

Abstract
The established correlation between neurodegenerative disorders and intracerebral deposition of polyglutamine aggregates motivates attempts to better understand their fibrillar structure. We designed polyglutamines with a few lysines inserted to overcome the hindrance of extreme insolubility and two D-lysines to limit the lengths of β-strands. One is 33 amino acids long (PolyQKd-33) and the other has one fewer glutamine (PolyQKd-32). Both form well-dispersed fibrils suitable for analysis by electron microscopy. Electron diffraction confirmed cross-β structures in both fibrils. Remarkably, the deletion of just one glutamine residue from the middle of the peptide leads to substantially different amyloid structures. PolyQKd-32 fibrils are consistently 10–20% wider than PolyQKd-33, as measured by negative staining, cryo-electron microscopy, and scanning transmission electron microscopy. Scanning transmission electron microscopy analysis revealed that the PolyQKd-32 fibrils have 50% higher mass-per-length than PolyQKd-33. This distinction can be explained by a superpleated β-structure model for PolyQKd-33 and a model with two β-solenoid protofibrils for PolyQKd-32. These data provide evidence for β-arch-containing structures in polyglutamine fibrils and open future possibilities for structure-based drug design.
Introduction
Polyglutamine (polyQ) sequences are found in a variety of innocuous proteins (1,2), but at least nine human proteins containing polyQ runs of longer than 20 residues are associated with severe hereditary neurodegenerative diseases, including Huntington’s disease, spinobulbar muscular atrophy, dentatorubral-pallidoluysian atrophy, and spinocerebellar ataxia (3). The disease-related proteins are not sequence-similar to each other outside of the polyQ regions. The pathogenic mechanisms of these diseases are not fully understood but one thing is definite—the brain tissues of patients contain deposits of the polyQ-rich proteins (4,5). Furthermore, longer polyQ tracts (>30 residues or so) have a stronger predisposition to aggregate, leading to a higher risk and earlier onset of disease (6,7). This linking of polyQ aggregation to neurodegenerative disorders serves as a powerful motivation to better understand the molecular architecture of these aggregates.
Over the past decade, progress has been made toward clarifying the three-dimensional structural arrangement of several other amyloid fibrils, due to application of new experimental techniques (8–11), but the structures of polyQ amyloids have remained elusive. The main obstacle has been the polymorphism and the high insolubility of these polypeptides, beginning even with molecules of <10 residues (12–14). Solubilization of these polyQ molecules and conferring the ability to form well-ordered fibrils requires the addition of flanking charged residues (12,15). Using these modified polyQ constructs, it was established that like other amyloids, the polyQs form cross-β structures (15), and electron microscopy (EM) images have revealed a variety of ribbon-like fibrils or bundles thereof (6,16). However, these constructs still cannot produce well-dispersed and well-ordered fibrils for high quality EM and image analysis.
A substantive step forward has been an account of the packing of glutamine side chains within β-structured aggregates. Initially, Perutz and colleagues (15) suggested that the side chains along with the β-structured backbone help to stabilize the aggregates and they introduced the term glutamine-zipper. Subsequent x-ray diffraction analysis established the arrangement of H-bonded glutamine side chains within β-crystalline assemblies (17,18). In them, the glutamine side chains form H-bonded ladders, which protrude from the planes of the β-sheets and interdigitate tightly to bring the β-sheets together. Within the β-sheets, the chains can be arranged in either a parallel or an antiparallel manner.
Notwithstanding, the overall folds of polyQ chains in amyloid fibrils remained poorly understood. At first, a water-filled nanotube model was suggested in which the β-sheets are strongly curved (19). However, such nanotubes should have a lumen visualizable by negative staining, which is not the case. In other work, it was shown that 45-residue polypeptides with repeats of PGQ9 and PGQ10 fibrillized as efficiently as Q45 (12). The corresponding PGQ7 and PGQ8 polypeptides assembled less readily, whereas introducing one additional proline residue (normally incompatible with a β-strand conformation (20)) in the center of the Q9 element of PGQ9 completely blocked assembly. Polypeptides with PDGQ9 repeats aggregated even more efficiently than PGQ9. These observations suggested that the disease-related polyQ-containing amyloids consist of alternating β-strands and turns with an optimal repeat length of 11–12 residues. It was also suggested that the polyQ chains have an antiparallel β-meander fold (12). A parallel β-helix model (now also called a β-solenoid (21)) was also proposed (12). Later, a parallel superpleated β-structure was proposed for several amyloid fibrils (e.g., Ure2p and Sup35p, α-synuclein, amylin) and, in particular, for polyQ regions of huntingtin (22). Such arrangements are stacks of polypeptide chains composed of several β-strands connected by loops and zigzagging in a planar serpentine fold. Unlike in β-meanders, in superpleated β-structures the β-strands are relatively rotated by 90° around their axes so that they interact via the side chains, not via the polypeptide backbones. When the serpentines stack, their β-strands are integrated into different β-sheets. The minimal motif for such a structure is a strand-loop-strand called a β-arch (11). Other studies accounted for the x-ray patterns of polyQ crystallites as slab-like β-structures, 2–3 nm thick in the β-strand direction. The slab thickness indicated that the polypeptide should form β-strands of ∼7–8 residues separated by reverse turns. It was suggested that polyQ β-crystallites may consist of β-arches (17,18). Recently, based on solid-state NMR spectroscopy data, it has been suggested that polyQ37 and polyQ54 peptides form amyloids compatible with the superpleated β-structure with the β-strands assembled in antiparallel fashion (23). An antiparallel arrangement of polyQ β-strands was also suggested by electron paramagnetic resonance spectroscopy (24).
