

Abstract
To investigate the early stages of cell-cell interactions occurring between living biological samples, imaging methods with appropriate spatiotemporal resolution are required. Among the techniques currently available, those based on optical trapping are promising. Methods to image trapped objects, however, in general suffer from a lack of three-dimensional resolution, due to technical constraints. Here, we have developed an original setup comprising two independent modules: holographic optical tweezers, which offer a versatile and precise way to move multiple objects simultaneously but independently, and a confocal microscope that provides fast three-dimensional image acquisition. The optical decoupling of these two modules through the same objective gives users the possibility to easily investigate very early steps in biological interactions. We illustrate the potential of this setup with an analysis of infection by the fungus Drechmeria coniospora of different developmental stages of Caenorhabditis elegans. This has allowed us to identify specific areas on the nematode’s surface where fungal spores adhere preferentially. We also quantified this adhesion process for different mutant nematode strains, and thereby derive insights into the host factors that mediate fungal spore adhesion.
Introduction
Cell-cell contacts are key elements of regulation in diverse biological systems, such as neuronal networks (1), immune synapses (2,3), or between host and pathogen (4). Delineating the molecular mechanisms involved in these processes is still challenging because there is a need to record the sequence of events occurring at the early steps with appropriate spatiotemporal resolution. In this context, the use of trapping techniques to manipulate and control the initial contact events between cells holds much promise. Among these techniques, optical tweezers can be used to manipulate biological systems without damage (5–9). Optical tweezers were first described in the seminal work of Ashkin et al. (10) in which the authors provided the first demonstration that a single laser beam focused through a lens can trap mesoscopic objects (10,11). In addition to their precision and speed, the main advantage of using optical tweezers lies in the limited mechanical stress they place on the manipulated object.
Techniques to image trapped objects in general suffer, however, from a lack of resolution, especially if one needs to maintain a trap and undertake three-dimensional (3D) imaging simultaneously. In part, this is because most optical trapping systems are built around epifluorescent or total internal reflection fluorescence microscopy systems for data acquisition (12). To overcome this issue, optical trapping systems have been developed that combine tweezers with a confocal or two-photon imaging module (13–17). Such systems significantly improve the spatial resolution but are still interdependent: the focal plane of imaging is directly linked to that of the traps. More recently, independent trapping and imaging modules have been combined but with two objectives in an upright-inverted configuration (18).
In this study, and because we needed to record from biological samples with 3D resolution, we have designed a versatile setup optimized for general biological applications on cell-cell interactions. It combines two independent modules, one for trapping and the other for imaging. The trapping module has been described previously (19). It is based on holographic optical tweezers (HOT) using a spatial light modulator (SLM), allowing different possibilities to move multiple trapped objects independently in three dimensions (19) as well as rapid and long-distance displacement of traps using speeds of up to 500 μm/s. The second module comprises a versatile fast confocal imaging system based on a Nipkow spinning disk system. Here, we provide a complete decoupling of the two modules by correcting the trap focalization during the scanning of 3D objects. It is noteworthy that the two modules use the same objective, thereby providing a simpler access to biological samples in comparison to a two-objective configuration (18).
As a proof of principle, we study the earliest step of infection of Caenorhabditis elegans by the fungus Drechmeria coniospora to illustrate the potential of this setup. For the first time, to our knowledge, we analyze precisely the initiation of adhesion between the fungal spores and the worms by testing the first contact of single spores to different regions of the worm and on different stages of development. Finally, by using multicolor acquisition and multiple traps at the same time, we show how this setup is suitable for following the subsequent steps of the infection and could be used to investigate the molecular signal transduction cascades that underlie the host innate immune response (reviewed in (20)).
Materials and Methods
Biological material
All C. elegans strains were maintained on nematode growth media and fed with Escherichia coli strain OP50, as described (21). In addition to the wild-type strain N2 that was obtained from the Caenorhabditis Genetics Center (CGC), the following mutants were used: bus-2(e2687) IV, bus-4(e2693) IV, bus-12(e2977) IV, bus-17(e2800) X (22), the kind gift of Jonathan Hodgkin, as well as the transgenic strains IG1256 (CHC-1::GFP; MYO-2::RFP) (23). Methods for infecting C. elegans with D. coniospora were as previously described (24).
The 3A9 T cell hybridoma is grown as described in the Supporting Material (see Method M1).
Sample preparation for imaging
Worms were anesthetized with levamisole (Fisher Scientific, Illkirch, France) in 50 mM NaCl at a concentration that depended on the worm’s developmental stage (between 0.01% and 0.005%). Following removal from levamisole, all worms recovered a normal mobility after 10 min to 6 h. For some experiments, the worm cuticle was labeled by incubation for 30 min at 20°C with a 10 μg/ml solution of DiD (Life Technologies, Saint-Aubin, France) in 50 mM NaCl.
When needed, D. coniospora spores (107/mL) were labeled with DiI (Life Technologies) at a final concentration of 20 μg/ml in 50 mM NaCl for 15 min at 20°C, and then washed in the same buffer.
All trapping experiments were done in the anesthetic solution. For the infection experiments, a sample holder was designed (Fig. S1) to avoid uncontrolled worm infection by spores diffusing within the medium. Two adjacent wells (one for the worms, the other for the spores) are connected by a small channel (1 × 0.5 × 0.1 mm) allowing spores to be dragged from one side to the other with the optical tweezers. Due to a strong adherence of the spores to the cover glass, we add spores repeatedly to the reservoir to keep some of them in suspension. In each experimental condition, the adhesion was evaluated for 10 spores, in a maximum of five attempts for each spore, on five worms, with no more than two spores per worm. The experiments were performed at 21°C.
