

Abstract
Despite the power of model systems to reveal basic immunologic mechanisms, critical differences exist between species that necessitate the direct study of human cells. Illustrating this point is the difference in phenotype between patients with Severe Combined Immune Deficiency (SCID) caused by mutations affecting the common gamma chain (γc) cytokine signaling pathway and mice with similar mutations. Although in both species, null mutations in either IL2RG (which encodes γc), or its direct downstream signaling partner JAK3, result in T and NK cell deficiency, an associated B cell deficiency is seen in mice but not in humans with these genetic defects. In this study, we applied recent data that have revised our understanding of the earliest stages of lymphoid commitment in human bone marrow (BM), to determine the requirement for signaling through IL2RG and JAK3 in normal development of human lymphoid progenitors. BM samples from SCID patients with IL2RG (n=3) or JAK3 deficiency (n=2), which produce similar “T-NK-B+” clinical phenotypes, were compared to normal BM and umbilical cord blood as well as BM from children on enzyme treatment for adenosine deaminase deficient SCID (n=2). In both IL2RG- and JAK3-SCID patients, the early stages of lymphoid commitment from HSC were present with development of Lymphoid-primed Multipotent Progenitors, common lymphoid progenitors and B cell progenitors, and normal expression patterns of IL7RA and TLSPR, and the DNA recombination genes DNTT and RAG1. Thus, in humans, signaling through the γc pathway is not required for pre-thymic lymphoid commitment or for DNA rearrangement.
Keywords: Human, Immunodeficiency Diseases, Lymphoid progenitors, Stem Cells, B Cells, T Cells, Cell Differentiation, Hematopoiesis, Bone Marrow, Severe Combined Immunodeficiency, Common Gamma Chain, Human Bone Marrow, Common Lymphoid Progenitor, Lymphoid-primed Multipotent Progenitor
Introduction
Severe Combined Immune Deficiency (SCID) is a rare genetic syndrome characterized by a profound deficiency in functional T lymphoid cells and variable degrees of B cell deficiency or dysfunction.(1) Multiple genetic mutations can cause this syndrome, several of which cause defects in cytokine signaling. Depending on the specific mutation, normal production of B lymphocytes and/or Natural Killer (NK) cells is also affected. The most common form of SCID, X-linked SCID (X-SCID),(2–7) is caused by null mutations in the IL2RG gene, whose protein product is a type I cytokine receptor known as the common gamma chain (γc) (7). γc is a cytokine receptor subunit that forms a complex with the ligand specific receptors for IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21, to provide a common signaling chain for these receptors. (8–13) Mutations of IL2RG therefore result in a complex immunologic phenotype due to an inability to differentiate or function in response to multiple lymphoid cytokines.(8) Human SCID due to deficiency in γc is characterized by an absence of peripheral T and NK cells, and present but functionally impaired B cells.(8, 14)
After cytokine ligand stimulation of one of its partner receptor chains, dimerization of γc activates the hematopoietic-restricted tyrosine kinase Janus-activated kinase 3 (JAK3)/signal transducer and activator of transcription (STAT) pathway.(15–17) Disruption of the gene encoding JAK3 causes an autosomal form of SCID with an otherwise identical clinical phenotype to X-SCID.(18–20)
Although IL2RG and JAK3 null mutations in mice produce the same profound deficiency of T and NK cells seen in humans, a major species-specific difference is seen in B cell development. Whereas humans with mutations in IL2RG or JAK3 have normal numbers of circulating B cells,(18, 21) mice with similar mutations are unable to develop B cells.(22–26). The lack of B cell development in mice with defective γc signaling has been specifically attributed to an inability to respond to IL-7, as mice deficient in IL7Rα, the IL-7 ligand binding partner to γc, are similarly unable to develop B cells (27). Further demonstrating that IL-7 requirements for B lymphopoiesis are different between the two species, patients with IL-7Rα defects have T cell deficiency but normal numbers of B cells (27, 28).
All stages of hematopoiesis, from early progenitors through many categories of mature lymphoid cells, have been examined in mice that are null for IL2RG, IL7Rα, or IL7(29); however, examination of how these genes affect lymphoid commitment in humans has been lacking, as patients with these mutations are rare, access to bone marrow (BM) is difficult and the identification of the early lymphoid progenitor stages in normal human BM has lagged behind that of mice(30).
The stepwise process of differentiation from multipotent hematopoietic stem cells (HSC) into functional lymphocytes initially proceeds through specific progenitor stages with progressively more limited lineage potential. Upregulation of CD10 expression on CD34+ progenitor cells has long been assumed to herald the onset of lymphoid commitment in human hematopoiesis.(31) We have recently identified a lymphoid progenitor stage in human BM (32) that precedes the previously described CD34+CD10+ common lymphoid progenitor (CLP) in lineage commitment from HSC. This progenitor lacks CD10 expression and is marked by high expression of the homing molecule L-selectin (CD62L). The CD34+linneg CD10neg CD45RA+CD62Lhi progenitors are functionally similar to the lymphoid-primed multipotent progenitor (LMPP) identified in murine BM,(33) in that they possess full lymphoid (T, B and NK), dendritic and some myeloid (mostly monocytic) potential, but lack the ability to generate the erythroid or megakaryocytic lineages. Transcriptional profiling and functional assays of the CD34+linneg CD10neg CD45RA+CD62Lhi LMPPs place them as intermediate in development between CD34+linneg CD38neg hematopoietic stem cells (HSC) and the mostly B cell committed CD34+linneg CD10+ CLP.(32) We now apply these recent insights into normal human lymphoid commitment (32) to analyses of BM from patients with SCID, to define the impact of γc- and JAK3-deficiency on development of the first stages of human lymphopoiesis.
Materials and Methods
Collection of bone marrow, cord blood and peripheral blood cells
Human BM was collected from patients with Severe Combined Immunodeficiency at University of California, Los Angeles (UCLA) and Children’s Hospital of Los Angeles (CHLA) under informed consent through protocols approved by the UCLA and CHLA Institutional Review Boards. Normal human adult BM was obtained from healthy donors as anonymous waste materials isolated from filters after BM harvest and from commercial sources (AllCells, Alameda CA). Umbilical cord blood (CB) was collected as anonymous waste material from normal deliveries at UCLA.
Cell Isolation and Analysis
Mononuclear cells were isolated by density gradient centrifugation using Ficoll-Paque (GE Healthcare). Samples were enriched for CD34+ cells by the magnetic-activated cell-sorting (MACS) system (Miltenyi Biotec) and frozen for later analysis. For flow cytometry analysis and isolation, CD34+ enriched samples were incubated with combinations of monoclonal antibodies specific for human antigens [allophycocyanin-indotricarbocyanine–conjugated (APC-Cy7) anti-CD34 (581; Biolegend); and PerCP-indotricarbocyanine 5.5 (PerCP-Cy5.5)–anti-CD45RA (HI100), allo-phycocyanin (APC)–anti-CD38 (HIT2), PE- indotricarbocyanine7 (PE-Cy7)–anti-CD10 (HI10a), PE–anti-CD62L (DREG-56), PE–anti-CD7 (M-T701), PE –anti-CD127 (aka IL7Ralpha) (HIL-7R-M21), FITC anti-CD7(M-T701), APC anti-CD19 (4G7 and SJ25C1), PE or APC-Cy7 anti-CD3 (SK7), FITC anti-CD56 (MY31), APC anti-CD4 (RPA-T4), PE-Cy7 anti-CD8 (RPA-T8), PE anti-CD24 (ML5), FITC anti-CD20 (L27), PE anti-CD20 (2H7), PerCP-Cy5.5 anti-IgM (MHM-88, Biolegend), FITC anti-CD27 (M-T271), APC anti-CD43 (IG10), FITC anti-CD45RO(UCHL1), APC anti-TSLPR (1F11/TSLPR (also known as 1F11 and AB81_85.1F11)) and FITC–labeled lineage-depletion antibodies anti-CD3 (SK7), anti- CD14 (M2E2), anti-CD19 (4G7), anti-CD56 (MY31) and anti-CD235a (GA-R2); all from Becton Dickinson unless noted].
