: Demethylation of trimethylated histone H3 lysine 4; HDAC3: Histone deacetylase 3; HDAC7: Histone deacetylase 7; JNK: Jun N-terminal kinase; MAP3K4: Mitogen-activated protein kinase kinase kinase 4; MAPKs: Mitogen-activated protein kinases; M-CSF: Macrophage colony-stimulating factor; MMP9: Matrix metalloproteinase-9; NF-κB: Receptor activator of nuclear factor-κB; NFKBIB: Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, beta; OCs: Osteoclasts; OBs: Osteoblasts; PPARG: Peroxisome proliferator-activated receptor gamma; RANKL: Receptor activator of nuclear factor-κB ligand; RBBP7: Retinoblastoma binding protein 7; TNF: Tumor necrosis factor; TRAP: Tartrate-resistant acid phosphatase.
Competing interests
The authors declared that there are no competing interests.
Authors’ contributions
The work presented here was carried out as a collaboration between all of the authors. RYP and HKS defined the research theme. FJT and WTC designed the methods and experiments, FJT and GHG carried out the laboratory experiments, analyzed the data, interpreted the results, and wrote the paper. CWH and MJT carried out the STRING and IPA analyses. RYP and JFS co-designed the dispersal and colonization experiments, and worked together on the collection of the data and their interpretation. FJT co-designed the experiments, and contributed to discussions of the analyses, interpretation, and presentation. All authors have contributed to, read, and approved of the manuscript.
Supplementary Material
CO reduced the number of TRAP(+) cells in bone marrow macrophages. Air+R, ordinary incubate condition (37°C, 5% CO2) with MCSF 20 ng and RANKL 20 ng added; CO+R, ordinary incubate condition plus CO, MCSF 20 ng and RANKL 20 ng.
{kind=link}
Positive control experiment with Calcitonin 30 nM and RANKL 20 ng in RAW cells.
{kind=link}
Contributor Information
Feng-Jen Tseng, Email: ptg.rdc@gmail.com.
Wei-Tso Chia, Email: 4926602@yahoo.com.tw.
Jia-Fwu Shyu, Email: shyujeff@ndmctsgh.edu.tw.
Guo-Hau Gou, Email: guohau.gou@gmail.com.
Huey-Kang Sytwu, Email: sytwu@ndmctsgh.edu.tw.
Ching-Wu Hsia, Email: welovearthurhsia@gmail.com.
Min-Jen Tseng, Email: biomjt@ccu.edu.tw.
Ru-Yu Pan, Email: pan1628@yahoo.com.
Acknowledgments
This project was supported by the National Defense Medical Center (MAB101-26-001 to Ru-Yu Pan).
References
- Rasmussen H. Ionic and hormonal control of calcium homeostasis. Am J Med. 1971;50:567–588. doi: 10.1016/0002-9343(71)90113-6. [DOI] [PubMed] [Google Scholar]
- Mundy GR, Guise TA. Hormonal control of calcium homeostasis. Clin Chem. 1999;45:1347–1352. [PubMed] [Google Scholar]
- Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, Kondo H, Richards WG, Bannon TW, Noda M, Clement K, Vaisse C, Karsenty G. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434:514–520. doi: 10.1038/nature03398. [DOI] [PubMed] [Google Scholar]
- Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- Ross FP, Teitelbaum SL. Alphavbeta3 and macrophage colony-stimulating factor: partners in osteoclast biology. Immunol Rev. 2005;208:88–105. doi: 10.1111/j.0105-2896.2005.00331.x. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251–264. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002;20:795–823. doi: 10.1146/annurev.immunol.20.100301.064753. [DOI] [PubMed] [Google Scholar]
- Kong Y-Y, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- Walsh MC, Choi Y. Biology of the TRANCE axis. Cytokine Growth Factor Rev. 2003;14:251–263. doi: 10.1016/S1359-6101(03)00027-3. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/S0092-8674(00)81569-X. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas CG, Haldane JS, Haldane JBS. The laws of combination of haemoglobin with carbon monoxide and oxygen. J Physiol. 1912;44:275–304. doi: 10.1113/jphysiol.1912.sp001517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Burg R. Toxicology Update. J Appl Toxicol. 1999;19:379–386. doi: 10.1002/(SICI)1099-1263(199909/10)19:5<379::AID-JAT563>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Sjostrand T. Endogenous production of carbon monoxide in man under normal and pathophysiological conditions. Scand J Clin Lab Invest. 1949;1:201–214. doi: 10.3109/00365514909069943. [DOI] [Google Scholar]
