

Abstract
Angiogenesis is a multistep process driven by a wide range of positive and negative regulatory factors. Extracellular matrix (ECM) plays a crucial role in the regulation of this process. The degradation of ECM, occurring in response to an angiogenic stimulus, leads to degradation or partial modification of matrix molecules, release of soluble factors, and exposure of cryptic sites with pro- and/or antiangiogenic activity. ECM molecules and fragments, resulting from proteolysis, can also act directly as inflammatory stimuli, and this can explain the exacerbated angiogenesis that drives and maintains several inflammatory diseases. In this review we have summarized some of the more recent literature data concerning the molecular control of ECM in angiogenesis in both physiological and pathological conditions.
1. Introduction
The extracellular matrix (ECM) is the noncellular component present within all tissues and organs, consisting of a variety of structural and signalling molecules secreted from differentiated mesenchymal cells including chondrocytes and fibroblasts and with biochemical, biomechanical, and structural properties critical for the development of organs.
The ECM provides mechanical adhesive support for the cellular constituents, directs their morphological organization, and influences physiological functions, by binding growth factors and interacting with cell-surface receptors. Two biochemically and morphologically differentiated entities have been identified: the interstitial matrix and the extracellular basement membranes (BMs). The first one is mainly composed of fibrillar and nonfibrillar collagens, elastic fibers, and glycosaminoglycan- (GAG-) containing noncollagenous glycoproteins (hyaluronan and proteoglycans) (Figure 1(a)). The BMs are highly specialized extracellular matrix sheets underlining epithelial or endothelial cells, consisting of collagen IV, laminins, entactin, and heparan sulfate proteoglycans (Figure 1(b)), which affect cell shape, gene expression, proliferation, migration, and apoptosis.
Figure 1.
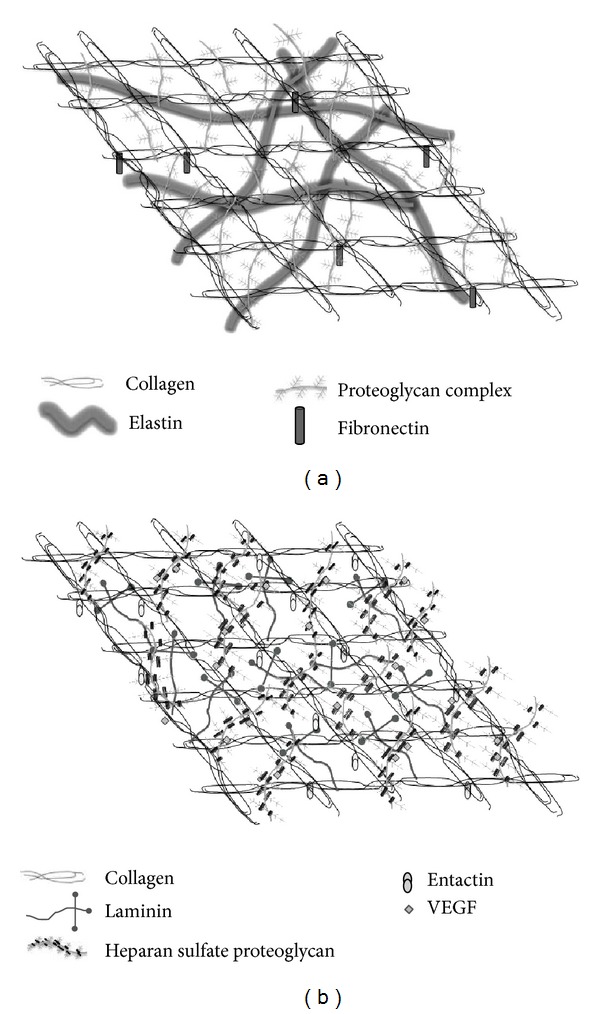
A schematic drawing of the extracellular matrix molecular organization. The interstitial matrix is mainly composed of collagen, fibronectin, elastin, and proteoglycans (a). The extracellular basement membrane mainly consists of collagen IV, laminin, entactin, and heparan sulfate proteoglycans which bind to VEGF (b).
The ECM is a highly dynamic structure, undergoing continuous remodelling, which consists in the deposition, degradation, and modification of its components. An abnormal ECM dynamic leads to pathological processes including tissue fibrosis and cancer.
The three-dimensional (3D) and computational in vitro studies [1, 2] clearly demonstrate that besides its remodelling ECM controls and regulates physiological and pathological angiogenesis [3] at several levels by several ways.
Angiogenesis has been studied by means of several in vitro and in vivo models, including endothelial cell cultures, chick embryo chorioallantoic membrane (CAM) assay [4], and ocular models [5]. Angiogenesis is a multistep process that generally begins when the endothelial cells switch from the “quiescent” to the “angiogenic phenotype” in response to angiogenic stimuli [6, 7] (Figure 2). Subsequently, enzymatic degradation of capillary BM occurs and vascular permeability increases leading to extravasation of blood proteins and their accumulation into interstitial collagen matrix to form a new, provisional ECM. Then, endothelial cells begin to proliferate, invade the ECM, and take part in the formation of an immature capillary structure and deposition of a new complex BM. Finally, pericytes are recruited, thereby providing stabilization for the new vessels. The soluble growth factors, membrane-bound proteins, cell-matrix and cell-cell interactions, and hemodynamic forces all act in concert to control and influence angiogenesis, and the balanced activity between specific angiogenic molecules which can initiate this process and specific inhibitory molecules which can stop it are thought to be critical for an optimal angiogenic response.
Figure 2.
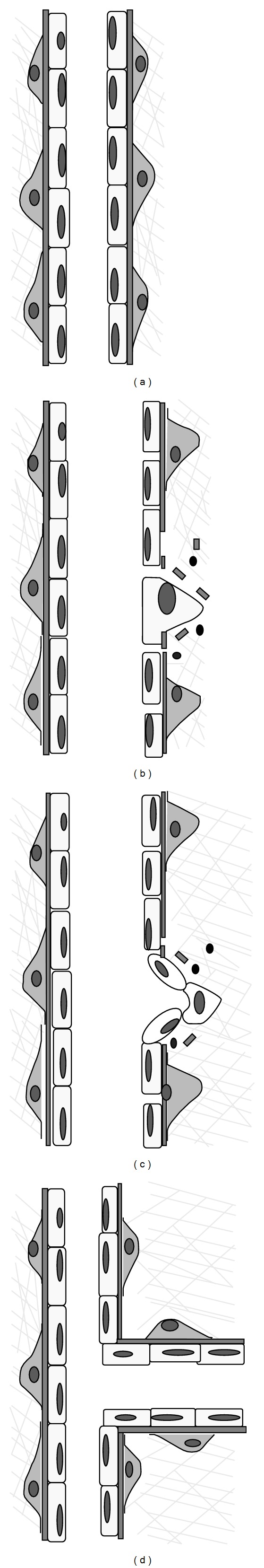
After stimulation with angiogenic factors of a quiescent vessel (a), the degradation of the basement membrane, pericyte detachment, and loosening of endothelial cell junctions occur (b). Endothelial cells begin to proliferate, migrate, and take part in formation of an immature capillary structure and deposition of a new complex basement membrane (c). Finally, pericytes are recruited thereby providing stabilization for the new vessel (d).