HSC and progenitor cells were analyzed and isolated on a FACSAria II (355-, 405-, 488-, 561- and 633-nm lasers; BD Immunocytometry Systems) as previously described.(32) The DNA-intercalating dye DAPI (4′,6-diamid- ino-2-phenylindole) was added for analysis of viability.
The full gating sequence for HSC (CD34+LinnegCD38neg) was 1) Appropriate forward scatter (FSC) vs side scatter (SSC) (“lymphoid” gate), 2) DAPI negative and Lineage Marker (CD3,CD14,CD19,CD56) negative, gate, and finally 3) CD34+ and CD38neg. The full gating sequence for LMPP (CD34+linnegCD34+CD10negCD45RA+CD62Lhi) was 1) FSC vs SSC (lymphoid gate), 2) DAPI negative and Lineage Marker negative,, 3) CD34+ and CD10neg, and finally 5) CD62Lhi and CD45RA+. The full gating sequence for CLP (CD34+linnegCD10+) was 1) FSC vs SSC (lymphoid gate), 2) DAPI negative and Lineage Marker negative, 3) CD34+ and finally 4) CD10 + and CD45RA+. A ‘no-antibody’ control defined negative gates. B1 cells were defined as DAPI− CD20+CD27+CD43+CD3− as previously described(34, 35); B1 cell gating was defined by negative controls using the florescent minus one (FMO) technique.(36)
Quantitative PCR analysis
After isolation of cells by flow cytometry, RNA was extracted with a Qiagen RNAEsay Microkit (Qiagen) and reverse-transcribed with Omniscript RT, OLIGO DT, random hexamers and RNAguard (Pharmacia Biotech). An ABI Viia7 was used for real-time PCR with Taqman Gene Expression Mastermix and TaqMan probe–based gene-expression analysis [probes: Hs00172121_m1 (RAG1), Hs00172743_m1 (DNTT), Hs01092694_m1 (EBF1), Hs00233515_m1 (CD2), Hs01062241_m1 (CD3E) Hs00984230_m1 (B2M) (Life Technologies)]. Each biologic sample was assayed in technical triplicate. The optimal reference gene for BM populations was previously determined to be Beta 2 Microglobulin (B2M) (32, 37), and B2M expression was detected in all populations and samples tested (Supplemental Figure 1). Quantitative PCR results were normalized through the use of the change-in-cycling-threshold methods (ΔΔCT).
Statistical analysis
Prism version 5 (GraphPad Software Inc) was used for statistical analysis and graphic generation. Flow cytometry data were analyzed with FlowJo software.
Results
Clinical and immunologic characteristics of subjects
Clinical data of subjects with SCID and healthy control subjects who provided BM samples are presented in Table I. BM samples from three male infants with IL2RG-deficient SCID (aged 2 – weeks 3 months), and one female and one male infant with JAK3-deficient SCID (both aged 3 months) were analyzed to assess the effects of γc pathway signaling defects. BM samples from three adults and a six year old child were used as healthy donor controls. For closer age-matched controls, we also examined marrow from two children (ages 3 months and 21 months) with Adenosine Deaminase (ADA) deficient (ADA-SCID) both of whom were on PEG-ADA enzyme replacement therapy with partial immune recovery at the time of BM collection and flow cytometry analysis(38). Analysis of umbilical cord blood was included as a reflection of normal newborn hematopoiesis.
Table I.
Patient Characteristics
| Cell Source | Clinical Genotype | Phenotype | Age at Analysis | Sex |
|---|---|---|---|---|
| Bone Marrow | IL2RG SCID (1) | T− B+ NK− | 3 months | M |
| Bone Marrow | IL2RG SCID (2) | T− B+ NK− | 2 weeks | M |
| Bone Marrow | IL2RG SCID (3) | T− B+ NK− | 8 weeks | M |
| Bone Marrow | JAK3 SCID (1) | T− B+ NK− | 3 months | F |
| Bone Marrow | JAK3 SCID (2) | T− B+ NK− | 3 months | M |
| Bone Marrow | ADA SCID (1) | T− B+ NK+a | 3 months | F |
| Bone Marrow | ADA SCID (2) | T− B− NK+ | 21 months | F |
| Bone Marrow | Normal | T+B+ NK+ | 6 yrs | Unknown |
| Bone Marrow | Normal | T+B+ NK+ | Adult (>20yrs) | Unknown |
| Bone Marrow | Normal | T+B+ NK+ | Adult (>20yrs) | Unknown |
| Bone Marrow | Normal | T+B+ NK+ | Adult (>20yrs) | Unknown |
| Cord Blood | Normal | T+B+ NK+ | Neonatal | Unknown |
| Cord Blood | Normal | T+B+ NK+ | Neonatal | Unknown |
| Cord Blood | Normal | T+B+ NK+ | Neonatal | Unknown |
Shown are details for each SCID patient sample as well as samples used as normal controls. “Clinical Genotype” refers to diagnosis of SCID genotype confirmed by clinical mutation studies. “Phenotype” refers to lineages affected based on initial analysis of peripheral blood. “Age at Analysis” refers to age at which bone marrow was sampled for these studies
IL2RG SCID = IL2Rgamma deficient Severe Combined Immune Deficiency aka X-SCID
JAK3 SCID =Janus Associated Kinase 3 deficient SCID
ADA SCID = Adenosine Deaminase deficient SCID
T = T cell
B = B cell
NK = Natural Killer cell
+ Cell type present in blood
− Cell type absent/markedly decreased in blood
ADA SCID (1) had started exogenous ADA enzyme therapy at time of initial peripheral blood phenotyping; both ADA-deficient patients (1) and (2) were on ADA enzyme therapy at time of marrow analysis
As expected based on findings in peripheral blood, BM aspirates from patients with IL2RG- and JAK3-deficient SCID showed an almost complete absence of contaminating T lymphoid cells (Fig. 1a). Of note, in three of the five patients whose peripheral blood was analyzed, maternal engraftment of T lymphocytes was detected by Fluorescent In Situ Hybridization. Consistent with maternal engraftment, the few CD3+ T cells detectable in SCID marrow aspirates were predominantly mature CD45RO+ cells, in contrast to normal adult BM and cord blood in which naive CD45RA+ T cells predominated (Fig. 1b). Concordant with their peripheral blood phenotype, NK cells were barely detectable (<1%) in the BM of SCID patients (Fig. 1c).
Fig. 1. Lack of IL2RG/JAK3 signaling results in a profound T and NK cell deficiency.

(a) Percentage of human BM CD34neg cells that are CD3+ T cells as detected by flow cytometry (b) Ratio of mature CD45R0+ cells to immature CD45RA+ cells amongst human BM CD3+ T cells as detected by flow cytometry (c) Percentage of human BM CD34neg cells that are CD56+ NK cells as detected by flow cytometry (IL2RG SCID n=2, JAK SCID n=2, NBM [normal BM] n=3 [Two adult, One 6 yo], NCB [normal cord blood] n=2. Bars represent the range of values from independent biologic samples.