- Sjostrand T. The formation of carbon monoxide by the decomposition of haemoglobin in vivo. Acta Physiol Scand. 1952;26:338–344. doi: 10.1111/j.1748-1716.1952.tb00915.x. [DOI] [PubMed] [Google Scholar]
- Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AMK, Soares MP. Carbon Monoxide Generated by Heme Oxygenase 1 Suppresses Endothelial Cell Apoptosis. J Exp Med. 2000;192:1015–1026. doi: 10.1084/jem.192.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache I, Otterbein LE, Alam J, Wiegand GW, Choi AMK. Heme oxygenase-1 inhibits TNF-alpha -induced apoptosis in cultured fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2000;278:L312–319. doi: 10.1152/ajplung.2000.278.2.L312. [DOI] [PubMed] [Google Scholar]
- Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J Biol Chem. 1997;272:32804–32809. doi: 10.1074/jbc.272.52.32804. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- Lin TH, Tang CH, Hung SY, Liu SH, Lin YM, Fu WM, Yang RS. Upregulation of heme oxygenase-1 inhibits the maturation and mineralization of osteoblasts. J Cell Physiol. 2010;222:757–768. doi: 10.1002/jcp.22008. [DOI] [PubMed] [Google Scholar]
- Collin-Osdoby P, Yu X, Zheng H. Osdoby P (Eds.): RANKL-Mediated Osteoclast Formation from Murine RAW 264.7 Cells. Totowa, New Jersey: Humana Press; 2003. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Sudo T, Saito T, Osada H, Tsujimoto M. Involvement of p38 Mitogen-activated Protein Kinase Signaling Pathway in Osteoclastogenesis Mediated by Receptor Activator of NF-kappa B Ligand (RANKL) J Biol Chem. 2000;275:31155–31161. doi: 10.1074/jbc.M001229200. [DOI] [PubMed] [Google Scholar]
- David J-P, Sabapathy K, Hoffmann O, Idarraga MH, Wagner EF. JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and -independent mechanisms. J Cell Sci. 2002;115:4317–4325. doi: 10.1242/jcs.00082. [DOI] [PubMed] [Google Scholar]
- Hotokezaka H, Sakai E, Kanaoka K, Saito K, Matsuo K, Kitaura H, Yoshida N, Nakayama K. U0126 and PD98059, specific inhibitors of MEK, accelerate differentiation of RAW264.7 cells into osteoclast-like cells. J Biol Chem. 2002;277:47366–47372. doi: 10.1074/jbc.M208284200. [DOI] [PubMed] [Google Scholar]
- Ryan DP, Matthews JM. Protein-protein interactions in human disease. Curr Opin Struct Biol. 2005;15:441–446. doi: 10.1016/j.sbi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Sato K, Balla J, Otterbein L, Smith RN, Brouard S, Lin Y, Csizmadia E, Sevigny J, Robson SC, Vercellotti G, Choi AM, Bach FH, Soares MP. Carbon Monoxide Generated by Heme Oxygenase-1 Suppresses the Rejection of Mouse-to-Rat Cardiac Transplants. J Immunol. 2001;166:4185–4194. doi: 10.4049/jimmunol.166.6.4185. [DOI] [PubMed] [Google Scholar]
- Fujita T, Toda K, Karimova A, Yan SF, Naka Y, Yet SF, Pinsky DJ. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med. 2001;7:598–604. doi: 10.1038/87929. [DOI] [PubMed] [Google Scholar]
- Ndisang JF, Gai P, Berni L, Mirabella C, Baronti R, Mannaioni PF, Masini E. Modulation of the immunological response of guinea pig mast cells by carbon monoxide. Immunopharmacology. 1999;43:65–73. doi: 10.1016/S0162-3109(99)00045-4. [DOI] [PubMed] [Google Scholar]
- Chora AA, Fontoura P, Cunha A, Pais TF, Cardoso S, Ho PP, Lee LY, Sobel RA, Steinman L, Soares MP. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest. 2007;117:438–447. doi: 10.1172/JCI28844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C-M, Lin H-H, Chiang M-T, Chang P-F, Chau L-Y. Systemic Expression of Heme Oxygenase-1 Ameliorates Type 1 Diabetes in NOD Mice. Diabetes. 2007;56:1240–1247. doi: 10.2337/db06-0495. [DOI] [PubMed] [Google Scholar]
- Ferrandiz ML, Maicas N, Garcia-Arnandis I, Terencio MC, Motterlini R, Devesa I, Joosten LA, van den Berg WB, Alcaraz MJ. Treatment with a CO-releasing molecule (CORM-3) reduces joint inflammation and erosion in murine collagen-induced arthritis. Ann Rheum Dis. 2008;67:1211–1217. doi: 10.1136/ard.2007.082412. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006;12:17–25. doi: 10.1016/j.molmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Wang Z-Q, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- Soysa NS, Alles N. NF-[kappa]B functions in osteoclasts. Biochem Biophys Res Commun. 2009;378:1–5. doi: 10.1016/j.bbrc.2008.10.146. [DOI] [PubMed] [Google Scholar]