However, further experimental evidence is still needed to explain how long polyQ runs are accommodated in amyloid fibrils. Native polyQ polypeptides have extremely poor solubility and structural polymorphism, and each polypeptide can assemble into various arrangements of β-structure due to lack of any limiting factor. These challenges have to be resolved to produce well-ordered polyQ fibrils suitable for high quality microscopy. In this study, we designed synthetic polyQ polypeptides adapted by the insertion of charged and D-amino acids to elevate solubility and most importantly to improve homogeneity by restricting the length of each β-strand. It was intended that each polypeptide would fold into three β-strands of a definite length and therefore form fibrils of consistent structure amenable to structural analysis. Our data show that they form amyloid fibrils and are consistent with superpleated and β-solenoid folds, with only the addition of a single Q in the central sequence favoring a switch between these two conformations.
Methods
Peptide synthesis
Polypeptides were synthesized using an Fmoc-continuous flow apparatus (Pioneer, Applied Biosystems, Foster City, CA) starting from preloaded Fmoc-Lys(tBoc)-PEG-PS resins at a 0.2 mmol scale. The coupling reactions were performed with 0.5 M of HATU (1-[Bis (dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate in the presence of 1 M DIEA (diisopropylethylamine). Protecting group removal and final cleavage from the resin were carried out with TFA/phenol/H2O/Triisopropylsilane (88/5/5/2.5%) during 4 h in both cases. The crude polypeptides were purified by reversed phase-high performance liquid chromatography on a C18 column (Interchrom UP5 WOD/25M Uptispere 300 5 ODB, 250 × 21.2 mm). Correctness of the synthesis was confirmed by electrospray ionization mass spectrometry.
In vitro fibril assembly
Lyophilized polypeptide was dissolved in an aqueous solution of 10 mM sodium phosphate or triethylamine (TEA) with 150 mM sodium chloride at pH in the range of 11–12, at a final protein concentration of 5–10 mg/mL. Fibrils typically formed within a few hours but samples were routinely incubated for at least 16 h at room temperature to ensure that formation of fibrils was complete. The best assembly conditions for EM and scanning transmission electron microscopy (STEM) were determined to be TEA buffer at pH 11.8.
Negative staining EM
Specimens were prepared by adsorbing 5-μl drops on to freshly glow-discharged continuous carbon film-bearing grids (Electron Microscopy Science, Hatfield, PA), rinsing with water, and staining with 1% w/w uranyl acetate, and then examined in a CM120 microscope (FEI, Hillsboro, OR) operated at 120 kV. Fibril diameters were measured from digitized negatives (at 1.41 Å/pixel), using Bsoft (25,26). Fibrils were segmented into boxes of 200 pixels along the fibrillar length and 150 pixels across, and profiles calculated by axial averaging. Individual profiles (103 segments of PolyQKd-32, 79 segments of PolyQKd-33) were aligned and averaged. Diameters were measured by applying the full width at half-maximum (FWHM) criterion for edge detection criterion. The experimental uncertainty quoted is the difference between values obtained for two independently analyzed half-data sets.
Electron diffraction
Fibrils in TEA buffer were desalted in a 0.5 mL microcentrifugal filter with 10k MWCO membrane (Millipore, Billerica, MA). Specimens were prepared by applying drops of 0.5–2 μl to freshly glow-discharged carbon-coated grids, washing with water, blotting, and air-drying. Diffraction patterns were taken from areas of 6 μm diameter with doses of 1–4 e−/Å2. Diffraction spacings were calibrated with evaporated thallium chloride (Electron Microscopy Science, Hatfield, PA).
Cryo-EM
4 μl drops of sample were adsorbed to freshly glow-discharged holey carbon film-bearing grids (Quantifoil, Grosslöbichau, Germany), blotted, and vitrified by plunging into liquid ethane on a Vitrobot cryo-station (FEI). These grids were examined in a CM200-FEG microscope (FEI) equipped with a Gatan 626 cryo-holder (Gatan, Pleasanton, CA). The images were recorded on film (SO-163, Kodak) and digitized at 1.67 Å/pixel with a Super CoolScan 9000 ED scanner (Nikon, Melville, NY). Fibrils from images with similar defocus values (∼2.7 μm) were selected, corrected for contrast transfer by phase-flipping, and computationally straightened using Bsoft (25,26) and RUBY-HELIX (27). Density profiles were obtained by averaging the densities along computationally straightened fibrils. 18 fibrils, totaling ∼1 μm in length, were used in this calculation from PolyQKd-32, and 13 from PolyQKd-33. Fibril diameters and estimated margins of error were measured from these data as described below (Results).