Dynamic HOT combined with independent fast 3D-confocal imaging setup
The fast confocal imaging module was combined with HOT around a motorized commercial double-stage inverted microscope (Nikon France SAS, Champigny-sur-Marne, France) (Fig. 1 A) (see also Table S1 for device references). The lower level of the stage microscope (M1) is used for confocal imaging, direct visualization, and brightfield imaging, the upper one (D1) for fluorescence recovery after photobleaching (FRAP) and fluorescence wide-field illumination. We added a third one (D2) for the HOT module.
Figure 1.
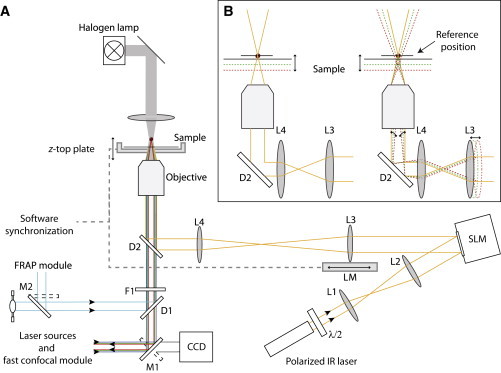
Overview and schematic of the experimental setup. (A) Instrument layout showing the illumination path for brightfield (gray), fluorescence excitation and emission (dark blue, green, and red) and FRAP and arc lamp (cyan) and the trapping IR beam (orange). Mirrors (M1 and M2), dichroic mirrors (D1 and D2), λ/2 wave plate (λ/2), lenses (L1–L4), spatial light modulator (SLM), linear motor (LM), IR filter (F1), and CCD camera (CCD). The gray dotted line represents the software synchronization between the trapping and imaging modules. Two EM-CDD cameras record filtered fluorescence signals coming from the spinning disk confocal module. (B) Representation of L3 motion effect on the trap focalization during 3D sample imaging. On the left side, the focal plane of the trapped object is not dissociated from the one of 3D imaging. On the right side, the displacement of L3 by LM is synchronized compensating the z-displacement of the imaging module and keeping the trap at its reference position. At other positions than the initial one, the IR laser becomes decollimated, this modifies the trapping force (Fig. S5).
The microscope is equipped with a halogen light, a differential interference contrast polarizer, a Wollaston prism, and an electromechanical shutter (Uniblitz, Polytec, Châtillon, France) for fast contrast imaging during multicolor acquisition. Two objectives (Nikon, CFI 4 ×, 0.1 NA) and (Nikon, CFI Plan 10 ×, 0.25 NA) are used for HOT alignment and sample location, the two others (Nikon, CFI Plan Fluor 40 × oil, 1.3 NA) and (Nikon, CFI Plan Fluor 100 × oil, 1.3 NA) for recording and trapping. A xy motorized stage (ASI, Optophase, Lyon, France) is combined with a piezo z-top plate for 3D acquisition (ASI). Four different light pathways (100% binocular, 100% left, 100% right, and a modified base port for 20% binocular and 80% right) are set on the microscope stage. The microscope environment is regulated by a temperature controller box (Digital Pixel, Optophase). The whole system is built on an optical breadboard (Melles Griot Industrie, Voisins-le-Bretonneux, France).
Trapping module
The trapping setup comprises two main optical components, a 1050 nm laser (IPG, Burbach, Germany) to create the trap and a programmable SLM (Hamamatsu Photonics France, Massy, France) to control in real time multiple traps in 3D. The direction of the infrared (IR) light polarization is tuned by a half-wave plate (λ/2 wave plate) (Thorlabs SAS France, Maisons-Laffite, France) to fit the SLM liquid crystal polar orientation. Two lenses L1 (f = 30 mm) and L2 (f = 120 mm) (Thorlabs) expand the laser beam diameter to overfill the SLM. The reflected laser beam is guided by an adjustable second telescope made of two lenses L3 (f = 150 mm) and L4 (f = 125 mm) (Thorlabs) to fill the back aperture of the trapping objectives and to minimize the loss of power. A perfect reproduction of the SLM hologram at the back focal plane of the objective required a 4f-configuration of the L3L4 telescope. The linear motor LM (Physik Instrumente, PI France SAS, Montrouge, France) controls the L3 position to dissociate the focal plane of imaging from the trap position. The light is reflected on the D2 IR dichroic mirror (Rocky Mountain Instrument, Lafayette, CO). The IR filter F1 (Thorlabs) is duplicated to block efficiently IR light on the collection path. Images from the charge-coupled device (CCD) camera (JAI, Stemmer imaging SAS, Suresnes, France) on the right side port of the microscope allow monitoring of traps on the sample, using custom-made software (25).
Imaging module
This setup, adapted by Visitron (Visitron Systems GmbH, Puchheim, Germany), is composed of three modules. The first one encompasses six laser sources: 405, 440, 473, 488, 561, and 640 nm lines. All lasers except the 473 nm laser line are combined (AMS Technologies SARL, Les Ulis, France) in the fiber of the spinning disk module for confocal imaging. The 473 nm laser line is collimated with part of the 405 nm laser in an independent fiber (AMS Technologies) for FRAP experiments. The second module i.e., the confocal spinning disk head (Yokogawa, Visitron Systems GmbH) is on the left side of the microscope. Two 512 × 512 pixels EM-CCD cameras (Hamamatsu) are set for simultaneous two-color detection. The third module (Visitron) dedicated to FRAP measurement has been implemented on the same port as the mercury lamp (Nikon) by using a double lamphouse adaptor M2. A real-time FRAP scanner module provides the possibility to perform classical spot and region of interest FRAP or FRAP on the fly measurements during trapping experiments.