Human lymphoid commitment is not dependent on IL2RG/JAK3 signaling
We have previously shown in human BM that IL2RG transcription is significantly up-regulated during differentiation of HSC into LMPP (CD34+ Linneg CD10neg CD45RA+CD62Lhi), and expression continues to increase between the LMPP and CLP (CD34+ Linneg CD10+CD45RA+) stages.(32) We thus investigated whether absence of IL2RG/JAK3 signaling would affect the generation of these early stages of human lymphoid commitment. The frequency of immunophenotypic HSC based on expression of the progenitor antigen CD34 and absence of CD38 and other lineage specific antigens (CD34+linneg CD38neg cells) was similar in normal BM and SCID BM samples (Fig. 2a, Supplemental Table 1). The CD10neg LMPP and CD10+ CLP populations were both readily detectable in BM from infants with IL2RG-deficient SCID and JAK3-deficient SCID as well as infants on treatment for ADA-deficient SCID (Figs. 2b and 2c, Supplemental Table 1). Thus although IL2RG is expressed in the earliest stages of human lymphoid commitment, signaling through IL2RG/JAK3 is not required to generate these progenitors. As we have previously noted, the profile of lymphoid progenitors in umbilical cord blood was markedly different to that of all normal and SCID BM samples, with no clear population of immunophenotypic LMPP and a relatively low frequency of immunophenotypic CLP.
Fig. 2. Lack of IL2RG/JAK3 signaling does not block early lymphoid commitment.
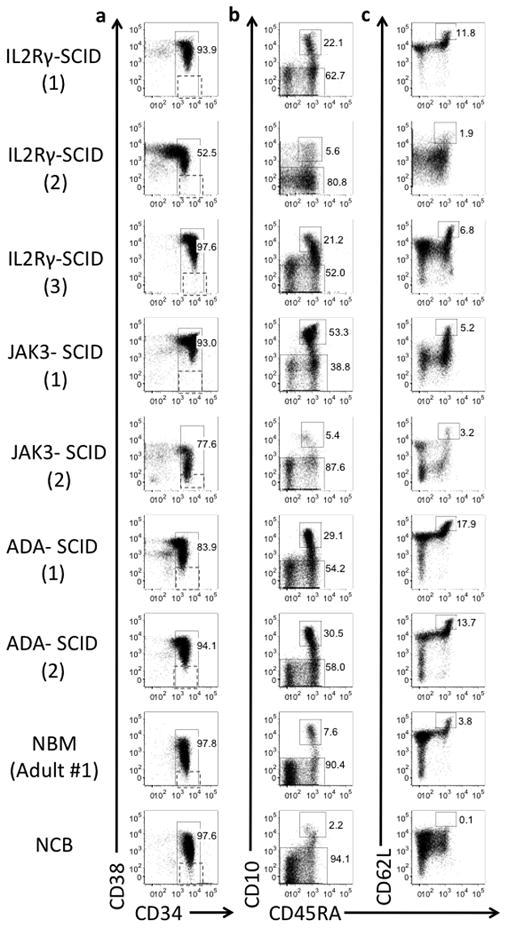
(a) CD34 and CD38 expression on CD34+ enriched DAPI negative, lineage negative (linneg) hematopoietic cells (lineage includes CD3, CD14, CD19, CD56 and Glycophorin a). Hematopoietic Stem Cell (HSC, defined as CD34+ DAPIneg linneg CD38neg) gating shown. (b) Common Lymphoid Progenitors (CLP, defined as CD34+ DAPIneg linneg CD45RA+ CD10+) are detected within CD34+Lin neg cells from all sources (c) Lymphoid-primed multipotent progenitors (LMPP, defined as CD34+ DAPIneg linneg CD10neg CD45RA+ CD62Lhi) are detected within CD34+CD10neg linneg hematopoietic cells from all BM samples but not normal CB. Numbers in parentheses for each SCID sample refer to information in Table 1.
IL7Rα and TSLPR are expressed normally in the absence of IL2RG/JAK3 signaling
Consistent with BM from healthy infants, CD34+ progenitor cells from all IL2RG- and JAK3-deficient SCID patients were heavily skewed toward B cell commitment, comprising mostly fully B cell-committed CD34+CD10+CD19+ progenitors, and to a lesser extent the more immature CD34+ CD10+ CD19− CLP population (which has predominantly B cell potential but can also generate small numbers of NK and T cells) (32, 39–41) (Fig. 3a). Surface expression of IL7Rα (the receptor chain which binds IL-7 and partners with the signaling domain IL2RG) was detected on CLPs from normal BM and BM from all SCID patients (IL2RG, JAK3 and treated ADA) (Fig. 3b). (42) IL7Rα’s alternative dimerization and signaling partner, Thymic Stromal Lymphopoietin Receptor (TSLPR, gene name CRLF2), was also detected on CLPs from all normal and SCID samples examined (Fig. 3c). Thus signaling through γc or its downstream partner JAK3 is not required for normal generation of B lymphoid progenitors. γc-deficient B lymphoid progenitors however possess normal expression of both surface IL7Rα and its alternative signaling partner TSLPR, and are thus capable of responding to TSLP.
Fig. 3. IL7Rα and TLSPR are expressed in the absence of IL2RG/JAK3 signaling.
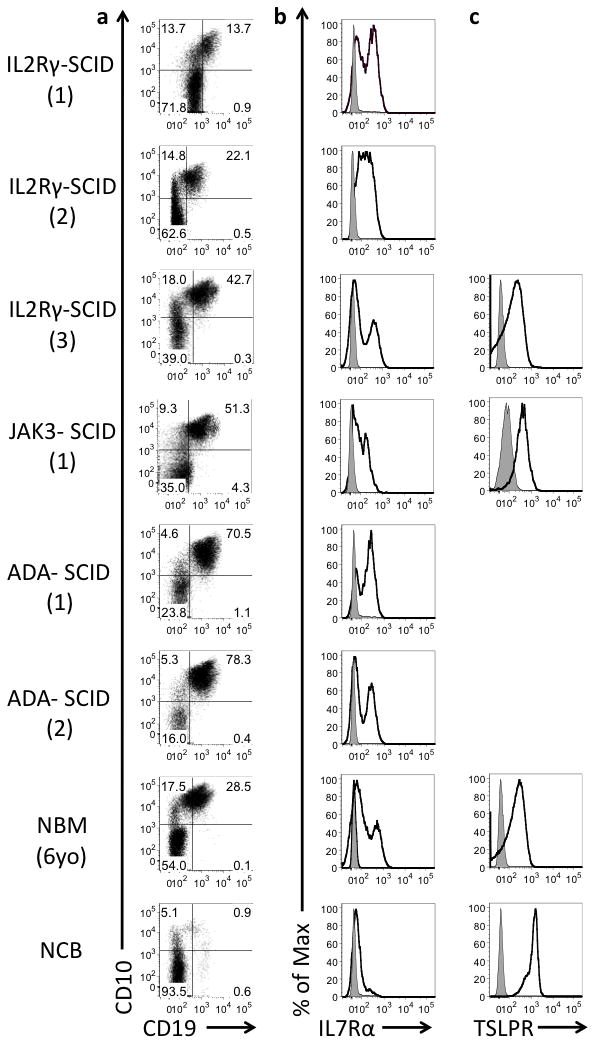
(a) CD10 and CD19 expression on CD34+ hematopoietic progenitor cells. Cell surface expression (shown as black histogram) of (b) IL7Rα and (c) TSLPR on cells gated as Common Lymphoid Progenitors (CD34+CD10+CD19neg). Unstained controls shown as gray). Numbers in parentheses for each SCID sample refer to information in Table 1.