- Grigoriadis A, Wang Z, Cecchini M, Hofstetter W, Felix R, Fleisch H, Wagner E. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–448. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Miyazaki T, Kadono Y, Takayanagi H, Miura T, Nishina H, Katada T, Wakabayashi K, Oda H, Nakamura K, Tanaka S. Possible Involvement of IκB Kinase 2 and MKK7 in Osteoclastogenesis Induced by Receptor Activator of Nuclear Factor κB Ligand. J Bone Miner Res. 2002;17:612–621. doi: 10.1359/jbmr.2002.17.4.612. [DOI] [PubMed] [Google Scholar]
- Wan Y, Chong LW, Evans RM, Wan Y, Chong L-W, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007;13:1496–1503. doi: 10.1038/nm1672. [DOI] [PubMed] [Google Scholar]
- Matsuo K, Owens JM, Tonko M, Elliott C, Chambers TJ, Wagner EF. Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat Genet. 2000;24:184–187. doi: 10.1038/72855. [DOI] [PubMed] [Google Scholar]
- Crotti TN, Flannery M, Walsh NC, Fleming JD, Goldring SR, McHugh KP. NFATc1 regulation of the human beta3 integrin promoter in osteoclast differentiation. Gene. 2006;372:92–102. doi: 10.1016/j.gene.2005.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J-i, Wagner EF, Mak TW, Kodama T, Taniguchi T. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/S1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Kogawa M, Wada S, Takayanagi H, Tsujimoto M, Katayama S, Hisatake K, Nogi Y. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J Biol Chem. 2004;279:45969–45979. doi: 10.1074/jbc.M408795200. [DOI] [PubMed] [Google Scholar]
- Yang J, Kiefer S, Rauchman M. Characterization of the gene encoding mouse retinoblastoma binding protein-7, a component of chromatin-remodeling complexes. Genomics. 2002;80:407–415. doi: 10.1006/geno.2002.6844. [DOI] [PubMed] [Google Scholar]
- Gao Z, He Q, Peng B, Chiao PJ, Ye J. Regulation of nuclear translocation of HDAC3 by IkappaBalpha is required for tumor necrosis factor inhibition of peroxisome proliferator-activated receptor gamma function. J Biol Chem. 2006;281:4540–4547. doi: 10.1074/jbc.M507784200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham L, Kaiser B, Romsa A, Schwarz T, Gopalakrishnan R, Jensen ED, Mansky KC. HDAC3 and HDAC7 have opposite effects on osteoclast differentiation. J Biol Chem. 2011;286:12056–12065. doi: 10.1074/jbc.M110.216853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui T, Hirose J, Tsutsumi S, Nakamura K, Aburatani H, Tanaka S. Epigenetic regulation of osteoclast differentiation: possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J Bone Miner Res. 2011;26:2665–2671. doi: 10.1002/jbmr.464. [DOI] [PubMed] [Google Scholar]
- Abell AN, Jordan NV, Huang W, Prat A, Midland AA, Johnson NL, Granger DA, Mieczkowski PA, Perou CM, Gomez SM, Li L, Johnson GL. MAP3K4/CBP-regulated H2B acetylation controls epithelial-mesenchymal transition in trophoblast stem cells. Cell Stem Cell. 2011;8:525–537. doi: 10.1016/j.stem.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will B, Zhou L, Vogler TO, Ben-Neriah S, Schinke C, Tamari R, Yu Y, Bhagat TD, Bhattacharyya S, Barreyro L, Heuck C, Mo Y, Parekh S, McMahon C, Pellagatti A, Boultwood J, Montagna C, Silverman L, Maciejewski J, Greally JM, Ye BH, List AF, Steidl C, Steidl U, Verma A. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood. 2012;120:2076–2086. doi: 10.1182/blood-2011-12-399683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi M, Elion EA. MAP kinase pathways. J Cell Sci. 2005;118:3569–3572. doi: 10.1242/jcs.02470. [DOI] [PubMed] [Google Scholar]
- Nakamura I, Kadono Y, Takayanagi H, Jimi E, Miyazaki T, Oda H, Nakamura K, Tanaka S, Rodan GA, Duong Le T. IL-1 regulates cytoskeletal organization in osteoclasts via TNF receptor-associated factor 6/c-Src complex. J Immunol. 2002;168:5103–5109. doi: 10.4049/jimmunol.168.10.5103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CO reduced the number of TRAP(+) cells in bone marrow macrophages. Air+R, ordinary incubate condition (37°C, 5% CO2) with MCSF 20 ng and RANKL 20 ng added; CO+R, ordinary incubate condition plus CO, MCSF 20 ng and RANKL 20 ng.
Positive control experiment with Calcitonin 30 nM and RANKL 20 ng in RAW cells.