Data acquisition
For brightfield or fluorescence image recording, the laser power, the integration time, and gain of the camera were adjusted to optimize the contrast. 3 μm latex beads (Polyscience Europe GmbH, Eppelheim, Germany) were used for LM calibration. For D. coniospora trapping, a laser power set at 60 mW at the focalization point allowed us to drag the spores at a maximum speed of 0.5 mm/s. With higher power, greater forces could be achieved, but at the potential cost of biological damage.
Results and Discussion
Decoupling HOT from fast 3D confocal imaging
Our aim was to combine HOT with fast 3D confocal imaging to observe biological samples before, during, and after the initial contact with trapped objects (Fig. 1 A). Our first experimental setup kept the image acquisition and the trap within the same focal plane (Fig. 1 B, left panel). The motion of the z-top plate supporting the samples permits the acquisition of 3D images but the trapped samples moved at the same time. As a consequence, it was not possible to dissociate the confocal plane from the trapping one, or to record a 3D stack of images of the trapped object (Fig. 1 B, left panel and Fig. 2 A). Here, we implemented a system to keep traps independent from the imaging module (Fig. 1 B, right panel and Fig. 2 B). The details of the setup are described in the Materials and Methods section.
Figure 2.

Effect of uncoupling trap from imaging on the 3D image reconstruction of a trapped object. In (A) and (B), on the left panels, a single latex bead (arrow) from the ones settled down on the cover glass is trapped, moved by the z-top plate and imaged in brightfield (scale bar, 10 μm); on the right panels, a CD45 membrane-stained T cell hybridoma (CD4 3A9 T cell) was trapped and observed by confocal imaging (stack of 20 images at 1 μm intervals). Scale bar, 15 μm. (A) The sample was moved by the z-top plate without synchronization (Fig. 1B, left panel). The trapped bead stays in the focal plane, whereas beads on the cover glass are defocused. As a consequence, a trapped object is constantly viewed through the identical focal plane all along the stack as illustrated on the right panel with a 3D-image of a cell. (B) Same experiments but with the trapped object maintained in its reference position by synchronization (Fig. 1B, right panel). The trapped bead appears defocused as the ones on the cover glass. On the right, 3D image reconstruction of a trapped cell becomes possible from the stack of images recorded at different altitudes. See also Movie S1 and Movie S2, on beads and cells, respectively.
We required the position of traps within the sample to be held static and therefore, changing the focalization distance of the IR laser. Time-lapse imaging required the distance of the traps to be adjusted sufficiently rapidly to match the maximum frame rate of the imaging system, for instance at 30 ms in full frame in the present case. Moreover, to reach appropriate spatiotemporal resolution for live imaging, one wants to record 10 focal planes in <1 s for 3D-image reconstruction of 10 μm thick objects.
One possible solution lies in refreshing the hologram of the SLM and synchronizing it with the detection frame rate. The calculation of hologram patterns is limited, however, to 2 to 5 Hz when performed in real time, a frequency incompatible with the required image acquisition rate. Although it would in principle be possible to optimize the SLM refreshing rate by performing the calculation of hologram patterns at the camera frame rate, this option is not easily applicable if one wants to trap samples subject to Brownian motion or uncontrolled movements. Alternatively, it is possible to move one of the HOT lenses, as long as it can operate in synchrony with the imaging module. As a first estimation, a one micrometer displacement of the sample from the focal plane would imply a lens movement of around 2 mm. Therefore, repeated lens displacement with a linear motor LM at a frequency of 30 Hz allows the synchronization of the trapping and imaging modules. In principle, the LM is run as a slave of the imaging system by external voltage control. Consequently, this latter option adequately fits the main constraints required to achieve the appropriate temporal resolution for live imaging.
Servo control of the lens displacement
Overfilling the SLM active area implied blocking L1 and L2 lenses at a specific distance. Moreover, our setup was not compatible with the addition of a motor to move L4 due to space limitation on the optical bench. Consequently, we only had the possibility of adding a motor to the L3 lens (Fig. 1 A). This LM has the capability to operate a 2 mm displacement in <30 ms (Fig. S2).
The LM has to compensate the sample holder displacement in real time by correcting the position of the L3 lens to keep the traps at their initial positions during the acquisition of a 3D stack of images. The displacement has to be calculated for a continuum of positions. We therefore determined empirically the relationship linking the position of the sample holder to that of L3, thanks to the procedure described in Fig. S3, wherein the characteristic shape of imaged latex beads in bright field (Fig. 3 A) is used to evaluate their relative position to the focal plane.
Figure 3.