STEM
Fibrils were assembled in TEA buffer, buffer exchanged to water, and then prepared for STEM dark-field imaging according to standard procedures of the Brookhaven STEM facility (28,29). Tobacco mosaic virus was included to serve as an internal mass-per-length standard (131.4 kDa/nm). Typically, a 2 μl drop of specimen was adsorbed for 1 min, the grid was washed 10 times with water, and then blotted and plunged into liquid nitrogen slush. The grid was freeze-dried overnight with gradual warming to −80°C and then transferred under vacuum into the STEM. Dark-field digital micrographs were recorded at raster steps of 1.0 and 2.0 nm per pixel. These images were analyzed using PCMass32 (available from the Brookhaven STEM resource, ftp.stem.bnl.gov) and Bsoft (25,26). Boxes 52 nm long and 10 nm wide were used for the measurements. Histograms were calculated from these data. In each case, a Gaussian distribution was fitted to the main peak using Kaleidagraph (Reading, PA). To obtain more finely sampled transverse density profiles, the data were converted to Vernier sampling (30,31) at 0.33 nm per bin with the Total1D tool in the PIC image processing software package (32). A box size of 20 pixels long (at 1 nm/pixel) by 25 pixels across was used for measurement (32 boxes for PolyQKd-32 and 30 for PolyQKd-33). Again, edges were defined using the FWHM method and the experimental uncertainty as the difference between the values obtained from two independently analyzed half-data sets.
Molecular modeling
Fibril models were built using the BIOPOLYMER module of INSIGHT II (Accelrys, San Diego, CA) (33). The resulting structures were subjected to 300 steps of minimization based on the steepest descent algorithm with the H-bond distance constraints set at K = 50, to improve the H-bond geometry. The next 500 steps of refinement were performed by using the conjugate gradients algorithm. To allay concern that the H-bond constraints generated significant tensions in the energy-minimized structure, the last calculation was performed without any restrictions. The DISCOVER module of INSIGHT II, the consistent valence force field, and the distance-dependent dielectric constant were used. The program PROCHECK (34) was used to check the quality of the modeled structure.
Results
Molecular design
The 33-residue polypeptide PolyQKd-33 was intended to have three β-strands separated by two bends (Fig. 1). To insert bends, we introduced D-lysine at positions 10 and 21. D-lysine forces a β-strand to bend via strong steric hindrance to continuing the strand. The putative bends divide the polypeptide into segments of length ∼8 residues (when two turns are allowed), suggested by previous work (12) to be optimal for polyQ-based β-strands. The second polypeptide (PolyQKd-32) differs only in the removal of one glutamine from the region between the two bends. If indeed this region were to form a β-strand, there should be a relative rotation of 180° between its ends, vis-a-vis PolyQKd-33, which might affect fibrillation. Finally, four additional L-Lys residues were inserted into the polyQ segments to make the polypeptides soluble at pH values below 11 (the pKa of Lys).
Figure 1.
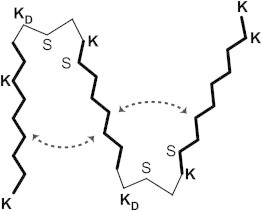
Schematic representation of the designed peptides. K, KD, and S denote location of L-lysine, D-lysine, and L-serine, respectively. The remaining positions are occupied by L-glutamines, which are not denoted by letters, for greater clarity. The peptide shown in the scheme is PolyQKd-33. The peptide PolyQKd-32 has one less L-Gln in the middle section. The angles between β-strands indicated by dashed lines are arbitrary.
PolyQKd peptides form well-dispersed fibrils at high pH
PolyQKd-32 and -33 have similar physical and biochemical behaviors. They are soluble in buffers at neutral pH, and fibril assembly was achieved on raising the pH to 11.5–12.0 in 10 mM sodium phosphate and 150 mM sodium chloride. The optimal pH for assembly was determined to be ∼11.8, at which fibrils formed gradually over several hours, with minimal aggregation. Similar results were obtained when assembly was performed in TEA, a volatile organic buffer that is suitable for STEM analysis.
In conventional EM images of negatively stained preparations, fibrils were typically observed to be long (>1μm), gently curved, and smooth-sided, and with no indication of an axial lumen (Fig. 2, A–D). Sometimes they pair into two or more closely associated fibrils that run together for considerable distances before separating. We measured the widths of the basic (single-stranded) fibrils to be 4.51 ± 0.23 nm for PolyQKd-33 and 5.22 ± 0.39 nm for PolyQKd-32.
Figure 2.
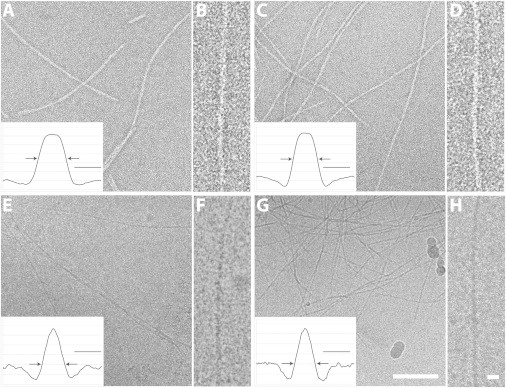
Images of in vitro-assembled PolyQKd-32 and PolyQKd-33 fibrils. Electron micrographs of negatively stained and vitrified PolyQKd-32 (A, B and E, F) and PolyQKd-33 fibrils (C, D and G, H), respectively with their corresponding averaged density profiles (arrows mark operationally defined edges). The scale bars shown in the density profiles are 5 nm. Scale bars indicate 100 nm in A, C, E, G and 10 nm in B, D, F, H.