B cell differentiation is maintained in the absence of IL2RG and JAK3 signaling
Despite the lack of functional γc signaling, IL2RG- and JAK3-deficient lymphoid progenitors were able to produce immunophenotypically late stages of B cell differentiation, as shown by expression of CD24, CD20 and surface immunoglobulin (IgM) (Figs. 4a, 4b, and 4c). Based on a recently described phenotype for human B-1 cells (CD20+CD3−CD43+CD27+)(34), few of the B cells in BM of SCID patients were of the B1 cell lineage (Figs. 4d and 4e).
Fig. 4. B cell differentiation is maintained in the absence of IL2RG/JAK3 signaling.
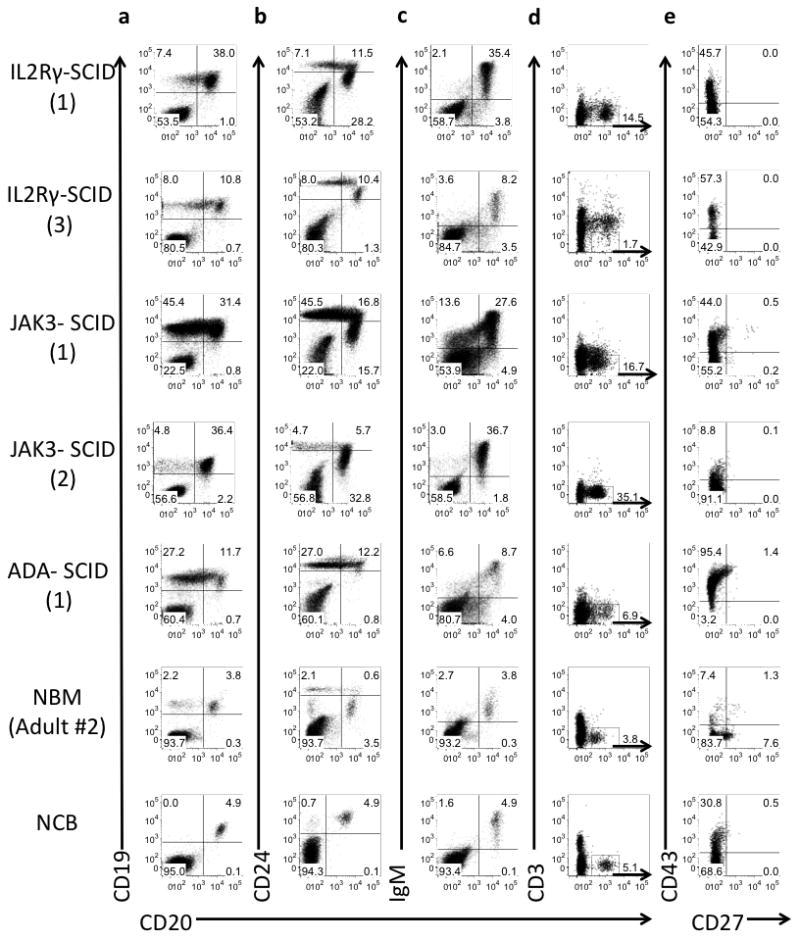
(a) CD19 and CD20, (b) CD24 and CD20, and (c) membrane bound IgM and CD20 expression on CD34neg hematopoietic cells. (d) CD3 and CD20 expression on CD34neg cells (e) B1 cells (CD43+CD27+) within CD3neg CD20+ gated B cells. Numbers in parentheses for each SCID sample refer to information in Table 1.
To determine if γc signaling is required for induction of expression of genes involved in DNA rearrangement, specific subsets of cells representing each stage of differentiation were isolated by flow cytometry and analyzed for gene expression by qPCR. Consistent with their normal counterparts, DNTT (aka TDT) (a DNA polymerase that catalyzes the addition of deoxynucleotides in pre-B and pre-T lymphocytes during early differentiation generating antigen receptor diversity by synthesizing non-germ line elements during VDJ rearrangement) and RAG1 (activator of immunoglobulin V-D-J recombination) were expressed normally in SCID BM, with absent expression in HSC, onset of expression at low levels at the LMPP stage and then further upregulation in CLP and B cell progenitors (Figs. 5a and 5b). EBF1, a transcription factor critical for B cell differentiation that is a direct target of Stat-5 mediated IL7R signaling, (43) and whose overexpression can partially rescue B lineage potential in IL7 KO mice (29, 44, 45), was upregulated in CLP and subsequent stages of B cell differentiation in both SCID BM and in normal BM (Fig. 5c). Of note, although EBF1 was not expressed in normal adult LMPPs in our previous studies (32) or in this study (in either the adult or the 6 year old subject), we were able to detect EBF1 expression in LMPPs from all IL2RG-deficient and JAK3-deficient SCID BM samples, demonstrating that expression of this B cell specific transcription factor occurred at an earlier stage of lymphoid differentiation in these IL2RG and JAK3 deficient SCID patients. However, it should be noted that in BM from infants with ADA-deficient SCID on treatment with PEG-ADA, LMPP also expressed EBF1, suggesting that the discrepancy of results with normal BM may be due to the very young age of the SCID patients rather than their genetic mutations.
Fig. 5. Gene expression for B cell commitment and DNA rearrangement is maintained in the absence of IL2RG/JAK3 signaling.

Quantitative PCR analysis of gene expression in HSC, LMPP, and CLP from BM of IL2RG-SCID (n=3) JAK3-SCID (n=1), ADA-SCID (n=2) and NBM (n=2, one adult and one 6 yo child), presented relative to expression of B-cell Progenitors (BP). (a) DNA nucleotidyltransferase (DNTT, aka TDT) (b) Recombination activating gene 1 (RAG1) (c) Early B Cell Factor 1 (EBF1) (d) Cluster of Differentiation 2 (CD2). Equivalent numbers of HSC (CD34+lin neg -CD38neg), LMPP (CD34+linneg CD45RA+CD10neg CD62Lhi), CLP (CD34+linneg CD10+) and BP (CD34+CD10+CD19+) were tested. Gene expression is shown in the sequence HSC, LMPP, CLP and BP, and * indicates that no expression was detectable in these populations. Each biologic sample was assayed in technical triplicate, Error bars represent SEM of the means of biologic samples.
We have previously shown that CD2, which encodes a T and NK cell specific cell surface adhesion molecule, is normally upregulated in LMPP but is not detectable in either HSC or CLP(32). Similar to normal BM, CD2 was also exclusively detected in the LMPP of BM from patients with SCID (two of three tested with IL2RG deficiency and the sole patient tested with JAK3 deficiency) (Fig. 5d), providing further support that the potential for T and NK differentiation is not restricted at the LMPP stage but disrupts later stages of lineage specific differentiation in the marrow and thymus (Fig. 6).
Fig. 6. Lymphoid lineage commitment in normal and γc-deficient BM.