Through adhesive interactions with integrins expressed on the endothelial cells surface, the ECM orchestrates complex signalling cascades within the cells and affects many fundamental aspects of their biology, including proliferation, migration, cytoskeletal organization, cell shape, survival, and ultimately blood vessel stabilization. Moreover, matrix molecules or fragments that show pro- and antiangiogenic activity (Table 1) are critical in the onset of angiogenesis and angiogenic cytokines which directly bind matrix and require proteolytic processing to become active [8].
Table 1.
ECM molecules and fragments with proangiogenic and antiangiogenic activity.
| Proangiogenic | Antiangiogenic |
|---|---|
| Intact molecules | Fragments |
| Collagen I | |
| Collagen III | Arresten |
| Collagen IV | Canstatin |
| Collagen XV | Tumstatin |
| Collagen XVIII | Restin |
| Fibrillin | Endostatin |
| Fibulin-1 | Anastellin |
| Fibrin/fibrinogen | Heparin binding fragments |
| Fibronectin | Endorepellin |
| Glypican-1 | Endostatin |
| Laminin-1 | Elastin derived peptides |
| Laminin 8 | Transient molecules |
| Perlecan | Thrombospondin-1 |
| Tenascin C | Thrombospondin-2 |
| Tenascin X | |
| Vitronectin | |
| Decorin | |
| Fragments | |
| Fragment E (fibrin) |
2. ECM Components Involved in Angiogenesis
2.1. ECM Molecules
In vivo studies in knockout mice for BMs genes (fibronectin, laminin, collagen IV, and perlecan) revealed significant cardiovascular dysfunctions [9–11]. In detail, several studies bear out the hypothesis that blood vessels formation and survival are connected with collagen synthesis and deposition in BM [12, 13]. Endothelial cell adhesion to ECM, via integrins-collagen I interaction, leads to activation and/or suppression of multiple signalling pathways. In human dermal microvascular endothelial cells, isolated from neonatal foreskins and anchored to collagen I, as well as in a mouse model of skin angiogenesis involving subdermal injection of Matrigel together with immortalized human cells stably transfected with VEGF165, the interaction of collagen I with α1β1, α2β1, αvβ3, and αvβ5 integrins on cell surfaces induces the activation of MAP kinase pathway which, in turn, supports endothelial cells survival and suppresses apoptosis [14]. In the same in vitro model, other investigators demonstrated that integrins α1β1 and α2β1 binding to collagen I induced suppression of cAMP-dependent PKA, following reorganization of actin fibers and changes in cell shape [15]. In mouse skin model that used VEGF-expressing cells together with packaging cells producing retroviruses encoding RhoA GTPase mutants, the endothelial cells adhesion to collagen I selectively induced activation of Src and Rho and suppression of Rac activity, which, in turn, disrupt intercellular junctions [16]. Interestingly, collagen I also contributes to coalescence of pinocytic intracellular vacuoles, which is an essential step for lumen formation [17, 18].
Type IV collagen, the main protein component of all BMs, has a crucial role in endothelial cell proliferation and cell behaviour [19]. An in vitro study that analysed angiogenic and nonangiogenic culture systems demonstrated the dependence of angiogenesis on secretion and subsequent extracellular deposition of collagen type IV [20].
Laminin of BM appears to be involved prevalently in the regulation of the late stages of angiogenesis: it is responsible for cessation of endothelial cells proliferation and pericytes recruitment and vessels stabilization through Notch signalling activation [21]. By using antibodies directed to laminin receptor, it has been demonstrated that laminin can activate proteinases and contribute to matrix degradation [22].
Fibronectin is a widely distributed glycoprotein and is a component of plasma in a soluble dimeric form and of cell surface and ECM in a dimeric and multimeric form and is localized in ECM underlying endothelial cells. The arginine-glycine-aspartic acid (RGD) motif was the first sequence of fibronectin found to possess cell-adhesive properties [23] and the use of the microfluidic shear devices suggested that endothelial cells adhere to fibronectin stronger than type I collagen [24]. The binding of RGD sequence to the integrins α5β1 and αvβ3 in endothelial cells initiates the polymerization of fibronectin locally synthesized [25], which, in turn, regulates cell growth as demonstrated in cultures of mouse embryonic cells that lack endogenous fibronectin [26], regulates cytoskeletal organization [27], and stabilizes cell-matrix adhesion [28]. Additionally, fibronectin controls endothelial cell survival during angiogenesis in vivo by suppressing the activity of PKA [29]. Plasma fibronectin incorporated in the serum-free collagen gel culture of rat aorta induced a selective dose-dependent elongation of microvessels, without affecting mitotic activity of cells [30].
2.2. ECM Fragments: Matrikines and Matricryptic Sites
In the early stage of angiogenesis, the degradation of ECM occurs in response to angiogenic stimuli. As a consequence, matrix molecules are degraded or partially modified, soluble factors are released, and cryptic sites are exposed.
In 1960, the presence of small peptides derived from degradation of connective tissue glycoproteins was demonstrated for the first time [31]. Maquart et al. [32] introduced the term “matrikines” to designate peptides produced from degradation of ECM components, as result of enzymatic activity of proteinases produced by connective tissue cells. The same term has been then adopted to describe peptides derived by partial proteolysis of ECM surrounding microvessels. Endothelial cells produce proteinases, whose activation is responsible for degradation and liberation of matrix fragments which, in turn, can regulate cellular activity by binding specific cells receptors and by activating intracellular signalling pathways.
The term “matricryptic sites” has been coined to describe the biologically active sites that are not exposed in the mature, secreted form of ECM molecules but which become exposed after structural or conformational alterations [33].