Cryo-EM (Fig. 2, E–H), gave similar impressions of fibril morphology. From these images, the fibril width was measured to be 3.86 ± 0.13 nm for PolyQKd-33 fibrils, whereas PolyQKd-32 fibrils were 4.18 ± 0.33 nm in width. Thus, both EM techniques consistently indicated a slightly larger diameter for PolyQKd-32 fibrils.
Electron diffraction confirmed the presence of cross-β structure. Specimens of both PolyQKd-32 and PolyQKd-33 fibrils yielded a strong diffraction ring at a spacing of 0.47 nm, the hallmark of cross-β structure (Fig. 3). We would point out that the PolyQKd electron diffraction patterns do not have the reflection at a spacing of ∼1.0 nm that sometimes is observed on the amyloid diffraction patterns. This reflection comes from lateral packing of β- sheets and is generally fainter and broader than the 0.47 nm meridional reflection. In many cases, the 1.0 nm reflection is observed only faintly if at all, unless there are many fibrils packed together and are regularly equispaced in bundles. That was not the case with our EM specimens.
Figure 3.
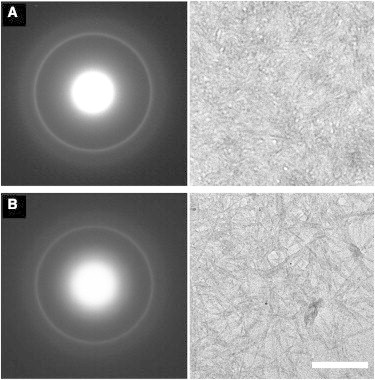
Electron diffraction patterns and corresponding electron micrographs of unstained fibrils. (A) PolyQKd-33 fibrils and (B) PolyQKd-32 fibrils. The scale bar of 200 nm applies to both EM images. The ring in the diffractograms is at a spacing of 0.47 nm.
Mass-per-length difference between PolyQKd-32 and PolyQKd-33 fibrils
To further characterize the fibril structures, we performed mass-per-length analysis (MPL) by STEM (35). Specimens were prepared by buffer exchange from assembly buffer (TEA) to water, adsorption to the carbon support film, further water-washing, and then freeze-drying. We observed that buffer-exchanged specimens had markedly cleaner and more uniform backgrounds than specimens prepared directly from TEA buffer at pH 11.8. For optimal results, the buffer-exchange to water has to be done gently and immediately before grid preparation. Any physical disturbance or protracted storage in water (>24 hours) causes fibril aggregation.
From the STEM images, we measured the MPL from many fibrils of both constructs: only fibrils that were clearly single stranded were included in this analysis (Fig. 4, Fig. 5, A and B ). In both cases, a symmetric unimodal distribution was obtained. From these data, we obtained a value of 12.93 ± 1.69 kDa/nm for the mean linear density of PolyQKd-32 fibrils and 9.57 ± 1.23 kDa/nm for the PolyQKd-33 fibrils (Fig. 5, C and D). In considering possible models of cross-β structures, it is convenient to consider MPL values expressed in terms of numbers of subunits per axial repeat (0.47 nm), given the molecular mass of the monomer (3954.3 and 4082.4 Da for PolyQKd-32 and -33, respectively). Thus, the MPL of PolyQKd-32 was determined to be 1.54 ± 0.20 subunits per axial repeat, and that of PolyQKd-33 fibrils as 1.10 ± 0.14 subunits per repeat.
Figure 4.

STEM images of in vitro assembled fibrils. STEM micrographs of (A) PolyQKd-32 and (B) PolyQKd-33 fibrils with clean background, typical of those used for mass-per-length measurements. Scale bar = 10 nm.
Figure 5.
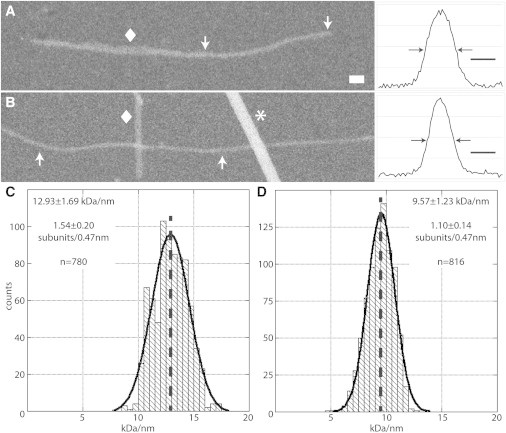
STEM microscopy and mass-per-length analysis. STEM micrographs of PolyQKd-32 (A) and PolyQKd-33 (B) fibrils with their corresponding transverse density profiles. Scale bar = 10 nm on micrographs and 5 nm on the density profiles (arrows mark operationally defined edges). Measurements were made only from uniform thin and apparently single-stranded regions, such as those delimited by white arrows. The regions marked by diamonds are thicker bundled fibril. Tobacco mosaic virus indicated by an asterisk was used as an internal calibration reference. (C) The distribution of PolyQKd-32 measurements has a mean at 1.54 ± 0.20 subunits per axial step (0.47nm), N = 780. (D) The distribution of PolyQKd-33 measurements has a mean at 1.10 ± 0.14 subunits per axial step, N = 816 (see also Fig. 4).