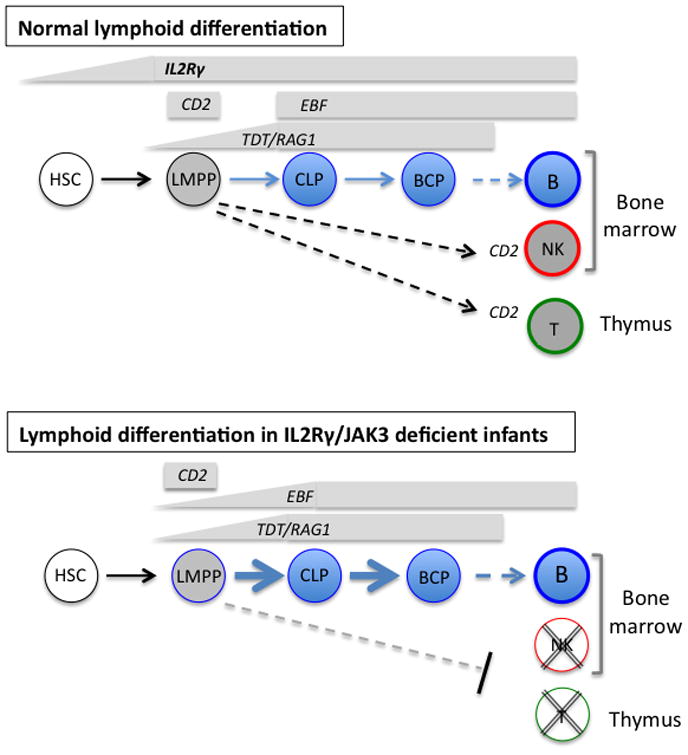
Shown are the lymphoid progenitor stages present in BM from healthy children and adults (upper) and from infants with IL2RG- or JAK3-deficient SCID (lower). Also shown is the pattern of expression of key genes at each stage of differentiation. IL2RG message is barely detectable in HSC and upregulated in LMPP and further in CLP. In both normal and SCID BM, TDT and RAG1 are first detected at the LMPP stage but significantly upregulated in CLP. In normal BM, EBF is first detected in CLP but in infants with defects in γc signaling and with ADA deficiency, EBF is detectable at the LMPP stage. CD2 expression in LMPP is not affected in T-NK-B+ SCID.
Discussion
Although the phenotype of lymphocytes in the peripheral blood of patients with SCID has been well described, studies of how the various genetic defects that lead to this life-threatening syndrome affect the earliest stages of lymphoid commitment have been lacking. Access to BM samples from untreated patients with SCID is challenging; these conditions are extremely rare and, unless detected early through newborn screening, infants typically present with life-threatening infections, requiring urgent hematopoietic stem cell transplantation or gene therapy to survive, after which the fundamental hematopoietic abnormalities are permanently corrected. In addition, although numerous studies on murine BM have been reported over the past two decades, there has relatively little information on the identification of normal human lymphoid progenitors. Of the studies on human lymphopoiesis that do exist, almost all have used umbilical cord blood rather than postnatal BM. It is now clear from the current report and from previous published data that important differences in immunophenotype and function exist between BM and CB stem cells and progenitors (32, 46–48). It should not be surprising that the types of progenitors that circulate transiently in neonatal CB, and the signaling requirements of those progenitors, are different from progenitors that reside in BM to provide steady state postnatal hematopoiesis. Thus measurement of progenitors in abnormal BM requires direct comparison with normal BM.
Age-matched controls are particularly challenging for studies on patients with SCID, almost all of whom are infants. Although HSCs maintain the potential to differentiate into lymphocytes throughout life, lymphoid output decreases relative to myeloid differentiation in later life (49). The frequency of B lymphoid progenitors is highest in infants, falling between early childhood (<10 years), and adulthood (10–60 years), with the lowest levels in elderly adults (>70 years) (50–53). In this report, we were able to include BM from a healthy six year old child, as well as BM samples from healthy adults as normal controls for comparison with BM from infants with IL2RG- and JAK3-deficient SCID. As additional controls, we included BM from two younger patients (3months and 22 months) on enzyme treatment for ADA-deficiency, a form of SCID in which γc signaling is normal, who showed partial recovery of T and B cell numbers by the time of marrow analysis.
Of note, although the LMPP and CLP immunophenotypes were very similar in normal and all types of SCID marrow, significant differences were seen between all the BM samples and those from cord blood, emphasizing the important influence of ontogeny and tissue source on immunophenotypic identification of lymphoid progenitors(47). Specifically, the CD34+linneg CD10neg CD45RA+CD62Lhi immunophenotype that is readily detectable in postnatal BM and identifies the earliest stage of lymphoid commitment (32) is not detectable in CB. In addition, in CB, almost all CD34+CD10+ progenitors co-express CD19 and thus the classic CD34+CD19−CD10+ CLP BM phenotype is very rare in this hematopoietic source.
Early B cell Factor 1 (EBF1), is a transcription factor critical for the early stages of B lineage commitment.(54) In mice, IL-7 has been shown to regulate expression of EBF1; lack of EBF expression is seen in IL7Ra−/− or IL7−/− mice and EBF1 overexpression can partially abrogate the B cell deficiency seen with these IL-7 signaling defects. (29, 44, 45, 55) In our previous studies and in the normal controls for the current report, we consistently found EBF upregulation occurs first at the CLP stage, when IL7Rα is also first expressed (32) with persistent expression in fully B cell committed CD34+CD19+ progenitors. Although LMPP in normal BM possess B cell potential, transcriptional evidence of B lymphoid commitment (EBF and PAX5) is not normally detectable at this earlier stage of lymphoid development(32). In contrast, in IL2RG- and JAK3- deficient SCID patients, EBF expression was upregulated earlier in lymphoid commitment, at the LMPP stage, a finding that could suggest an early skewing toward B lymphoid commitment in γc deficient multipotent progenitors which lack T and NK potential. However it is also possible that this difference is a function of age rather than signaling effects, as LMPPs in the ADA patients also expressed EBF. It would thus appear that the B lineage predominance that occurs in normal infancy originates at the LMPP stage.
We have shown that, consistent with its earlier stage of lymphoid commitment, the LMPP population has greater NK and T cell potential than CD34+linnegCD10+ “CLPs”, a population which is mostly committed to the B cell lineage(32). Consistent with its greater T and NK potential, CD3e and CD2, two markers shared between cells of these lineages, are exclusively expressed in CD34+10negCD62Lhi LMPP(32). Experimental restrictions make it difficult to definitively prove the identity of the BM precursors that normally seed the human thymus, but from numerous mouse models and more limited studies of human thymic progenitors, it appears that more than one marrow progenitor, and even HSC, can migrate to the thymus and initiate T cell differentiation.(56–62) It is likely that the human CD34+10negCD62Lhi LMPP population represents one such lymphoid progenitor that seeds the human thymus, given the finding of a similar immunophenotypic subset among human thymocytes and strong in vitro T cell potential. (31, 32, 40, 41). The presence in IL2RG- and JAK3-deficient BM of apparently normal LMPPs that express CD2 suggests that the impact of γc-signaling on T cell production is first felt within the thymus rather than within the thymus seeding progenitors.
The species differences in B cell production with γc and IL7Rα signaling defects has been assumed to be at least partly due to differences in the dependence of IL-7 signaling for B lymphopoiesis(21–23, 28, 30). Nonetheless the biology underlying this species difference has not been defined. Although adult murine B cell development is IL-7 dependent, the B-1 subset of cells that develop during murine fetal and neonatal life are produced (albeit at subnormal levels) in mice with IL-7 defects(63). Murine B-1 cells are a population of spontaneously IgM-secreting B cells of fetal origin that arise from a distinct hematopoietic progenitor to the adult B-2 cells and which do not require T cell help to function (64, 65). A potential explanation for the apparent species differences in IL2RG-, JAK3- and IL7Rα-deficient SCID is that the B cells circulating in human infants with these forms of SCID represent the human equivalent to murine IL-7 independent B-1 cells. Until recently this hypothesis could not be tested as no immunophenotype has been available to identify human B-1 cells. In our studies we took advantage of the recently described CD20+CD27+CD43+ immunophenotype which has been identified as a B-1 cell population in human umbilical CB and peripheral blood. (34, 35) We found that very few of the B cells in SCID BM expressed this B-1 phenotype, suggesting that either circulating B cells in these patients are not derived from a distinctive, IL-7 independent, fetal origin, or that the published immunophenotype does not identify marrow B-1 cells in these patients.