Two classes of proteolytic enzymes appear to be mainly involved in the matrix degradation: plasminogen activator (PA)/plasmin system and matrix metalloproteinases (MMPs). The plasminogen activator/plasmin system is an enzymatic cascade involved in the control of fibrin degradation, matrix turnover, and cell invasion. The physiological activators urokinase-type PA or tissue-type PA mediate the conversion of the inactive plasma zymogen, plasminogen, to the serine protease plasmin. The latter belongs to the large serine proteinase family and can act directly or indirectly, by cleaving numerous ECM proteins, including fibronectin, laminin, thrombospondin, and von Willebrand factor [34], by activating MMPs [35], or by liberating growth factors and cytokines sequestered within the ECM [36, 37].
MMPs belong to the family of zinc endopeptidases and can exist in both membrane-bound (MT-MMPs) and soluble forms. The latter are secreted as inactive proenzymes and their activation occurs in the extracellular compartment. MMPs are produced by a variety of cells, including epithelial cells, fibroblasts, inflammatory cells, and endothelial cells, and their activity is inhibited by the family of tissue inhibitors of metalloproteinase (TIMPs) [38]. At least, five MMPs are involved in angiogenesis: MMP-1, -2, -3, -7, and -9 are upregulated in endothelial cells in a variety of physiological and pathological settings [35].
Many of matrikines and matricryptic sites, resulting from PA/plasmin system and MMPs activity, are important physiologic angiogenesis inhibitors. Among these, endostatin has been extensively studied. It is a proteolytic fragment of the C-terminal noncollagenous domain of collagen XVIII, isolated from the conditioned media of a nonmetastatic murine hemangioendothelioma cell line [39]. Its role as a local inhibitor of angiogenesis has been demonstrated in vitro and in vivo [39, 40]; nevertheless, the evidence that lack of endostatin does not affect angiogenesis in major organs suggests that this fragment is not a critical regulator of angiogenesis [41]. Endostatin acts by inhibiting endothelial cells proliferation and migration [39] and by blocking G1/S phase transition and apoptosis of cells [42, 43]. Immunoblotting analysis of endostatin treated murine brain endothelial cells revealed that endostatin induces activation of phosphatases or other regulatory signalling proteins, which may interfere with FGF effects on endothelial cells; nevertheless, this occurs only if endostatin binds to the endothelial cell surface via heparan-sulfate proteoglycans [44, 45]. Moreover, endostatin inhibits VEGF-induced endothelial cell migration in a dose-dependent manner [46].
Tumstatin, canstatin, and arresten are derived from degradation of α3, α2, and α1 chain, respectively, of type IV collagen. Tumstatin, generated by MMP-9 proteolysis [47], binds to endothelial cells via αvβ 3 integrin [48]. Its antiangiogenic activity is restricted to amino acids 54–132 within the 244 amino acid complete sequence [49]. In vitro studies showed that tumstatin peptide negatively regulates endothelial cells proliferation and induces apoptosis [50] through inhibition of protein synthesis [51]. Canstatin in an antiangiogenic fragment derived from NC1 domain of the α2 chain of type IV collagen [52]. Recombinant canstatin selectively inhibits endothelial cells proliferation and tube formation in a dose-dependent manner [53]. Moreover, in human umbilical vein endothelial cells, canstatin inhibits the phosphorylation of Akt and focal adhesion kinase, induces Fas ligand expression, and activates caspase-dependent apoptotic pathways [54]; all these activities are mediated by interaction with αvβ 3 and αvβ 5 integrins on the surface of endothelial cells [55]. Arresten binds to α1β1 integrin and heparan sulphate proteoglycans and exerts its antiangiogenic effect in endothelial cells via inhibition of MAPK signalling [56, 57]. Arresten inhibits, in a dose-dependent manner, the proliferation of mouse retinal endothelial cells cultured on type IV collagen and stimulated with fibroblast growth factor-2 (FGF-2) [58]. In vivo, arresten significantly inhibits neovascularization in Matrigel plug assays [59].
Tetrastatin, pentastatin, and hexastatin are derived from degradation of type IV collagen, from α4, α5, and α6 chain, respectively. Tetrastatin and pentastatin lack antiangiogenic activity in the CAM assay [48]; nevertheless, in vitro they affect angiogenesis by interacting with human endothelial cells and potently inhibiting their migration [60]. On the other hand, hexastatin administration resulted in inhibition of angiogenesis in the CAM [48] and in Matrigel plug assay [61]. Additionally, through the non-RGD-dependent αvβ3 binding sites, hexastatin significantly inhibits endothelial cell proliferation [61].
Endorepellin is derived from perlecan [62] and by interacting with the α2β1 integrin receptor triggers a signalling cascade that leads to disruption of the endothelial actin cytoskeleton [63]. Endorepellin affects angiogenesis by interacting with the VEGF receptor through its laminin G-like, leading to the downstream of VEGF signalling [64, 65].
2.3. Angiogenic Factors
The role of VEGF as major inducer of angiogenesis is well recognized [66]. VEGF induces expression of α1β1 and α2β1 integrins in microvascular endothelial cells [67], endothelial cell migration, and proliferation [68, 69]. VEGF is not stored intracellularly but it bounds the cell surface or ECM and various MMPs [70] and PA [71] can generate diffusible, non-heparin-binding fragments. Two classes of VEGF binding sites have been identified on fibronectin: one constitutively available and the other whose availability is modulated by the conformational state of fibronectin, which, in turn, depends on heparin interaction [72, 73].
FGF-2 stimulates survival, proliferation, migration, and differentiation of endothelial cells both in vitro and in vivo and binds with high affinity to heparan sulfate proteoglycans located on the surface of most cells and within the ECM [74]. Heparan sulfate proteoglycans, particularly perlecan, modulate the binding of FGF-2 to its specific tyrosine kinase receptors [75] and protect the growth factor from proteolytic degradation by extracellular proteinases [76].
The proangiogenic activity of transforming growth factor beta 1 (TGF-β1) has been demonstrated in new-born mice [77] and in the CAM assay [78] and it has been confirmed in TGF-β1 knockout mice [79]. However, in vitro studies suggested that activity of TGF-β1 is strictly dependent on composition and organization of the ECM: TGF-β1 treatment elicits the formation of calcium and magnesium dependent tube-like structures, mimicking angiogenesis, in three-dimensional cultures [80], whereas it induces endothelial cells apoptosis in two-dimensional cultures [81]. TGF-β1 upregulates the VEGF expression and the interaction with its receptor in endothelial cells and this mechanism induces endothelial cell apoptosis and angiogenesis in vitro and in vivo [82, 83].