For the STEM images acquired, the electron probe, which is ∼0.3 nm in diameter, sampled the specimen at intervals of 2 nm per pixel and some at 1 nm per pixel. This sampling rate ensures minimal mass loss and is therefore suitable for determining MPLs accurately (29). However, it is too coarse for precise measurement of the diameters of fibrils as thin as these polyQ fibrils. To obtain averaged transverse density profiles that were more finely sampled (0.33 nm/pixel), the data were resorted computationally by Vernier sampling (31), Fig. 5, A and B, and the respective widths were measured. According to these data, the PolyQKd-32 fibrils (5.94 ± 0.66 nm) are slightly but significantly thicker than the PolyQKd-33 fibrils (4.62 ± 0.33 nm) confirming the trend already detected by negative staining and cryo-EM.
Structural interpretation
To interpret the experimental data, we used the following precepts. First, in a fibril, each polypeptide should have the same conformation and engage in equivalent interactions (10,11). Second, there should be bends at the positions of the D-amino acids and, as a result, the building-blocks should have three β-strands with two bends. Taken in the context of cross-β arrangements, these constraints lead us to three possible basic models: i), β-meanders; ii), β-solenoids; or iii), β-serpentines (Fig. 6).
Figure 6.
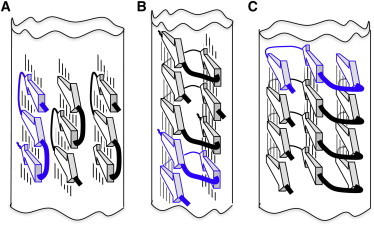
Models of amyloid fibrils built from three β-structured modules, each with three strands (arrows) and two turns. Lines between β-strands denote H-bonds. (A) β-meander model having three side-by-side β-sheets. Its width is between 2.7 and 3.7 nm and expected MPL is one molecule per cross section. In principle, β-meanders from the neighboring β-sheets can be axially displaced. (B) A β-solenoid model in a parallel conformation with estimated width of 1.8 to 3.7 nm and 2/3 molecule per fibril cross section. (C). A superpleated β-structure model in a parallel conformation with width of 2.9 to 3.7 nm and one molecule per fibril cross section.
We interpret the STEM data as showing that the PolyQKd-33 fibrils have one subunit per axial step, as do many other amyloids (35,36). The small discrepancy is discussed further below. The average fibril diameter observed by negative staining, cryo-EM, and STEM is in the range of 4 to 6 nm in each case. These data are well explained by a superpleated model with three β-sheets. As discussed further below, these β-sheets might be parallel (Fig. 6 C) or antiparallel (Fig. 7). Modeling shows a cross section resembling a 2.9 × 3.7 nm rectangle with 4.7 nm in diagonal (see Fig. 8 B). These dimensions are in good agreement with the experimental measurements for the average diameter: 4.5 nm by negative staining, 3.9 nm by cryo-EM, and 4.6 nm by STEM (Table 1). This concurrence further supports this model.
Figure 7.
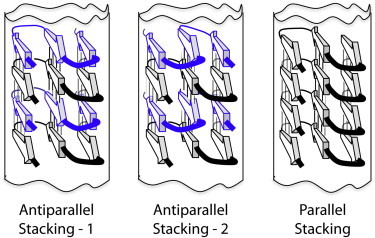
Superpleated β-structure models of PolyQ amyloid fibrils built from three β-structured modules each with three strands (arrows) and two turns. Lines between β-strands denote H-bonds. The polyQ chains can be arranged in two antiparallel (left, and center) or a parallel manner (right). Molecular modeling favors antiparallel stacking type 2 (data not shown) where positively charged Lys is located at greater distances and this diminishes the electrostatic repulsions. Moreover, in the antiparallel arrangement type 1, β-arcs that are packed one over the other have steric tensions.
Figure 8.
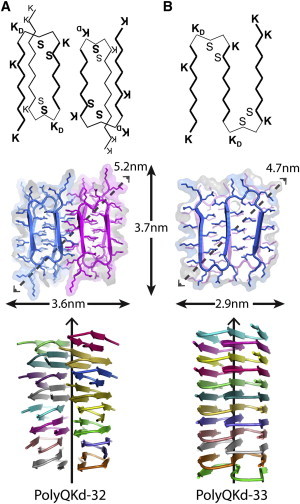
Ribbon representations (axial and lateral views) of structural models of PolyQKd-32 and PolyQKd-33 fibrils. Sequence motifs of β-arcs fit well blbbl conformation (41) (where b is β-conformation and l is left-handed α-conformation of residues). First, the D-lysine in the 10th and 21st positions of each β-arc should favor required left-handed α-conformation. Second, because uncompensated charges inside the protein strongly destabilize the structure, D-lysines in the 10th and 21st positions and L-lysines in the 13th and 24th positions should be on the surface of the engineered fibrils when the bends have the blbbl-arc. Third, for the inside positions of such β-arcs (positions 11, 14 and 22, 25) small serine was selected because this residue is frequently located there in the known blbbl-arcs. The modeled structures do not have steric tensions and donors and acceptors of H-bonds interact with each other or having the possibility to bind water molecules. In accordance with PROCHECK output, the overall average G-factor of the models are ∼0.50, a value typical for a good quality model.