FLT3 ligand (FL) (66) and TSLP (67, 68) have been suggested as activating pathways that induce B lymphopoiesis in the absence of IL-7. FLT3 expression has been used to discriminate murine LMPP from HSC by flow cytometry. However in humans, although FLT3 mRNA is increased in LMPP, FLT3 protein is detectable at similar levels in HSC and LMPP(32). As both RNA and protein expression of FLT3 are significantly down-regulated in CLP and B cell progenitors(32), it is likely that if FL induced signaling is responsible for B lymphoid differentiation in the setting of γc-deficiency, this effect is predominantly at the LMPP stage, rather than on more B cell committed progenitors such as CD34+CD10+ CLP and CD34+CD19+ B cell progenitors.
TSLP binds to IL-7Rα, and activates Stat5 signaling through the TSLPR chain, independently from γc.(69, 70) As with γc deficiency, B cell deficiency is seen in mice but not humans with IL-7Rα deficiency. However, TSLP knockout mice have normal lymphopoiesis demonstrating that TSLP is redundant in mice when γc signaling is normal. Nonetheless, murine studies show that TSLP has a non-redundant role in the setting of γc deficiency; TSLPR/γc double knockout mice exhibit more severe lymphopenia than γc−/− mice, and TSLP administration can partially rescue lymphopenia in γc deficient mice(71). It has been postulated the normal B cell numbers seen in human γc deficiency could be due to alternative signaling from TSLP through the IL-7Ra/TSLPR complex. Although our data do not answer this possibility directly, the finding that surface TSLPR and IL-7Rα were readily detectable on CLP and B cell progenitors demonstrates that TSLP induced signaling in these lymphoid progenitors is at least possible.
Although B cells in patients with IL2RG-SCID have undergone normal V(D)J immunoglobulin rearrangement, (72, 73) most JH remain in germ-line configuration. The T cell defects present in all types of SCID would be expected to contribute to the functional defects of B cells. However clinical data of immune reconstitution following transplantation show that an indirect effect on B cells is insufficient to fully explain the functional defects in γc-deficient B cells. In a large follow-up study of patients with SCID who had undergone non-myeloablative transplantation(74), in the subset of patients with γc mutations, immunoglobulin deficiency was not corrected in the absence of donor B cell chimerism even if T cell reconstitution was present. Thus intrinsic functional defects exist in γc-deficient B cells. Of the cytokines that enlist the γc associated receptors, IL-4 deficiency is known to be required for mature B cell function and thus impaired signaling through IL-4Ra/γc dimers could account for the B cell functional defects in human X-SCID (75, 76).
In summary, in view of the normal generation of both LMPP and CLP in the presence of ILR2G- and JAK3-deficiency, and the subsequent robust production of B cells, we conclude that γc signaling is not required for lymphoid commitment in humans. The normal expression of DNTT and RAG1 demonstrate that γc signaling is also not required for DNA recombination. The profound lymphoid defects seen in these patients therefore result from blocks to differentiation downstream from the multipotent lymphoid progenitor stage, as NK cells differentiate from more committed progenitors in the marrow and as T cell commitment is initiated and sustained in the thymus.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH) National Heart, Lung and Blood Institute NRSA (1F30HL110458) (LAK), NIH (P01 HL073104, DBK & GMC), California Institute of Regenerative Medicine (CIRM) (RM1-01707 (GMC), U01 AI100801, 1 U01 AI087628 and R01 FD003005-01 (DBK), NIH T32HL066992 (CS), CIRM CSUN UCLA Bridges to Stem Cell Research Program (TB1-01183) (SZ), NIH/NCATS UCLA CTSI Grant (UL1TR000124), Hyundai Hope on Wheels Research Grant Award and St. Baldrick’s Foundation Scholar Career Development Award (180637) (SDO). We gratefully acknowledge the expert technical assistance of the Broad Stem Cell Research Center Flow Cytometry Core and the CFAR Virology Core Lab & Tissue Culture/PCR Facility at UCLA.
References
- 1.Yee A, De Ravin SS, Elliott E, Ziegler JB. Severe combined immunodeficiency: a national surveillance study. Pediatr Allergy Immunol. 2008;19:298–302. doi: 10.1111/j.1399-3038.2007.00646.x. [DOI] [PubMed] [Google Scholar]
- 2.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625–655. doi: 10.1146/annurev.immunol.22.012703.104614. [DOI] [PubMed] [Google Scholar]
- 3.Fischer A, Cavazzana-Calvo M, De Saint Basile G, DeVillartay JP, Di Santo JP, Hivroz C, Rieux-Laucat F, Le Deist F. Naturally occurring primary deficiencies of the immune system. Annu Rev Immunol. 1997;15:93–124. doi: 10.1146/annurev.immunol.15.1.93. [DOI] [PubMed] [Google Scholar]
- 4.Stephan JL, Vlekova V, Le Deist F, Blanche S, Donadieu J, De Saint-Basile G, Durandy A, Griscelli C, Fischer A. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr. 1993;123:564–572. doi: 10.1016/s0022-3476(05)80951-5. [DOI] [PubMed] [Google Scholar]
- 5.Schumacher RF, Notarangelo LD. Severe combined immunodeficiencies of the common gamma-chain/JAK3 signaling pathway. Isr Med Assoc J. 2002;4:131–135. [PubMed] [Google Scholar]
- 6.Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125:S182–194. doi: 10.1016/j.jaci.2009.07.053. [DOI] [PubMed] [Google Scholar]
- 7.Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, McBride OW, Leonard WJ. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993;73:147–157. doi: 10.1016/0092-8674(93)90167-o. [DOI] [PubMed] [Google Scholar]
- 8.Leonard WJ. Cytokines and immunodeficiency diseases. Nat Rev Immunol. 2001;1:200–208. doi: 10.1038/35105066. [DOI] [PubMed] [Google Scholar]
- 9.Russell SM, Keegan AD, Harada N, Nakamura Y, Noguchi M, Leland P, Friedmann MC, Miyajima A, Puri RK, Paul WE, et al. Interleukin-2 receptor gamma chain: a functional component of the interleukin-4 receptor. Science. 1993;262:1880–1883. doi: 10.1126/science.8266078. [DOI] [PubMed] [Google Scholar]
- 10.Noguchi M, Nakamura Y, Russell SM, Ziegler SF, Tsang M, Cao X, Leonard WJ. Interleukin-2 receptor gamma chain: a functional component of the interleukin-7 receptor. Science. 1993;262:1877–1880. doi: 10.1126/science.8266077. [DOI] [PubMed] [Google Scholar]
- 11.Asao H, Okuyama C, Kumaki S, Ishii N, Tsuchiya S, Foster D, Sugamura K. Cutting edge: the common gamma-chain is an indispensable subunit of the IL-21 receptor complex. J Immunol. 2001;167:1–5. doi: 10.4049/jimmunol.167.1.1. [DOI] [PubMed] [Google Scholar]
- 12.Kondo M, Takeshita T, Ishii N, Nakamura M, Watanabe S, Arai K, Sugamura K. Sharing of the interleukin-2 (IL-2) receptor gamma chain between receptors for IL-2 and IL-4. Science. 1993;262:1874–1877. doi: 10.1126/science.8266076. [DOI] [PubMed] [Google Scholar]
- 13.Kimura Y, Takeshita T, Kondo M, Ishii N, Nakamura M, Van Snick J, Sugamura K. Sharing of the IL-2 receptor gamma chain with the functional IL-9 receptor complex. Int Immunol. 1995;7:115–120. doi: 10.1093/intimm/7.1.115. [DOI] [PubMed] [Google Scholar]