3. ECM as a Link between Angiogenesis and Inflammation
Angiogenesis and inflammation are distinct processes that can occur independently of each other, although in some cases they are codependent. Angiogenesis can stimulate and intensify the inflammatory response by providing nutrients and oxygen in inflammatory sites, and some angiogenic factors exert proinflammatory activity. Conversely, in chronic inflammation, inflammatory cells produce cytokines and growth factors that may affect endothelial cell functions.
Angiogenesis and inflammation are associated with nuclear factor kappa-B (NF-κB) and angiopoietin- (Ang-) Tie2 signalling pathways. NF-κB is an inducible transcription factor, whose activation regulates the expression of genes for proinflammatory cytokines, chemokines, and enzymes that generate mediators of inflammation. There is also evidence that this transcription factor is involved in the regulation of migration, proliferation and survival of endothelial cells [84], and expression of MMPs [85]. Ang-1 is secreted and incorporated into and sequestered by the ECM. By binding Tie-2 receptor expressed in endothelial cells, it does not affect the cells proliferation; however, it stimulates endothelial cells migration, sprouting, and survival and promotes recruitment of the pericytes and smooth muscle cells [86]. Mice lacking of Ang-1 and Tie-2 showed defective remodelling and maturation of the vasculature, whereas the transgenic mice overexpressing Ang-1 displayed increased vascularization suggesting a role in the formation of blood vessels during development [87]. Additionally, Ang-1 has been shown to inhibit vascular permeability and exert anti-inflammatory effects [88]. It has been demonstrated that Ang-1 can act also in nonendothelial cells, including monocytes via p38 and Erk1/2 phosphorylation and macrophages inducing their switch toward a proinflammatory phenotype [89]. Unlike Ang-1, its antagonist, Ang-2, is not incorporated into ECM and disrupts blood vessel formation in the mouse embryo [90]. Stored in endothelial Weibel-Palade bodies, Ang-2 is rapidly released in response to exogenous stimuli and promotes inflammation [91].
Angiogenesis and inflammation share ECM remodelling which, in turn, directly and indirectly influences both processes. ECM molecules and fragments, resulting from proteolysis, can act directly as inflammatory stimuli; they can influence immune cell activation and survival and proper tissue repair as well as each step of angiogenesis, as previously described.
Inflammatory cells produce a large amount of MMPs, which activate cytokines and chemokines through cleavage, regulate inflammatory cells response, and notably contribute to ECM degradation. Analysis of RNA levels expression in mononuclear cells isolated from venous blood of normal volunteers revealed that they highly express several forms of MMPs [92]. Expression of MMPs can be selectively induced by binding of bacteria to toll-like receptor 2 in monocytes and CD14, toll-like receptor 2, and toll-like receptor 1 in macrophages and is strictly dependent on the stage of cellular differentiation [93, 94].
The role of inflammation in tumor development is not less negligible: an inflammatory microenvironment can increase mutation rates and enhance the proliferation of cancer cells, contributing to tumor initiation, and innate and adaptive immune system cells can infiltrate the tumor sites, release growth and survival factors, and facilitate angiogenesis, tumor growth, invasion, and metastasis by themselves or by inducing other effector molecules. The increase of several immune cell-derived factors, such as interleukin-1 (IL-1), IL-6, IL-8, IL-10, and tumor necrosis factor alpha (TNF-α), has been described in tumor microenvironment: they sustain inflammatory process, contribute to tumor progression, and also exert proangiogenic activity [95, 96].
Osteopontin (OPN), a multifunctional ECM phosphoprotein-containing and RGD integrin binding domain, is limited to the bone, kidney, and epithelial linings and is secreted in body fluids including milk, blood, and urine in normal conditions, whereas it is upregulated at sites of inflammation and tissue remodelling. Chakraborty et al. [97] provided both in vitro and in vivo experimental evidences that OPN regulates Brk/NF-κB/ATF-4 signalling cascades which, in turn, upregulates the VEGF expression and tumor angiogenesis through autocrine and paracrine mechanisms in breast cancer system.
Inflammatory cells release MMPs involved in the release of angiogenic factors, such as VEGF and FGF-2, and cryptic antiangiogenic factors. VEGF derived from matrix stores, as a result of MMP-9, is implicated in the angiogenic switch and tumor growth [98].
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation of synovial joints and destruction of cartilage and bone, as well as by systemic extra-articular inflammation. The synovium that normally is relatively acellular and with scattered blood vessels, results hyperplastic, rich of inflammatory cells and high vascularized in RA. The new vessels besides provide nutrients and oxygen, also sustain the inflammation by being a source of cytokines, chemokines, and proteases [99], facilitate the ingress of inflammatory cells into the synovium, and, therefore, stimulate pannus formation. At the same time, infiltrated inflammatory cells stimulate angiogenic process, by upregulating proangiogenic factors [100, 101] and activating NF-κB. Upregulation of Angs-Tie 2 [102] and VEGF [103] detected in chronic inflamed synovium and serum of RA patients emphasizes the interdependence of inflammation and angiogenesis in RA. VEGF may act as a proinflammatory mediator and as an angiogenic stimulator in RA joints: it induces the production of chemokines by endothelial cells, such as MCP-1 and IL-8 [104], which, in turn, recruit monocytes in synovial membranes. Several clinical studies demonstrated high levels of MMPs, directly involved in ECM degradation, in the systemic circulation and synovial fluid of patients with RA and their correlation with clinical activity [105, 106].
Osteoarthritis (OA) is a disease characterized by degeneration of cartilage and its underlying bone within a joint as well as bony overgrowth. Even if OA has been commonly described as “noninflammatory” disorder, in order to distinguish it from inflammatory arthritis, evidence is now accumulating that synovitis occurs and exacerbates structural damage [107, 108]. The inflammation contributes directly to angiogenesis, observed in the synovium of osteoarthritic joints [107], and these two processes contribute to pain and damage. Hypoxia, a common feature of the inflamed synovial environment, and IL-1 stimulate VEGF expression in synovial fibroblasts [109]. Also TNF-α, which promotes angiogenesis in vivo [110] and regulates the expression of MMP-9 and MMP-1 [111], is involved in OA. Thus, angiogenesis may exacerbate inflammation in OA, by facilitating inflammatory cell infiltration [111].