Table 1.
Summary of measurements by all EM methods
| Diameter (nm) | MPL (subunits /0.47nm) | |||
|---|---|---|---|---|
| Methods | Negative staining EM | Cryo-EM | STEM | STEM |
| PolyQKd-32 | 5.22 ± 0.39 | 4.18 ± 0.33 | 5.94 ± 0.66 | 1.54 ± 0.20 |
| PolyQKd-33 | 4.51 ± 0.23 | 3.86 ± 0.13 | 4.62 ± 0.33 | 1.10 ± 0.14 |
| Difference | 13.6 (%) | 7.7 (%) | 22.2 (%) | n/a |
As for the other candidates, the closest β-solenoid model, with two β-strands per turn giving two subunits in three steps of the 0.47 nm repeat (Fig. 6 B), predicts an MPL of 0.67 subunits per axial repeat. This does not satisfy the STEM data. An alternative model, with three long β-sheets packed side by side and formed by stacks of H-bonded β-meanders (Fig. 6 A) does fit the MPL and width data, but does not explain why lateral growth of the stack should be limited to three β-meanders. Moreover, the lysines in the central β-sheet would be buried in close proximity in the fibril core and their mutual electrostatic repulsion makes this model even less plausible.
For PolyQKd-32 fibrils, the STEM MPL measurements give 1.54 ± 0.20 peptides per 0.47 nm axial repeat. These data cannot be explained by models with protofibrils having superpleated β-structures as they give only integral numbers of molecules per axial repeat (1, 2, etc.). They can, in principle, be explained by a β-meander model with a 4-sheet stack giving 1.33 subunits per axial repeat and an estimated width of ∼4 nm, but, as explained previously, the absence of limits to lateral growth and the burying of lysines make this model improbable. The data also disagree with a single β-solenoid protofibril model with 0.67 subunits per cross section, i.e., 1.5 turns of the solenoid per subunit (Fig. 6 B), but a model with two such closely associated β-solenoid protofibrils and 1.33 peptides per cross section agrees with the MPL data to within experimental error. Furthermore, the predicted dimensions of such model are ∼3.6 × 3.7 nm in cross section with 5.2 nm on diagonal (Fig. 8 A), fitting the experimental data (5.2 nm by negative stain; 4.2 nm in cryo-EM; 5.9 nm by STEM, Table 1).
The models proposed provide a simple explanation for the major change in MPL of the fibrils that accompanies insertion of a single amino acid. Both models have β-arches and zipper packing of glutamine side chains and differ only in the overall arrangement of the β-arches.
Discussion
Experimental uncertainty—EM measurements of fibril thickness
In each case—negative staining transmission electron microscopy, cryo-EM, and STEM of unstained freeze-dried specimens—noise in the calculated density profile was suppressed by extensive averaging. However, the three kinds of measurements are subject to different sources of experimental uncertainty. Moreover, the actual numbers obtained for fibril width necessarily depend on the edge detection criterion employed. In negative staining, the principal uncertainty relates to how far the protein structure penetrates into the stain bed that has accumulated along the edges and may be complicated by some positive staining. For these measurements we used the FWHM criterion. The same criterion was applied to the STEM data where apparent width is likely to be exaggerated somewhat by curvature in the filament segments analyzed and imperfect alignment of the sampling boxes with the fibril edges. With cryo-EM data, there is residual negative interference fringes along the edges despite the contrast transfer function correction applied and we define, operationally, the edges as the points at which the profile first falls to the background level defined outside the interference fringes.
Experimental uncertainty—numbers of subunits per axial step
A STEM determination of MPL involves making measurements from a sizeable set of fibril segments, compiling a histogram, and—provided that the resulting distribution is symmetrical and there are no other indications of multiple species—fitting a Gaussian curve. The position of its mean is the MPL determination for that data set. In principle, the experimental uncertainty could be made arbitrarily small by adding more and more data. However, there are numerous potential sources of systematic error that may vary somewhat from experiment to experiment, making it difficult to achieve an absolute degree of reproducibility. These factors have been reviewed by Goldsbury et al. (35), who concluded that a residual discrepancy of 5–10% between the measured value and the actual value is typical.
Relationship between the designed peptides and native polyQ containing amyloids
Bare polyQ polypeptides are not found in vivo (2): rather, the polyQ tracts are parts of longer proteins with other domains, which are likely to interfere by steric hindrance with the addition of subunits to the ends of growing fibrils and thus with the assembly of well-ordered fibrils (37). Furthermore, native polyQ-containing proteins do not contain any D-amino acids in their polyQ tracts that would restrict the lengths of β-strands. These considerations suggest that the fibrillar organization in native polyQ-containing amyloids is in general less regular than in the fibrils we have engineered, but we posit that they polymerize accordingly to much the same set of interactions as the fibrils that we have studied in vitro.