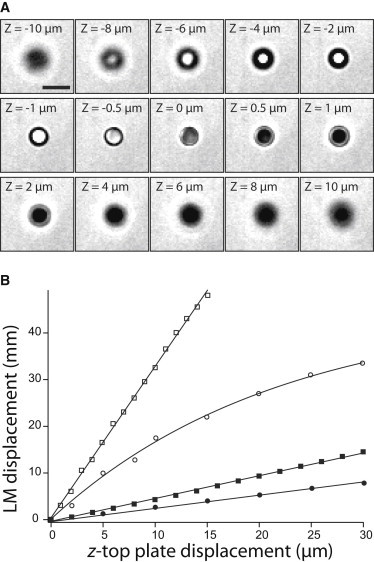
Calibration of the lens displacement compensating the z-top plate motion. (A) Brightfield images of a latex bead (3 μm in diameter) at different altitudes. Out of focus position changes the image of the bead with a white or black centroid above and below the reference plane, respectively. These well-defined changes in the bead shape are used to control L3 position thanks to additional beads settled on the cover glass. 40 × objective and scale bar, 5 μm. (B) Calibration data reported for different combinations of objectives (40 × or 100 × objective) and L3 lenses (100 or 125 mm focal length): 40 ×/100 mm (solid circle), 40 ×/125 mm (solid square), 100 ×/100 mm (open circle) and 100 ×/125 mm (open square). All of the experimental data were fitted with Eq. 1 except the 100 ×/100 mm data fitted with Eq. 2. Fitting parameters are configured in the synchronization module of the Visiview software (Fig. S4).
The overlap of the imaging system and traps’ focal planes defined the reference positions of the two actuators, the z-top plate controlling the sample position and the LM controlling the L3, respectively. This reference initial position was achieved by tuning the L3 position to optimally trap a reference bead that had settled on the cover glass. Next, we moved the sample holder by steps along the optical axis and controlled L3 to maintain the trapped bead on the cover glass. The position was validated once the shape of the trapped bead matched that of nontrapped settled beads on the CCD image. By iteration, we drew an experimental curve of the L3 displacement as a function of the sample holder position on the optical axis (Fig. 3 B). The linear equation Eq. 1 efficiently fits the experimental data from which a and b parameters were estimated for a given set of lenses and objective. Once the values are filled up within the Visiview software (Fig. S4), the L3 position is automatically calculated to compensate the sample displacement in real time
| (1) |
General consideration on the optical setup
Some biological applications might require a range of xy magnification, a z-stack accessible range for optimized trapping forces, and depth of field exploration. For instance, the maximal 50 mm LM course limits the thickness of the z-stack accessible range to 15 and 40 μm with the 100 × and 40 × objectives, respectively (Fig. 3 B). This means that higher magnification requires more L3 translation to compensate a given z-top plate displacement, but reduces the image sampling. Moreover, the longer the focal lenses are, the longer the L3 translation required to compensate the same z-top plate displacement (Fig. 3 B).
In fact, a different combination of optical lenses and objective will critically influence the servo control of L3 displacement and in some cases, the fitting model linking the displacements between actuators, when we are in the nonoptimal 4f-configuration. This occurred with the combination of the 100 × objective with a 100 mm L4 focal length for which a nonlinear curve was observed (Fig. 3 B). In this special case, an exponential equation, Eq. 2, fits efficiently the data:
| (2) |
A L3/L4 set of 150/100 mm focal lengths fits more perfectly the back aperture of the objective and implies smaller LM displacement. We have been constrained, however, by a space limitation on the optical bench to use a L3/L4 set of 150/125 mm focal lengths.
Moreover, the movement of L3 slightly decollimates the IR laser beam at the back aperture of the objective. We have evaluated this effect on the trapping force F by measuring it with the drag force method on a spherical object (Fig. S5), and quantifying it using Stokes’ law for the frictional force, Eq. 3:
| (3) |
in which η is the viscosity constant of the medium, r the radius of the bead, v the maximum velocity (25,26). The variations of F result from two effects: 1), the angle of incidence of the photons coming out varies with the distance of trapping from the objective. Consequently, the closer the trap, the higher the trapping force; 2), the diameter of the beam filling the back aperture of the objective varies with the movement of the L3 lens. It increases or decreases when the trapping plane is above or below the imaging focal plane, respectively. This has a direct effect on the quantity of photons reaching the trapped bead, and consequently causes a change in the trapping force (Fig. S5). But from our own observations, this did not affect the ability to trap objects such as bacteria, beads, spores or eukaryotic cells along the required z-axis range.
In the following biological experiments, we have performed the observations with the optical setup described in the Materials and Methods section, the 40 × objective and the laser power set at 60 mW in the trapping plane. Such parameters allow us to move samples at the maximum speed of the motors. The force applied to objects such as spores cannot be calculated due to their complex shape and their unknown refractive index.
Trapping movement possibilities with spores for different imaging modes
Depending on the biological models and questions, users can have different needs. To provide the most versatile solution, the system was developed to increase the number of possible manipulations for the trapped sample. First, using the motors of the sample holder with a fixed trapped sample in the imaging field and with the displacement of the sample on the cover glass (Fig. 4 A) allows users to move the trapped sample for long distances, in the centimeter range for motors and in the micrometer range along the optical axes. These fast displacements give the possibility to bring two biological objects together from different sides, preventing the risk of unwanted contacts. But using this displacement, one is not able to move the different trapped objects independently in 3D.
Figure 4.
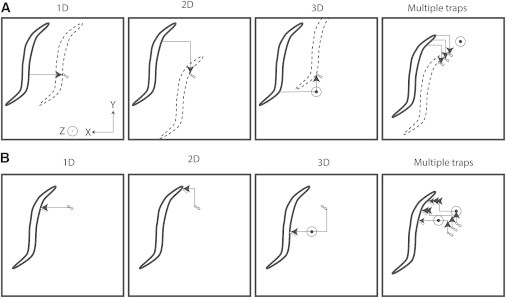
Multiple spatiotemporal trajectories to analyze spore-worm interactions. (A) Sedated worms can be moved by the xyz stage of the microscope to contact a stationary trapped spore(s). In the case of multiple traps, the spores were moved together with identical displacement. (B) Alternatively, trapped spore(s) can be moved in all directions by HOT. Moreover, each trap is independent both in space and time. Directional movements are indicated by red lines in the xy plane and by a circle in the z axis; the direction by an arrow; multiple arrows represent different arrival times. Movies S3–S5 illustrates the different modes of analysis of D. coniospora spore adhesion on C. elegans.