Psoriasis is a chronic inflammatory disease of skin and small joints characterized by excessive growth of the epidermal keratinocytes, inflammatory cell accumulation, and excessive dermal angiogenesis. Psoriasis is characterized by the overproduction of interferon gamma (INF-γ), TNF-α, and IL-17 [112]. TNF-α induced upregulation of IL-24 and activation of signal transducer and activator of transcription 3 (STAT3) signalling in mice keratinocytes [113]. IL-9 contributes to the development of psoriatic lesions, by inducing Th17-related inflammation and by promoting angiogenesis [114]. Keratinocytes in the psoriatic skin lesions are a source of proangiogenic cytokines, such as VEGF, whose levels have been shown to be dramatically elevated in human psoriatic skin [115] and correlate with increased levels of inflammatory cytokines and MMPs [116] and with degree of psoriasis severity [117]. Additionally, keratinocytes isolated from psoriatic skin show a strongly reduced expression of thrombospondin-1, an endogenous inhibitor of angiogenesis. Enzyme immunoassay [118] and immunohistochemistry analysis [119] revealed that keratinocytes in psoriasis express and produce MMP-1 and MMP-19, which are decreased by anti-TNF-alpha [120].
Ocular angiogenesis is an important cause for severe loss of vision in several disorders, such as age-related macular degeneration, diabetic retinopathy, retinal artery or vein occlusion, and retinopathy of prematurity. Even if the mechanism that leads to abnormal growth of new vessels in eyes remains to be elucidated, it is related to inflammation. VEGF, as well as IL-6, IL-8, and IL-10, monocyte chemoattractant protein-1 levels are higher in patients with ocular diseases, when compared with normal subjects, and significantly decreased after administration of bevacizumab, a humanized anti-VEGF monoclonal IgG1 antibody [121, 122]. VEGF intravitreal injection induced upregulation of intercellular adhesion molecule in endothelial cells and, consequently, the local adhesion of leucocytes and vascular permeability [123]. Injection of FGF into the vitreous cavity did not show the same effect of FGF mobilized by degradation of the matrix by proteolytic enzymes [123]. Conversely, macrophage depletion in mice reduced the choroidal neovascularization and was associated with decreased macrophage infiltration and VEGF protein [124].
4. Concluding Remarks
Overall, the literature data analyzed in this review demonstrate that, in addition to providing basic support for cells, ECM is also a critical component that allows the intercellular crosstalk and stores several important mediators which, in turn, regulate several cellular and connective components functions, including angiogenesis.
The progressive increase of knowledge concerning the cellular and extracellular mechanisms that regulate angiogenesis in normal and pathological conditions may provide the potential basis for the development of new therapeutic approaches against abnormal angiogenesis that contributes to the pathogenesis of several disorders.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Fang C, Man YG, Cuttitta F, et al. Novel phenotypic fluorescent three-dimensional co-culture platforms for recapitulating tumor in vivo progression and for personalized therapy. Journal of Cancer. 2013;4(9):755–763. doi: 10.7150/jca.7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edgar LT, Sibole SC, Underwood CJ, Guilkey JE, Weiss JA. A computational model of in vitro angiogenesis based on extracellular matrix fibre orientation. Computer Methods in Biomechanics and Biomedical Engineering. 2013;16(7):790–801. doi: 10.1080/10255842.2012.662678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folkman J. Tumor angiogenesis: therapeutic implications. New England Journal of Medicine. 1971;285(21):1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 4.Ribatti D. Chicken chorioallantoic membrane angiogenesis model. Methods in Molecular Biology. 2012;843:47–57. doi: 10.1007/978-1-61779-523-7_5. [DOI] [PubMed] [Google Scholar]
- 5.Montezuma SR, Vavvas D, Miller JW. Review of the ocular angiogenesis animal models. Seminars in Ophthalmology. 2009;24(2):52–61. doi: 10.1080/08820530902800017. [DOI] [PubMed] [Google Scholar]
- 6.Ribatti D, Nico B, Crivellato E. Morphological and molecular aspects of physiological vascular morphogenesis. Angiogenesis. 2009;12(1):101–111. doi: 10.1007/s10456-008-9125-1. [DOI] [PubMed] [Google Scholar]
- 7.Bastaki M, Nelli EE, Dell'Era P, et al. Basic fibroblast growth factor-induced angiogenic phenotype in mouse endothelium. A study of aortic and microvascular endothelial cell lines. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(3):454–464. doi: 10.1161/01.atv.17.3.454. [DOI] [PubMed] [Google Scholar]
- 8.van Hinsbergh VWM, Koolwijk P. Endothelial sprouting and angiogenesis: matrix metalloproteinases in the lead. Cardiovascular Research. 2008;78(2):203–212. doi: 10.1093/cvr/cvm102. [DOI] [PubMed] [Google Scholar]
- 9.Thyboll J, Kortesmaa J, Cao R, et al. Deletion of the laminin α4 chain leads to impaired microvessel maturation. Molecular and Cellular Biology. 2002;22(4):1194–1202. doi: 10.1128/MCB.22.4.1194-1202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Astrof S, Crowley D, Hynes RO. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Developmental Biology. 2007;311(1):11–24. doi: 10.1016/j.ydbio.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pöschl E, Schlötzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131(7):1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 12.Maragoudakis ME, Missirlis E, Karakiulakis GD, Sarmonica M, Bastakis M, Tsopanoglou N. Basement membrane biosynthesis as a target for developing inhibitors of angiogenesis with anti-tumor properties. Kidney International. 1993;43(1):147–150. doi: 10.1038/ki.1993.24. [DOI] [PubMed] [Google Scholar]
- 13.Sweeney SM, DiLullo G, Slater SJ, et al. Angiogenesis in collagen I requires α2β1 ligation of a GFP*GER sequence and possibly p38 MAPK activation and focal adhesion disassembly. Journal of Biological Chemistry. 2003;278(33):30516–30524. doi: 10.1074/jbc.M304237200. [DOI] [PubMed] [Google Scholar]
- 14.Perruzzi CA, de Fougerolles AR, Koteliansky VE, Whelan MC, Westlin WF, Senger DR. Functional overlap and cooperativity among αv and β1 integrin subfamilies during skin angiogenesis. Journal of Investigative Dermatology. 2003;120(6):1100–1109. doi: 10.1046/j.1523-1747.2003.12236.x. [DOI] [PubMed] [Google Scholar]