- 14.Buckley RH. Primary immunodeficiency diseases due to defects in lymphocytes. N Engl J Med. 2000;343:1313–1324. doi: 10.1056/NEJM200011023431806. [DOI] [PubMed] [Google Scholar]
- 15.Witthuhn BA, Silvennoinen O, Miura O, Lai KS, Cwik C, Liu ET, Ihle JN. Involvement of the Jak-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature. 1994;370:153–157. doi: 10.1038/370153a0. [DOI] [PubMed] [Google Scholar]
- 16.Johnston JA, Kawamura M, Kirken RA, Chen YQ, Blake TB, Shibuya K, Ortaldo JR, McVicar DW, O’Shea JJ. Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2. Nature. 1994;370:151–153. doi: 10.1038/370151a0. [DOI] [PubMed] [Google Scholar]
- 17.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36:515–528. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O’Shea JJ, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID) Nature. 1995;377:65–68. doi: 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- 19.Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, Migone TS, Noguchi M, Markert ML, Buckley RH, O’Shea JJ, Leonard WJ. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995;270:797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- 20.O’Shea JJ, Husa M, Li D, Hofmann SR, Watford W, Roberts JL, Buckley RH, Changelian P, Candotti F. Jak3 and the pathogenesis of severe combined immunodeficiency. Mol Immunol. 2004;41:727–737. doi: 10.1016/j.molimm.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 21.Sugamura K, Asao H, Kondo M, Tanaka N, Ishii N, Ohbo K, Nakamura M, Takeshita T. The interleukin-2 receptor gamma chain: its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu Rev Immunol. 1996;14:179–205. doi: 10.1146/annurev.immunol.14.1.179. [DOI] [PubMed] [Google Scholar]
- 22.Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, Drago J, Noguchi M, Grinberg A, Bloom ET, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity. 1995;2:223–238. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 23.DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci U S A. 1995;92:377–381. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Doherty PC, Grosveld GC, Ihle JN. Defective lymphoid development in mice lacking Jak3. Science. 1995;270:800–802. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- 25.Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, Miyake K, Nakauchi H, Shirasawa T, Saito T. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity. 1995;3:771–782. doi: 10.1016/1074-7613(95)90066-7. [DOI] [PubMed] [Google Scholar]
- 26.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270:794–797. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 27.Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, Meyer JD, Davison BL. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med. 1994;180:1955–1960. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. Nat Genet. 1998;20:394–397. doi: 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- 29.Kikuchi K, Kasai H, Watanabe A, Lai AY, Kondo M. IL-7 specifies B cell fate at the common lymphoid progenitor to pre-proB transition stage by maintaining early B cell factor expression. J Immunol. 2008;181:383–392. doi: 10.4049/jimmunol.181.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parekh C, Crooks GM. Critical differences in hematopoiesis and lymphoid development between humans and mice. J Clin Immunol. 2013;33:711–715. doi: 10.1007/s10875-012-9844-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galy A, Travis M, Cen D, Chen B. Human T, B, natural killer, and dendritic cells arise from a common bone marrow progenitor cell subset. Immunity. 1995;3:459–473. doi: 10.1016/1074-7613(95)90175-2. [DOI] [PubMed] [Google Scholar]
- 32.Kohn LA, Hao QL, Sasidharan R, Parekh C, Ge S, Zhu Y, Mikkola HK, Crooks GM. Lymphoid priming in human bone marrow begins before expression of CD10 with upregulation of L-selectin. Nat Immunol. 2012;13:963–971. doi: 10.1038/ni.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adolfsson J, Mansson R, Buza-Vidas N, Hultquist A, Liuba K, Jensen CT, Bryder D, Yang L, Borge OJ, Thoren LA, Anderson K, Sitnicka E, Sasaki Y, Sigvardsson M, Jacobsen SE. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121:295–306. doi: 10.1016/j.cell.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med. 2011;208:67–80. doi: 10.1084/jem.20101499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffin DO, Rothstein TL. Human b1 cell frequency: isolation and analysis of human b1 cells. Front Immunol. 2012;3:122. doi: 10.3389/fimmu.2012.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roederer M. Compensation in flow cytometry. Curr Protoc Cytom Chapter. 2002;1(Unit 1 14) doi: 10.1002/0471142956.cy0114s22. [DOI] [PubMed] [Google Scholar]
- 37.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan B, Wara D, Bastian J, Hershfield MS, Bohnsack J, Azen CG, Parkman R, Weinberg K, Kohn DB. Long-term efficacy of enzyme replacement therapy for adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID) Clin Immunol. 2005;117:133–143. doi: 10.1016/j.clim.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Ichii M, Oritani K, Yokota T, Zhang Q, Garrett KP, Kanakura Y, Kincade PW. The density of CD10 corresponds to commitment and progression in the human B lymphoid lineage. PLoS One. 2010;5:e12954. doi: 10.1371/journal.pone.0012954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Six EM, Bonhomme D, Monteiro M, Beldjord K, Jurkowska M, Cordier-Garcia C, Garrigue A, Dal Cortivo L, Rocha B, Fischer A, Cavazzana-Calvo M, Andre-Schmutz I. A human postnatal lymphoid progenitor capable of circulating and seeding the thymus. J Exp Med. 2007;204:3085–3093. doi: 10.1084/jem.20071003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, Dick JE. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11:585–593. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]
- 42.Ryan DH, Nuccie BL, Ritterman I, Liesveld JL, Abboud CN, Insel RA. Expression of interleukin-7 receptor by lineage-negative human bone marrow progenitors with enhanced lymphoid proliferative potential and B-lineage differentiation capacity. Blood. 1997;89:929–940. [PubMed] [Google Scholar]
- 43.Roessler S, Gyory I, Imhof S, Spivakov M, Williams RR, Busslinger M, Fisher AG, Grosschedl R. Distinct promoters mediate the regulation of Ebf1 gene expression by interleukin-7 and Pax5. Mol Cell Biol. 2007;27:579–594. doi: 10.1128/MCB.01192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dias S, Silva H, Jr, Cumano A, Vieira P. Interleukin-7 is necessary to maintain the B cell potential in common lymphoid progenitors. J Exp Med. 2005;201:971–979. doi: 10.1084/jem.20042393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kikuchi K, Lai AY, Hsu CL, Kondo M. IL-7 receptor signaling is necessary for stage transition in adult B cell development through up-regulation of EBF. J Exp Med. 2005;201:1197–1203. doi: 10.1084/jem.20050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hao QL, Zhu J, Price MA, Payne KJ, Barsky LW, Crooks GM. Identification of a novel, human multilymphoid progenitor in cord blood. Blood. 2001;97:3683–3690. doi: 10.1182/blood.v97.12.3683. [DOI] [PubMed] [Google Scholar]