Second, we can use the SLM to move trapped objects without changing the field of view on the cover glass (Fig. 4 B). This fine displacement gives a high precision in time and space of the manipulation, of <5 Hz and one micrometer in all directions. This also allows one to move independently >10 different trapped objects in the optical plane, or in three different focal planes. These two different ways to move objects can be used at the same time and during imaging, independently of the imaging mode (two-dimensional (2D) or 3D). Although we show here the versatility of the system using fungal spores, different biological objects can be trapped, like functionalized beads or different types of cells, using either objects in suspension or ones that are adherent to a surface.
Characterization of fungal spore adhesion to C. elegans
Nematodes are characterized by the presence of a tough exoskeleton, called the cuticle. It serves as a barrier to infection. Certain pathogens, however, such as the nematophagous fungus D. coniospora infect C. elegans and other species of nematode via their cuticle. D. coniospora produces adhesive nonmotile spores (conidia) that are able to attach very strongly to the worm’s cuticle. The spores have a teardrop-shape of 8 μm long, with a spherical head of <1 μm in diameter crowned by the adhesive bud (27). Following attachment, spores can germinate to produce invasive hyphae that penetrate the cuticle and grow throughout the epidermis (27,28). In C. elegans, infection with D. coniospora provokes a rapid innate immune response in the epidermis involving the expression of a large number of defense genes (20,24,29,30). If this host response has been well described, less is known about the very first step of the adhesion of the spore to the worm’s cuticle, and the molecular nature of this interaction. It has been previously shown that the adhesion of spores is not homogeneous on the adult, and that they adhere preferentially to the tip of the head and to the vulva (28,31). The results of studies using other species of nematode suggested that the interaction with the cuticle involves spore protein(s), rather than carbohydrates, localized to the adhesive bud. These were proposed to bind to protein(s) excreted from the sensory organs of the nematodes (32). Using C. elegans, it was shown that spore adhesion was reduced if worms were first treated with Pronase E, consistent with a requirement of proteinaceous material (31). All these studies were done using a concentrated solution of spores and by observing a population of randomly infected worms after several hours of infection.
As a test of the versatility of the system described previously, we decided to investigate precisely the adhesion of the fungus D. coniospora to C. elegans, by carefully controlling the contact of single spores to different regions of the worm and at different stages of development. To do so, we first developed a simple two-well chamber to keep spores separated from the worms until required (Fig. S1). We then used the HOT to move individual fungal spores into contact with the surface of individual worms. By using two simultaneous traps on a single spore, we were able to orient the spore’s contact with the worm. As expected, we observed that the adhesive bud was the only part of the spore that adhered (data not shown). Using a single trap, spores were orientated within the optical axis. By adjusting the z-position of the adhesive bud to bring it into the focal plane of the imaging system, we could show that the head, vulva, and tail region of the adult hermaphrodite worm as well as the male fan allow an immediate adhesion (Fig. 5 and Fig. S6). Once a spore had adhered, we were never able to detach it by pulling with the HOT. If spores adhered efficiently to the head at all stages, they did so to the body only at the late larval (L4) and young adult stages (Fig. 5).
Figure 5.

D. coniospora adhesion on C. elegans at different stages of development. The adhesion tests were performed on different areas of hermaphrodites or males at different stages of development as delineated in Fig. S6. A test is positive 1), if adhesion occurred during the course of HOT contacts between spore and worm and 2), if the association was stable enough not to be dissociated by HOT. In each case, the black column represent the number of spores that stuck to the worm following a single contact, the gray column is the cumulative number of adhered spores following between two and five trials; the white column represents the number of unsuccessful adhesion tests (i.e., following five trials). In most cases, a failure to adhere upon first contact as the result of an unsuitable orientation of the spore. aOnly a precursor of the vulva at this stage. bUncontrolled motion of the worm tail prevent the experiment.
The outermost layer of the nematode cuticle is called the surface coat, and is predominantly made of glycoproteins. It has been shown that its composition in glycans varies with the developmental stage and differs over the surface of the nematode (33). To test whether glycans could also be involved in the first contact adhesion of the fungal spore with the cuticle of the worms, we took advantage of known mutants in genes encoding different classes of one of the major enzymes responsible for the transfer of carbohydrate to protein, galactosyltransferase (bus-2, bus-4, and bus-17), as well as a sugar transporter (bus-12). These bus mutants were originally isolated in screens for mutants to resistant the pathogenic bacterium Microbacterium nematophilum. Their reduced susceptibility to M. nematophilum infection has been shown to result from a reduced adhesion of bacteria to the worm cuticle (22,34,35). The subsequent molecular and biochemical characterization of the bus mutants revealed that M. nematophilum attaches to the cuticle via specific glycans (34,35). We used the HOT system to test the adhesion of D. coniospora fungal spores to these mutants in the same way we tested the wild-type. Unexpectedly, we found that the body region of bus-2, bus-12 and bus-17 adult worms is more adhesive than that of the wild-type (Fig. 6). Probably as a consequence, the three mutants are more susceptible to D. coniospora infection (not shown). A detailed biochemical analysis of the composition in glycans in the bus-2 mutant worms has shown that the level of core-1 glycans is decreased, whereas that of core-2 fucosyl O-glycans is increased (36). In the same study the authors showed that there is an increased staining of the bus-2 mutant cuticle with a α-linked L-fucose-specific lectin (Ulex europaeus agglutinin I), suggesting that there is indeed an increased exposure of fucose on the bus mutant cuticle. Interestingly, this fucose-specific lectin also stains the vulva of wild-type worms (36), a preferential site for the adhesion of D. coniospora spores. Together, these results raise the possibility that fucosyl glycans are a target for the binding of D. coniospora spores.