- 15.Whelan MC, Senger DR. Collagen I initiates endothelial cell morphogenesis by inducing actin polymerization through suppression of cyclic AMP and protein kinase A. Journal of Biological Chemistry. 2003;278(1):327–334. doi: 10.1074/jbc.M207554200. [DOI] [PubMed] [Google Scholar]
- 16.Hoang MV, Whelan MC, Senger DR. Rho activity critically and selectively regulates endothelial cell organization during angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):1874–1879. doi: 10.1073/pnas.0308525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamei M, Brian Saunders W, Bayless KJ, Dye L, Davis GE, Weinstein BM. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature. 2006;442(7101):453–456. doi: 10.1038/nature04923. [DOI] [PubMed] [Google Scholar]
- 18.Stratman AN, Saunders WB, Sacharidou A, et al. Endothelial cell lumen and vascular guidance tunnel formation requires MT1-MMP-dependent proteolysis in 3-dimensional collagen matrices. Blood. 2009;114(2):237–247. doi: 10.1182/blood-2008-12-196451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madri JA. Extracellular matrix modulation of vascular cell behaviour. Transplant Immunology. 1997;5(3):179–183. doi: 10.1016/s0966-3274(97)80035-4. [DOI] [PubMed] [Google Scholar]
- 20.Bahramsoltani M, Slosarek I, de Spiegelaere W, Plendl J. Angiogenesis and collagen type IV expression in different endothelial cell culture systems. Anatomia, Histologia, Embryologia. 2014;43(2):103–115. doi: 10.1111/ahe.12052. [DOI] [PubMed] [Google Scholar]
- 21.Estrach S, Cailleteau L, Franco CA, et al. Laminin-binding integrins induce Dll4 expression and notch signaling in endothelial cells. Circulation Research. 2011;109(2):172–182. doi: 10.1161/CIRCRESAHA.111.240622. [DOI] [PubMed] [Google Scholar]
- 22.Khusal R, ca Costa Dias B, Moodley K, et al. In vitro inhibition of angiogenesis by antibodies directed against the 37kDa/67kDa laminin receptor. PLoS ONE. 2013;8(3) doi: 10.1371/journal.pone.0058888.e58888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309(5963):30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- 24.Young EWK, Wheeler AR, Simmons CA. Matrix-dependent adhesion of vascular and valvular endothelial cells in microfluidic channels. Lab on a Chip. 2007;7(12):1759–1766. doi: 10.1039/b712486d. [DOI] [PubMed] [Google Scholar]
- 25.Bourdoulous S, Orend G, MacKenna DA, Pasqualini R, Ruoslahti E. Fibronectin matrix regulates activation of RHO and CDC42 GTPases and cell cycle progression. Journal of Cell Biology. 1998;143(1):267–276. doi: 10.1083/jcb.143.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Molecular Biology of the Cell. 2002;13(10):3546–3559. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sottile J, Hocking DC, Swiatek PJ. Fibronectin matrix assembly enhances adhesion-dependent cell growth. Journal of Cell Science. 1998;111(19):2933–2943. doi: 10.1242/jcs.111.19.2933. [DOI] [PubMed] [Google Scholar]
- 28.Hocking DC, Sottile J, Langenbach KJ. Stimulation of integrin-mediated cell contractility by fibronectin. Journal of Biological Chemistry. 2000;275(14):10673–10682. doi: 10.1074/jbc.275.14.10673. [DOI] [PubMed] [Google Scholar]
- 29.Kim S, Bakre M, Yin H, Varner JA. Inhibition of endothelial cell survival and angiogenesis by protein kinase A. Journal of Clinical Investigation. 2002;110(7):933–941. doi: 10.1172/JCI14268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicosia RF, Bonanno E, Smith M. Fibronectin promotes the elongation of microvessels during angiogenesis in vitro. Journal of Cellular Physiology. 1993;154(3):654–661. doi: 10.1002/jcp.1041540325. [DOI] [PubMed] [Google Scholar]
- 31.Robert L, Robert B. Structural glycoproteins, their metabolism in normal and pathological connective tissue. Bibliotheca Nutritio et Dieta. 1969;13:123–130. doi: 10.1159/000385158. [DOI] [PubMed] [Google Scholar]
- 32.Maquart FX, Siméon A, Pasco S, Monboisse JC. Regulation of cell activity by the extracellular matrix: the concept of matrikines. Journal de la Societe de Biologie. 1999;193(4-5):423–428. [PubMed] [Google Scholar]
- 33.Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. American Journal of Pathology. 2000;156(5):1489–1498. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thrombosis Research. 2000;98(4):323–332. doi: 10.1016/s0049-3848(99)00242-x. [DOI] [PubMed] [Google Scholar]
- 35.Davis GE, Pintar Allen KA, Salazar R, Maxwell SA. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. Journal of Cell Science. 2001;114(5):917–930. doi: 10.1242/jcs.114.5.917. [DOI] [PubMed] [Google Scholar]
- 36.Saksela O, Moscatelli D, Sommer A, Rifkin DB. Endothelial cell-derived heparan sulfate binds basic fibroblast growth factor and protects it from proteolytic degradation. Journal of Cell Biology. 1988;107(2):743–751. doi: 10.1083/jcb.107.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.George SJ, Johnson JL, Smith MA, Jackson CL. Plasmin-mediated fibroblast growth factor-2 mobilisation supports smooth muscle cell proliferation in human saphenous vein. Journal of Vascular Research. 2001;38(5):492–501. doi: 10.1159/000051082. [DOI] [PubMed] [Google Scholar]
- 38.Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. European Journal of Cell Biology. 1997;74(2):111–122. [PubMed] [Google Scholar]
- 39.O'Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 40.Marneros AG, Olsen BR. The role of collagen-derived proteolytic fragments in angiogenesis. Matrix Biology. 2001;20(5-6):337–345. doi: 10.1016/s0945-053x(01)00151-2. [DOI] [PubMed] [Google Scholar]
- 41.Fukai N, Eklund L, Marneros AG, et al. Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO Journal. 2002;21(7):1535–1544. doi: 10.1093/emboj/21.7.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dhanabal M, Ramchandran R, Waterman MJF, et al. Endostatin induces endothelial cell apoptosis. Journal of Biological Chemistry. 1999;274(17):11721–11726. doi: 10.1074/jbc.274.17.11721. [DOI] [PubMed] [Google Scholar]
- 43.Dhanabal M, Volk R, Ramchandran R, Simons M, Sukhatme VP. Cloning, expression, and in vitro activity of human endostatin. Biochemical and Biophysical Research Communications. 1999;258(2):345–352. doi: 10.1006/bbrc.1999.0595. [DOI] [PubMed] [Google Scholar]
- 44.Dixelius J, Larsson H, Sasaki T, et al. Endostatin-induced tyrosine kinase signaling through the Shb adaptor protein regulates endothelial cell apoptosis. Blood. 2000;95(11):3403–3411. [PubMed] [Google Scholar]