- 47.Payne KJ, Crooks GM. Immune-cell lineage commitment: translation from mice to humans. Immunity. 2007;26:674–677. doi: 10.1016/j.immuni.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 48.Hao QL, Shah AJ, Thiemann FT, Smogorzewska EM, Crooks GM. A functional comparison of CD34 + CD38 − cells in cord blood and bone marrow. Blood. 1995;86:3745–3753. [PubMed] [Google Scholar]
- 49.Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, Weissman IL. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuranda K, Vargaftig J, de la Rochere P, Dosquet C, Charron D, Bardin F, Tonnelle C, Bonnet D, Goodhardt M. Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell. 2011;10:542–546. doi: 10.1111/j.1474-9726.2011.00675.x. [DOI] [PubMed] [Google Scholar]
- 51.Rossi MI, Yokota T, Medina KL, Garrett KP, Comp PC, Schipul AH, Jr, Kincade PW. B lymphopoiesis is active throughout human life, but there are developmental age-related changes. Blood. 2003;101:576–584. doi: 10.1182/blood-2002-03-0896. [DOI] [PubMed] [Google Scholar]
- 52.Caldwell CW, Poje E, Helikson MA. B-cell precursors in normal pediatric bone marrow. Am J Clin Pathol. 1991;95:816–823. doi: 10.1093/ajcp/95.6.816. [DOI] [PubMed] [Google Scholar]
- 53.Andreoni C, Rigal D, Bonnard M, Bernaud J. Phenotypic analysis of a large number of normal human bone marrow sample by flow cytometry. Blut. 1990;61:271–277. doi: 10.1007/BF01732876. [DOI] [PubMed] [Google Scholar]
- 54.Lin H, Grosschedl R. Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature. 1995;376:263–267. doi: 10.1038/376263a0. [DOI] [PubMed] [Google Scholar]
- 55.Tsapogas P, Zandi S, Ahsberg J, Zetterblad J, Welinder E, Jonsson JI, Mansson R, Qian H, Sigvardsson M. IL-7 mediates Ebf-1-dependent lineage restriction in early lymphoid progenitors. Blood. 2011;118:1283–1290. doi: 10.1182/blood-2011-01-332189. [DOI] [PubMed] [Google Scholar]
- 56.Ceredig R. Fates and potentials of thymus-seeding progenitors. Nat Immunol. 2012;13:309–310. doi: 10.1038/ni.2265. [DOI] [PubMed] [Google Scholar]
- 57.Luc S, Luis TC, Boukarabila H, Macaulay IC, Buza-Vidas N, Bouriez-Jones T, Lutteropp M, Woll PS, Loughran SJ, Mead AJ, Hultquist A, Brown J, Mizukami T, Matsuoka S, Ferry H, Anderson K, Duarte S, Atkinson D, Soneji S, Domanski A, Farley A, Sanjuan-Pla A, Carella C, Patient R, de Bruijn M, Enver T, Nerlov C, Blackburn C, Godin I, Jacobsen SE. The earliest thymic T cell progenitors sustain B cell and myeloid lineage potential. Nat Immunol. 2012;13:412–419. doi: 10.1038/ni.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wada H, Masuda K, Satoh R, Kakugawa K, Ikawa T, Katsura Y, Kawamoto H. Adult T-cell progenitors retain myeloid potential. Nature. 2008;452:768–772. doi: 10.1038/nature06839. [DOI] [PubMed] [Google Scholar]
- 59.Bell JJ, Bhandoola A. The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature. 2008;452:764–767. doi: 10.1038/nature06840. [DOI] [PubMed] [Google Scholar]
- 60.Perry SS, Welner RS, Kouro T, Kincade PW, Sun XH. Primitive lymphoid progenitors in bone marrow with T lineage reconstituting potential. J Immunol. 2006;177:2880–2887. doi: 10.4049/jimmunol.177.5.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schlenner SM, Rodewald HR. Early T cell development and the pitfalls of potential. Trends Immunol. 2010;31:303–310. doi: 10.1016/j.it.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 62.Hao QL, George AA, Zhu J, Barsky L, Zielinska E, Wang X, Price M, Ge S, Crooks GM. Human intrathymic lineage commitment is marked by differential CD7 expression: identification of CD7- lympho-myeloid thymic progenitors. Blood. 2008;111:1318–1326. doi: 10.1182/blood-2007-08-106294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Milne CD, Paige CJ. IL-7: a key regulator of B lymphopoiesis. Semin Immunol. 2006;18:20–30. doi: 10.1016/j.smim.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 64.Montecino-Rodriguez E, Leathers H, Dorshkind K. Identification of a B-1 B cell-specified progenitor. Nat Immunol. 2006;7:293–301. doi: 10.1038/ni1301. [DOI] [PubMed] [Google Scholar]
- 65.Dorshkind K, Montecino-Rodriguez E. Fetal B-cell lymphopoiesis and the emergence of B-1-cell potential. Nat Rev Immunol. 2007;7:213–219. doi: 10.1038/nri2019. [DOI] [PubMed] [Google Scholar]
- 66.Jensen CT, Kharazi S, Boiers C, Cheng M, Lubking A, Sitnicka E, Jacobsen SE. FLT3 ligand and not TSLP is the key regulator of IL-7-independent B-1 and B-2 B lymphopoiesis. Blood. 2008;112:2297–2304. doi: 10.1182/blood-2008-04-150508. [DOI] [PubMed] [Google Scholar]
- 67.Vosshenrich CA, Cumano A, Muller W, Di Santo JP, Vieira P. Thymic stromal-derived lymphopoietin distinguishes fetal from adult B cell development. Nat Immunol. 2003;4:773–779. doi: 10.1038/ni956. [DOI] [PubMed] [Google Scholar]
- 68.Vosshenrich CA, Cumano A, Muller W, Di Santo JP, Vieira P. Pre-B cell receptor expression is necessary for thymic stromal lymphopoietin responsiveness in the bone marrow but not in the liver environment. Proc Natl Acad Sci U S A. 2004;101:11070–11075. doi: 10.1073/pnas.0402919101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, Ziegler SF, Leonard WJ, Lodish HF. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol. 2000;1:59–64. doi: 10.1038/76923. [DOI] [PubMed] [Google Scholar]
- 70.Park LS, Martin U, Garka K, Gliniak B, Di Santo JP, Muller W, Largaespada DA, Copeland NG, Jenkins NA, Farr AG, Ziegler SF, Morrissey PJ, Paxton R, Sims JE. Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: Formation of a functional heteromeric complex requires interleukin 7 receptor. J Exp Med. 2000;192:659–670. doi: 10.1084/jem.192.5.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leonard WJ. TSLP: finally in the limelight. Nat Immunol. 2002;3:605–607. doi: 10.1038/ni0702-605. [DOI] [PubMed] [Google Scholar]
- 72.Abe T, Tsuge I, Kamachi Y, Torii S, Utsumi K, Akahori Y, Ichihara Y, Kurosawa Y, Matsuoka H. Evidence for defects in V(D)J rearrangements in patients with severe combined immunodeficiency. J Immunol. 1994;152:5504–5513. [PubMed] [Google Scholar]
- 73.Minegishi Y, Okawa H, Sugamura K, Yata J. Preferential utilization of the immature JH segment and absence of somatic mutation in the CDR3 junction of the Ig H chain gene in three X-linked severe combined immunodeficiency patients. Int Immunol. 1994;6:1709–1715. doi: 10.1093/intimm/6.11.1709. [DOI] [PubMed] [Google Scholar]
- 74.Buckley RH, Win CM, Moser BK, Parrott RE, Sajaroff E, Sarzotti-Kelsoe M. Post-transplantation B cell function in different molecular types of SCID. J Clin Immunol. 2013;33:96–110. doi: 10.1007/s10875-012-9797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuhn R, Rajewsky K, Muller W. Generation and analysis of interleukin-4 deficient mice. Science. 1991;254:707–710. doi: 10.1126/science.1948049. [DOI] [PubMed] [Google Scholar]
- 76.Harris DP, Goodrich S, Mohrs K, Mohrs M, Lund FE. Cutting edge: the development of IL-4-producing B cells (B effector 2 cells) is controlled by IL-4, IL-4 receptor alpha, and Th2 cells. J Immunol. 2005;175:7103–7107. doi: 10.4049/jimmunol.175.11.7103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.