Figure 6.
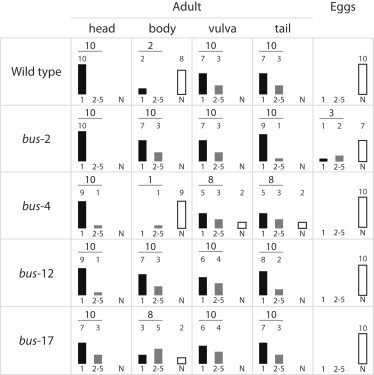
D. coniospora adhesion on adults or eggs of different mutants of C. elegans. Results are expressed as in Fig. 5.
We then tested the 3D imaging during the trapping period and the adhesion of the spore. As a proof of concept, we used a transgenic strain of C. elegans that contains two different fluorescent reporters, one labeling the apical epidermis in GFP, the other an internal structure called the pharynx in RFP. We also stained the worm cuticle and sensory neurons with DiD and the fungal spores with DiI. We were able to drag and image in 3D the adhesion of a fungal spore in multiple colors (three in this case), allowing a spatial resolution of the different subcellular entities of the worm (Fig. 7). This opens the possibility in the future to decipher the activation of the signaling pathways known to be acting during the infection process and to follow the subcellular events that occur after the first contact between the pathogen and its host.
Figure 7.
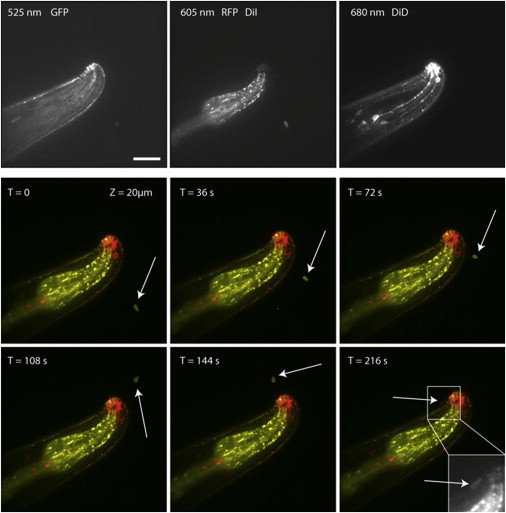
Early adhesion of a single spore on C. elegans by 3D multicolor confocal imaging. An adult nematode carrying GFP and RFP markers in the apical epidermis and pharynx, respectively, is visualized after staining its cuticle and neurons with DiD. A fungal spore stained with DiI (arrow) is trapped to contact the head area of the worm by HOT. The system allows the sample visualization in 3D before and during the adhesion process. Inset at T = 216 s: magnified DiI/mRFP fluorescence image of the contacted area showing the spore along the worm. See Movie S6. Scale bar, 20 μm.
Conclusion
In this work, we developed a setup combining dynamic optical tweezers with a fast 3D imaging confocal head in which we have made the two modules optomechanically independent from each other. This involved compensating in real time the displacement along the optical axis during 3D imaging to keep the trapped object in its initial position. This was done by an LM moving a lens in the light path of the trapping laser beam. We synchronized this motor with the z-scanning and the cameras. It provides access to all of the confocal imaging functionalities such as 3D multicolor imaging or FRAP measurements, while allowing multiple and independent manipulation of a large diversity of biological samples (Movie S6 and Movie S7).
In comparison with other HOT/confocal imaging setups (13,17,18) (Table 1), our system extends the possibility to use HOT for the analysis of cell-cell interactions on very different biological systems. This is first due to the possibility to extend imaging from 2D (13,17) to 3D ((18) and this work) while trapping objects. Moreover, as compared to (18), the present system uses a single objective for trapping and imaging, affording simple and efficient acquisition for routine experimentation. Although the trapping force is not rigorously constant during z-scanning due to slight decollimation of the IR laser beam, the traps conserve sufficient force to drag multiple objects with a large range of speed.
Table 1.
Characteristics of different setups combining HOTs with a confocal module
| Microscope | Tam et al. (17) |
Oddos et al. (13) |
Yevnin et al. (18) |
This work |
|---|---|---|---|---|
| Inverted 1 objective | Inverted 1 objective | Upright/inverted 2 objectives | Inverted 1 objective | |
| Imaging module | ||||
| Confocal head | Spinning | Scanning | Spinning | Spinning |
| Frame rate | ∼30 ms | ∼300 ms | ∼30 ms | ∼30 ms |
| Trapping module | ||||
| Directionality | No | 2D (xy) | 3D (xyz) | 3D (xyz) |
| Number of trap | 1 | 2 | Up to 10 | Up to 10 |
| Independent traps | – | 1 | Multiple | Multiple |
| Independent from the imaging module | No | No | Yes | Yes |
We previously used our HOT module with classical 2D imaging for many different applications such as the study of protein recruitment, cell-cell adhesion (T. Trombik, V. Rouger, S. Piatek, S. Mailfert, S. Ugolini, E. Vivier, D. Marguet, unpublished data), or nanotube formation between cells (37).