- 45.Sasaki T, Larsson H, Kreuger J, et al. Structural basis and potential role of heparin/heparan sulfate binding to the angiogenesis inhibitor endostatin. EMBO Journal. 1999;18(22):6240–6248. doi: 10.1093/emboj/18.22.6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamaguchi N, Anand-Apte B, Lee M, et al. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO Journal. 1999;18(16):4414–4423. doi: 10.1093/emboj/18.16.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamano Y, Zeisberg M, Sugimoto H, et al. Physiological levels of tumstatin, a fragment of collagen IV α3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via αVβ3 integrin. Cancer Cell. 2003;3(6):589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petitclerc E, Boutaud A, Prestayko A, et al. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. Journal of Biological Chemistry. 2000;275(11):8051–8061. doi: 10.1074/jbc.275.11.8051. [DOI] [PubMed] [Google Scholar]
- 49.Maeshima Y, Manfredi M, Reimerli C, et al. Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. Journal of Biological Chemistry. 2001;276(18):15240–15248. doi: 10.1074/jbc.M007764200. [DOI] [PubMed] [Google Scholar]
- 50.Zhang G-M, Zhang Y-M, Fu S-B, et al. Effects of cloned tumstatin-related and angiogenesis-inhibitory peptides on proliferation and apoptosis of endothelial cells. Chinese Medical Journal. 2008;121(22):2324–2330. [PubMed] [Google Scholar]
- 51.Maeshima Y, Sudhakar A, Lively JC, et al. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002;295(5552):140–143. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- 52.Kamphaus GD, Colorado PC, Panka DJ, et al. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. Journal of Biological Chemistry. 2000;275(2):1209–1215. doi: 10.1074/jbc.275.2.1209. [DOI] [PubMed] [Google Scholar]
- 53.He G-A, Luo J-X, Zhang T-Y, Wang F-Y, Li R-F. Canstatin-N fragment inhibits in vitro endothelial cell proliferation and suppresses in vivo tumor growth. Biochemical and Biophysical Research Communications. 2003;312(3):801–805. doi: 10.1016/j.bbrc.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 54.Panka DJ, Mier JW. Canstatin inhibits Akt activation and induces Fas-dependent apoptosis in endothelial cells. Journal of Biological Chemistry. 2003;278(39):37632–37636. doi: 10.1074/jbc.M307339200. [DOI] [PubMed] [Google Scholar]
- 55.Magnon C, Galaup A, Mullan B, et al. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with alphavbeta3 and alphavbeta5 integrins. Cancer Research. 2005;65(10):4353–4361. doi: 10.1158/0008-5472.CAN-04-3536. [DOI] [PubMed] [Google Scholar]
- 56.Sudhakar A, Nyberg P, Keshamouni VG, et al. Human α1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by α1β1 integrin. Journal of Clinical Investigation. 2005;115(10):2801–2810. doi: 10.1172/JCI24813. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Colorado PC, Torre A, Kamphaus G, et al. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Research. 2000;60(9):2520–2526. [PubMed] [Google Scholar]
- 58.Boosani CS, Nalabothula N, Sheibani N, Sudhakar A. Inhibitory effects of arresten on bFGF-induced proliferation, migration, and matrix metalloproteinase-2 activation in mouse retinal endothelial cells impact of arresten on mouse retinal Endothelial Cells. Current Eye Research. 2010;35(1):45–55. doi: 10.3109/02713680903374208. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Nyberg P, Xie L, Sugimoto H, et al. Characterization of the anti-angiogenic properties of arresten, an α1β1 integrin-dependent collagen-derived tumor suppressor. Experimental Cell Research. 2008;314(18):3292–3305. doi: 10.1016/j.yexcr.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karagiannis ED, Popel AS. Identification of novel short peptides derived from the α4, α5, and α6 fibrils of type IV collagen with anti-angiogenic properties. Biochemical and Biophysical Research Communications. 2007;354(2):434–439. doi: 10.1016/j.bbrc.2006.12.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mundel TM, Yliniemi A-M, Maeshima Y, Sugimoto H, Kieran M, Kalluri R. Type IV collagen α6 chain-derived noncollagenous domain 1 (α6(IV)NC1) inhibits angiogenesis and tumor growth. International Journal of Cancer. 2008;122(8):1738–1744. doi: 10.1002/ijc.23269. [DOI] [PubMed] [Google Scholar]
- 62.Mongiat M, Sweeney SM, San Antonio JD, Fu J, Iozzo RV. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. Journal of Biological Chemistry. 2003;278(6):4238–4249. doi: 10.1074/jbc.M210445200. [DOI] [PubMed] [Google Scholar]
- 63.Bix G, Fu J, Gonzalez EM, et al. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through α2β1 integrin. Journal of Cell Biology. 2004;166(1):97–109. doi: 10.1083/jcb.200401150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goyal A, Poluzzi C, Willis CD, Smythies J, Shellard A, Neill T. Endorepellin affects angiogenesis by antagonizing diverse vascular endothelial growth factor receptor 2 (VEGFR2)-evoked signaling pathways: transcriptional repression of hypoxia-inducible factor 1a and VEGFA and concurrent inhibition of nuclear factor of activated T cell 1 (NFAT1) activation. Journal of Biological Chemistry. 2012;287(52):43543–43556. doi: 10.1074/jbc.M112.401786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Willis CD, Poluzzi C, Mongiat M, Iozzo RV. Endorepellin laminin-like globular 1/2 domains bind Ig3-5 of vascular endothelial growth factor (VEGF) receptor 2 and block pro-angiogenic signaling by VEGFA in endothelial cells. FEBS Journal. 2013;280(10):2271–2284. doi: 10.1111/febs.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ribatti D, Vacca A, Presta M. The discovery of angiogenic factors: a historical review. General Pharmacology: Vascular System. 2000;35(5):227–231. doi: 10.1016/s0306-3623(01)00112-4. [DOI] [PubMed] [Google Scholar]
- 67.Senger DR, Claffey KP, Benes JE, Perruzzi CA, Sergiou AP, Detmar M. Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(25):13612–13617. doi: 10.1073/pnas.94.25.13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Senger DR, Ledbetter SR, Claffey KP, et al. Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the alphavbeta3 integrin, osteopontin, and thrombin. American Journal of Pathology. 1996;149(1):293–305. [PMC free article] [PubMed] [Google Scholar]