Parallel or antiparallel arrangements
As discussed previously, a superpleated model with three β-sheets complies with all current constraints on the PolyQKd-33 fibril. At this time, we see no compelling argument in favor of a parallel or an antiparallel arrangement. On one hand, a study of three polyQ-containing polypeptides by solid-state NMR spectroscopy favored antiparallel structures (23). However, those constructs were of different lengths and had different non-Q components than the fibrils studied here and the fibrils were all much wider than those assembled in this study. We have also performed molecular modeling that favors antiparallel stacking type 2 (Fig. 7) because the positively charged Lys residues are spaced further apart, diminishing the electrostatic repulsions (A. Kajava, unpublished results).
On the other hand, there are an already substantial number of amyloid fibrils in which parallel β-sheets have been demonstrated. Examples include the yeast prion filaments Ure2p (38) and Sup35p (39) whose fibrillizing domains are rich in Q and N, and the fungal prion Het-s (40). Moreover the model for the PolyQKd-32 fibril, a polypeptide so like PolyQKd-32 in many respects, has an unambiguously parallel arrangement.
Conclusions
Numerous in vitro studies have used synthetic polypeptides to investigate polyQ aggregation. The resulting aggregates can vary in structure from clumps to gels to fibrillar networks. The best way to mimic the natural fibril arrangement with synthetic polypeptides is to design modifications that favor polymerization into fibrils rather than clumps or gels. In this study, we found a way to do so, designing polypeptides that form well dispersed amyloid fibrils suitable for microscopy studies. Our experimental data show that the deletion of just one glutamine residue from the middle of the 33-residue peptide leads to a substantially different amyloid structure. This effect is explained in a straightforward way by two β-arch-containing models. The present results provide insight into the molecular architecture of PolyQ fibrils, and further support the conception of β-arch commonality in amyloid fibril structures. They also open new possibilities for structure-based design of drugs inhibiting fibril growth.
Acknowledgments
We thank Dr. L. N. Marekov for mass-spectrometry, Dr. A. Dearborn for helpful discussions, and T. Phung for adapting the PIC software package to run on a Mac workstation.
This work was supported in part by the intramural research program of National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and by the NCI under contract HHSN261200800001E, and by an Overseas Scholarship Scheme Phase II grant by the Higher Education Commission of Pakistan to A.B.A.
Footnotes
Rick K. Huang and Ulrich Baxa contributed equally to this work.
Contributor Information
Alasdair C. Steven, Email: stevena@mail.nih.gov.
Andrey V. Kajava, Email: andrey.kajava@crbm.cnrs.fr.
References
- 1.Karlin S., Burge C. Trinucleotide repeats and long homopeptides in genes and proteins associated with nervous system disease and development. Proc. Natl. Acad. Sci. USA. 1996;93:1560–1565. doi: 10.1073/pnas.93.4.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jorda J., Kajava A.V. Protein homorepeats sequences, structures, evolution, and functions. Adv. Protein Chem. Struct. Biol. 2010;79:59–88. doi: 10.1016/S1876-1623(10)79002-7. [DOI] [PubMed] [Google Scholar]
- 3.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 4.Davies S.W., Turmaine M., Bates G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 5.Becher M.W., Kotzuk J.A., Ross C.A. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 1998;4:387–397. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 6.Scherzinger E., Lurz R., Wanker E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 7.Martindale D., Hackam A., Hayden M.R. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 8.Antzutkin O.N., Balbach J.J., Tycko R. Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of beta-sheets in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2000;97:13045–13050. doi: 10.1073/pnas.230315097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Margittai M., Langen R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Q. Rev. Biophys. 2008;41:265–297. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- 10.Nelson R., Eisenberg D. Structural models of amyloid-like fibrils. Adv. Protein Chem. 2006;73:235–282. doi: 10.1016/S0065-3233(06)73008-X. [DOI] [PubMed] [Google Scholar]
- 11.Kajava A.V., Baxa U., Steven A.C. Beta arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 2010;24:1311–1319. doi: 10.1096/fj.09-145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thakur A.K., Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. Proc. Natl. Acad. Sci. USA. 2002;99:17014–17019. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka M., Machida Y., Nukina N. Expansion of polyglutamine induces the formation of quasi-aggregate in the early stage of protein fibrillization. J. Biol. Chem. 2003;278:34717–34724. doi: 10.1074/jbc.M209852200. [DOI] [PubMed] [Google Scholar]