Now, we are able to perform 3D imaging for the same type of applications. Here, we demonstrate the interest of this method by analyzing the infection process of C. elegans by D. coniospora. The experiments reported here by using fluorescent transgenic strains of C. elegans highlight the possibility of using our setup to follow in real time the molecular cascades that accompany the early steps of infection. In future work, we will aim to use the possibilities of 3D imaging and multiple trapping to understand how C. elegans worms are able to integrate simultaneous or sequential infections by D. coniospora. To overcome undesired effects due to worm anesthesia, we aim to add a microfluidic system to mechanically constrain the worm during the infection process (38).
Acknowledgments
The authors thank Federico Belloni for initiating the HOT project, Sophie Ugolini and Eric Vivier for general discussion and advice, Helmut Wurm, Peter Waltinger, and Gunter Köhn for their advice and technical assistance, Nassima Chouaki Benmansour for providing T cells, and all members of the two teams for constant support. We thank Jonathan Hodgkin for providing the bus mutants and the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs (P40 OD010440), for the wild-type strain.
This work was supported by institutional grants from Inserm and CNRS, and by specific grants from the Fondation pour la Recherche Médicale (FRM) FRM-DEQ-20090515412, the Agence Nationale de la Recherche (ANR) ANR-10-BLAN-1214 (nanoDIGICODE), ANR-10-INBS-04 (France BioImaging), ANR-11-LABX-0054 (Investissements d’Avenir – Labex INFORM), ANR-11-IDEX-0001-02 (Investissements d’Avenir–A∗MIDEX), ANR-12-BSV3-0001 (FUN-EL), and the European Research Council ERC-2010-AdG 20100317. V.R. was awarded fellowships from the Ligue Nationale Contre le Cancer.
Footnotes
Guillaume Bordet and Carole Couillault’s present address is Institut de Biologie du Développement de Marseille, Aix Marseille Université, CNRS, UMR 7288, Case 907, 13009 Marseille, France.
Contributor Information
Nathalie Pujol, Email: pujol@ciml.univ-mrs.fr.
Didier Marguet, Email: marguet@ciml.univ-mrs.fr.
Supporting Material
References
- 1.Yamada S., Nelson W.J. Synapses: sites of cell recognition, adhesion, and functional specification. Annu. Rev. Biochem. 2007;76:267–294. doi: 10.1146/annurev.biochem.75.103004.142811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grakoui A., Bromley S.K., Dustin M.L. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 3.Monks C.R., Freiberg B.A., Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 4.Stuart L.M., Ezekowitz R.A. Phagocytosis and comparative innate immunity: learning on the fly. Nat. Rev. Immunol. 2008;8:131–141. doi: 10.1038/nri2240. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y., Botvinick E.L., Chien S. Visualizing the mechanical activation of Src. Nature. 2005;434:1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 6.Khalil A.S., Ferrer J.M., Belcher A.M. Single M13 bacteriophage tethering and stretching. Proc. Natl. Acad. Sci. USA. 2007;104:4892–4897. doi: 10.1073/pnas.0605727104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Z., Anvari B., Brownell W.E. Membrane tether formation from outer hair cells with optical tweezers. Biophys. J. 2002;82:1386–1395. doi: 10.1016/S0006-3495(02)75493-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S.T., Takeuchi K., Reinherz E.L. The alphabeta T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 2009;284:31028–31037. doi: 10.1074/jbc.M109.052712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohanty S., Mohanty K., Gupta P. Dynamics of interaction of RBC with optical tweezers. Opt. Express. 2005;13:4745–4751. doi: 10.1364/opex.13.004745. [DOI] [PubMed] [Google Scholar]
- 10.Ashkin A., Dziedzic J.M., Chu S. Observation of a single-beam gradient force optical trap for dielectric particles. Opt. Lett. 1986;11:288–290. doi: 10.1364/ol.11.000288. [DOI] [PubMed] [Google Scholar]
- 11.Ashkin A. Optical trapping and manipulation of neutral particles using lasers. Proc. Natl. Acad. Sci. USA. 1997;94:4853–4860. doi: 10.1073/pnas.94.10.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grashoff C., Hoffman B.D., Schwartz M.A. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263–266. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oddos S., Dunsby C., French P.M. High-speed high-resolution imaging of intercellular immune synapses using optical tweezers. Biophys. J. 2008;95:L66–L68. doi: 10.1529/biophysj.108.143198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinrich M., Tian A., Baumgart T. Dynamic sorting of lipids and proteins in membrane tubes with a moving phase boundary. Proc. Natl. Acad. Sci. USA. 2010;107:7208–7213. doi: 10.1073/pnas.0913997107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goksör M., Enger J., Hanstorp D. Optical manipulation in combination with multiphoton microscopy for single-cell studies. Appl. Opt. 2004;43:4831–4837. doi: 10.1364/ao.43.004831. [DOI] [PubMed] [Google Scholar]
- 16.Hoffmann A., Meyer zu Hörste G., Greulich K.D. Optical tweezers for confocal microscopy. Appl. Phys. B. 2000;71:747–753. doi: 10.1046/j.1365-2818.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- 17.Tam J.M., Castro C.E., Vyas J.M. Control and manipulation of pathogens with an optical trap for live cell imaging of intercellular interactions. PLoS ONE. 2010;5:e15215. doi: 10.1371/journal.pone.0015215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yevnin M., Kasimov D., Roichman Y. Independent and simultaneous three-dimensional optical trapping and imaging. Biomed. Opt. Express. 2013;4:2087–2094. doi: 10.1364/BOE.4.002087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belloni F., Monneret S. Quadrant kinoform: an approach to multiplane dynamic three-dimensional holographic trapping. Appl. Opt. 2007;46:4587–4593. doi: 10.1364/ao.46.004587. [DOI] [PubMed] [Google Scholar]
- 20.Engelmann I., Pujol N. Innate immunity in C. elegans. Adv. Exp. Med. Biol. 2010;708:105–121. doi: 10.1007/978-1-4419-8059-5_6. [DOI] [PubMed] [Google Scholar]
- 21.Stiernagle, T. 2006. Maintenance of C. elegans. WormBook: the online review of C. elegans Biology:1–11. [DOI] [PMC free article] [PubMed]
- 22.Gravato-Nobre M.J., Stroud D., Hodgkin J. Glycosylation genes expressed in seam cells determine complex surface properties and bacterial adhesion to the cuticle of Caenorhabditis elegans. Genetics. 2011;187:141–155. doi: 10.1534/genetics.110.122002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dierking K., Polanowska J., Pujol N. Unusual regulation of a STAT protein by an SLC6 family transporter in C. elegans epidermal innate immunity. Cell Host Microbe. 2011;9:425–435. doi: 10.1016/j.chom.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Pujol N., Zugasti O., Ewbank J.J. Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 2008;4:e1000105. doi: 10.1371/journal.ppat.1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belloni F. Aix-Marseille University; Marseille, France: 2007. Interactive holographic optical tweezers for the dissection of membrane signaling processes. PhD thesis. [Google Scholar]
- 26.Capitanio M., Romano G., Finzi L. Calibration of optical tweezers with differential interference contrast signals. Rev. Sci. Instrum. 2002;73:1687–1696. [Google Scholar]
- 27.Dijksterhuis J., Veenhuis M., Harder W. Ultrastructural study of adhesion and initial stages of infection of nematodes by conidia of Drechmeria coniospora. Mycol. Res. 1990;94:1–8. [Google Scholar]
- 28.Jansson H.B., von Hofsten A., von Mecklenburg C. Life cycle of the endoparasitic nematophagous fungus Meria coniospora: a light and electron microscopic study. Antonie van Leeuwenhoek. 1984;50:321–327. doi: 10.1007/BF00394645. [DOI] [PubMed] [Google Scholar]
- 29.Couillault C., Pujol N., Ewbank J.J. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat. Immunol. 2004;5:488–494. doi: 10.1038/ni1060. [DOI] [PubMed] [Google Scholar]
- 30.Engelmann I., Griffon A., Ewbank J.J. A comprehensive analysis of gene expression changes provoked by bacterial and fungal infection in C. elegans. PLoS ONE. 2011;6:e19055. doi: 10.1371/journal.pone.0019055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jansson H.B. Adhesion of conidia of Drechmeria coniospora to Caenorhabditis elegans wild type and mutants. J. Nematol. 1994;26:430–435. [PMC free article] [PubMed] [Google Scholar]
- 32.Jansson H.B. Adhesion to nematodes of conidia from the nematophagous fungus Drechmeria coniospora. J. Gen. Microbiol. 1993;139:1899–1906. [Google Scholar]
- 33.Davies K.G., Curtis R.H. Cuticle surface coat of plant-parasitic nematodes. Annu. Rev. Phytopathol. 2011;49:135–156. doi: 10.1146/annurev-phyto-121310-111406. [DOI] [PubMed] [Google Scholar]
- 34.Gravato-Nobre M.J., Nicholas H.R., Hodgkin J. Multiple genes affect sensitivity of Caenorhabditis elegans to the bacterial pathogen Microbacterium nematophilum. Genetics. 2005;171:1033–1045. doi: 10.1534/genetics.105.045716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yook K., Hodgkin J. Mos1 mutagenesis reveals a diversity of mechanisms affecting response of Caenorhabditis elegans to the bacterial pathogen Microbacterium nematophilum. Genetics. 2007;175:681–697. doi: 10.1534/genetics.106.060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palaima E., Leymarie N., Cipollo J.F. The Caenorhabditis elegans bus-2 mutant reveals a new class of O-glycans affecting bacterial resistance. J. Biol. Chem. 2010;285:17662–17672. doi: 10.1074/jbc.M109.065433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rainy N., Chetrit D., Kloog Y. H-Ras transfers from B to T cells via tunneling nanotubes. Cell Death Dis. 2013;4:e726. doi: 10.1038/cddis.2013.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilleland C.L., Rohde C.B., Yanik M.F. Microfluidic immobilization of physiologically active Caenorhabditis elegans. Nat. Protoc. 2010;5:1888–1902. doi: 10.1038/nprot.2010.143. [DOI] [PubMed] [Google Scholar]
- 39.Allen P.M., Unanue E.R. Differential requirements for antigen processing by macrophages for lysozyme-specific T cell hybridomas. J. Immunol. 1984;132:1077–1079. [PubMed] [Google Scholar]
- 40.Babbitt B.P., Matsueda G., Allen P.M. Antigenic competition at the level of peptide-Ia binding. Proc. Natl. Acad. Sci. USA. 1986;83:4509–4513. doi: 10.1073/pnas.83.12.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.