- 14.Chen S., Ferrone F.A., Wetzel R. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perutz M.F., Johnson T., Finch J.T. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen S., Berthelier V., Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 17.Sikorski P., Atkins E. New model for crystalline polyglutamine assemblies and their connection with amyloid fibrils. Biomacromolecules. 2005;6:425–432. doi: 10.1021/bm0494388. [DOI] [PubMed] [Google Scholar]
- 18.Sharma D., Shinchuk L.M., Kirschner D.A. Polyglutamine homopolymers having 8–45 residues form slablike beta-crystallite assemblies. Proteins. 2005;61:398–411. doi: 10.1002/prot.20602. [DOI] [PubMed] [Google Scholar]
- 19.Perutz M.F., Finch J.T., Lesk A. Amyloid fibers are water-filled nanotubes. Proc. Natl. Acad. Sci. USA. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou P.Y., Fasman G.D. Empirical predictions of protein conformation. Annu. Rev. Biochem. 1978;47:251–276. doi: 10.1146/annurev.bi.47.070178.001343. [DOI] [PubMed] [Google Scholar]
- 21.Kajava A.V., Steven A.C. Beta-rolls, beta-helices, and other beta-solenoid proteins. Adv. Protein Chem. 2006;73:55–96. doi: 10.1016/S0065-3233(06)73003-0. [DOI] [PubMed] [Google Scholar]
- 22.Kajava A.V., Baxa U., Steven A.C. A model for Ure2p prion filaments and other amyloids: the parallel superpleated beta-structure. Proc. Natl. Acad. Sci. USA. 2004;101:7885–7890. doi: 10.1073/pnas.0402427101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider R., Schumacher M.C., Baldus M. Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy. J. Mol. Biol. 2011;412:121–136. doi: 10.1016/j.jmb.2011.06.045. [DOI] [PubMed] [Google Scholar]
- 24.Bugg C.W., Isas J.M., Langen R. Structural features and domain organization of huntingtin fibrils. J. Biol. Chem. 2012;287:31739–31746. doi: 10.1074/jbc.M112.353839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heymann J.B. Bsoft: image and molecular processing in electron microscopy. J. Struct. Biol. 2001;133:156–169. doi: 10.1006/jsbi.2001.4339. [DOI] [PubMed] [Google Scholar]
- 26.Heymann J.B., Belnap D.M. Bsoft: image processing and molecular modeling for electron microscopy. J. Struct. Biol. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Metlagel Z., Kikkawa Y.S., Kikkawa M. Ruby-Helix: an implementation of helical image processing based on object-oriented scripting language. J. Struct. Biol. 2007;157:95–105. doi: 10.1016/j.jsb.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 28.Sen A., Baxa U., Steven A.C. Mass analysis by scanning transmission electron microscopy and electron diffraction validate predictions of stacked beta-solenoid model of HET-s prion fibrils. J. Biol. Chem. 2007;282:5545–5550. doi: 10.1074/jbc.M611464200. [DOI] [PubMed] [Google Scholar]
- 29.Wall J.S., Simon M.N., Vinogradov S.N. Mass mapping of large globin complexes by scanning transmission electron microscopy. Methods Enzymol. 2008;436:487–501. doi: 10.1016/S0076-6879(08)36027-3. [DOI] [PubMed] [Google Scholar]
- 30.Steven A.C., Simpson T.A., Wall J.S. Radial density profiles of macromolecular filaments determined from dark-field scanning transmission electron micrographs. Improvements in technique and some applications. Ann. N. Y. Acad. Sci. 1986;483:188–201. doi: 10.1111/j.1749-6632.1986.tb34520.x. [DOI] [PubMed] [Google Scholar]
- 31.Steven A.C., Hainfeld J.F., Wall J.S. Radial distributions of density within macromolecular complexes determined from dark-field electron micrographs. Proc. Natl. Acad. Sci. USA. 1984;81:6363–6367. doi: 10.1073/pnas.81.20.6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trus B.L., Kocsis E., Steven A.C. Digital image processing of electron micrographs: the PIC system-III. J. Struct. Biol. 1996;116:61–67. doi: 10.1006/jsbi.1996.0011. [DOI] [PubMed] [Google Scholar]
- 33.Dayringer H.E., Tramontano A., Fletterick R.J. Interactive program for visualization and modelling of proteins, nucleic acids and small molecules. J. Mol. Graph. 1986;4:82–87. [Google Scholar]
- 34.Laskowski R.A., McArthur M.W., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:282–291. [Google Scholar]
- 35.Goldsbury C., Baxa U., Müller S.A. Amyloid structure and assembly: insights from scanning transmission electron microscopy. J. Struct. Biol. 2011;173:1–13. doi: 10.1016/j.jsb.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baxa U., Taylor K.L., Steven A.C. Architecture of Ure2p prion filaments: the N-terminal domains form a central core fiber. J. Biol. Chem. 2003;278:43717–43727. doi: 10.1074/jbc.M306004200. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi T., Katada S., Onodera O. Polyglutamine diseases: where does toxicity come from? what is toxicity? where are we going? J. Mol. Cell Biol. 2010;2:180–191. doi: 10.1093/jmcb/mjq005. [DOI] [PubMed] [Google Scholar]
- 38.Kryndushkin D.S., Wickner R.B., Tycko R. The core of Ure2p prion fibrils is formed by the N-terminal segment in a parallel cross-β structure: evidence from solid-state NMR. J. Mol. Biol. 2011;409:263–277. doi: 10.1016/j.jmb.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shewmaker F., Wickner R.B., Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel beta-sheet structure. Proc. Natl. Acad. Sci. USA. 2006;103:19754–19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wasmer C., Lange A., Meier B.H. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 41.Hennetin J., Jullian B., Kajava A.V. Standard conformations of beta-arches in beta-solenoid proteins. J. Mol. Biol. 2006;358:1094–1105. doi: 10.1016/j.jmb.2006.02.039. [DOI] [PubMed] [Google Scholar]