- 69.Wang S, Li X, Parra M, Verdin E, Bassel-Duby R, Olson EN. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(22):7738–7743. doi: 10.1073/pnas.0802857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee S, Jilan SM, Nikolova GV, Carpizo D, Luisa Iruela-Arispe M. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. Journal of Cell Biology. 2005;169(4):681–691. doi: 10.1083/jcb.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keyt BA, Berleau LT, Nguyen HV. The carboxyl-terminal domain (111–165) of vascular endothelial growth factor is critical for its mitogenic potency. Journal of Biological Chemistry. 1996;271(13):7788–7795. doi: 10.1074/jbc.271.13.7788. [DOI] [PubMed] [Google Scholar]
- 72.Mitsi M, Forsten-Williams K, Gopalakrishnan M, Nugent MA. A catalytic role of heparin within the extracellular matrix. Journal of Biological Chemistry. 2008;283(50):34796–34807. doi: 10.1074/jbc.M806692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mitsi M, Hong Z, Costello CE, Nugent MA. Heparin-mediated conformational changes in fibronectin expose vascular endothelial growth factor binding sites. Biochemistry. 2006;45(34):10319–10328. doi: 10.1021/bi060974p. [DOI] [PubMed] [Google Scholar]
- 74.Ishai-Michaeli R, Svahn CM, Weber M, et al. Importance of size and sulfation of heparin in release of basic fibroblast growth factor from the vascular endothelium and extracellular matrix. Biochemistry. 1992;31(7):2080–2088. doi: 10.1021/bi00122a027. [DOI] [PubMed] [Google Scholar]
- 75.Aviezer D, Levy E, Safran M, et al. Differential structural requirements of heparin and heparan sulfate proteoglycans that promote binding of basic fibroblast growth factor to its receptor. Journal of Biological Chemistry. 1994;269(1):114–121. [PubMed] [Google Scholar]
- 76.Saksela O, Rifkin DB. Release of basic fibroblast growth factor-heparan sulfate complexes from endothelial cells by plasminogen activator-mediated proteolytic activity. Journal of Cell Biology. 1990;110(3):767–775. doi: 10.1083/jcb.110.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roberts AB, Sporn MB, Assoian RK. Transforming growth factor type β: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(12):4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang EY, Moses HL. Transforming growth factor β1-induced changes in cell migration, proliferation, and angiogenesis in the chicken chorioallantoic membrane. Journal of Cell Biology. 1990;111(2):731–741. doi: 10.1083/jcb.111.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-β1 knock out mice. Development. 1995;121(6):1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 80.Merwin JR, Anderson JM, Kocher O, van Itallie CM, Madri JA. Transforming growth factor beta1 modulates extracellular matrix organization and cell-cell junctional complex formation during in vitro angiogenesis. Journal of Cellular Physiology. 1990;142(1):117–128. doi: 10.1002/jcp.1041420115. [DOI] [PubMed] [Google Scholar]
- 81.Frater-Schroder M, Risau W, Hallmann R. Tumor necrosis factor type α, a potent inhibitor of endothelial cell growth in vitro, is angiogenic in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(15):5277–5281. doi: 10.1073/pnas.84.15.5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P. Transforming growth factor-beta 1 (TGF-β1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. Journal of Cellular Physiology. 2009;219(2):449–458. doi: 10.1002/jcp.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ferrari G, Pintucci G, Seghezzi G, Hyman K, Galloway AC, Mignatti P. VEGF, a prosurvival factor, acts in concert with TGF-β1 to induce endothelial cell apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(46):17260–17265. doi: 10.1073/pnas.0605556103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kisseleva T, Song L, Vorontchikhina M, Feirt N, Kitajewski J, Schindler C. NF-κB regulation of endothelial cell function during LPS-induced toxemia and cancer. Journal of Clinical Investigation. 2006;116(11):2955–2963. doi: 10.1172/JCI27392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ko H-M, Kang J-H, Choi J-H, Park SJ, Bai S, Im S-Y. Platelet-activating factor induces matrix metalloproteinase-9 expression through Ca2+- or PI3K-dependent signaling pathway in a human vascular endothelial cell line. FEBS Letters. 2005;579(28):6451–6458. doi: 10.1016/j.febslet.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 86.Xu Y, Yu Q. Angiopoietin-1, unlike angiopoietin-2, is incorporated into the extracellular matrix via its linker peptide region. Journal of Biological Chemistry. 2001;276(37):34990–34998. doi: 10.1074/jbc.M103661200. [DOI] [PubMed] [Google Scholar]
- 87.Suri C, Jones PF, Patan S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87(7):1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 88.Pizurki L, Zhou Z, Glynos K, Roussos C, Papapetropoulos A. Angiopoietin-1 inhibits endothelial permeability, neutrophil adherence and IL-8 production. British Journal of Pharmacology. 2003;139(2):329–336. doi: 10.1038/sj.bjp.0705259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seok SH, Heo JI, Hwang JH, et al. Angiopoietin-1 elicits pro-inflammatory responses in monocytes and differentiating macrophages. Molecules and Cells. 2013;35(6):550–556. doi: 10.1007/s10059-013-0088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277(5322):55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 91.Fiedler U, Reiss Y, Scharpfenecker M, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-α and has a crucial role in the induction of inflammation. Nature Medicine. 2006;12(2):235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 92.Bar-Or A, Nuttall RK, Duddy M, et al. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain. 2003;126(12):2738–2749. doi: 10.1093/brain/awg285. [DOI] [PubMed] [Google Scholar]
- 93.Gebbia JA, Coleman JL, Benach JL. Selective induction of matrix metalloproteinases by Borrelia burgdorferi via toll-like receptor 2 in monocytes. Journal of Infectious Diseases. 2004;189(1):113–119. doi: 10.1086/380414. [DOI] [PubMed] [Google Scholar]
- 94.Elass E, Aubry L, Masson M, et al. Mycobacterial lipomannan induces matrix metalloproteinase-9 expression in human macrophagic cells through a toll-like receptor 1 (TLR1)/TLR2- and CD14-dependent mechanism. Infection and Immunity. 2005;73(10):7064–7068. doi: 10.1128/IAI.73.10.7064-7068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kai H, Kitadai Y, Kodama M, et al. Involvement of proinflammatory cytokines IL-1β and IL-6 in progression of human gastric carcinoma. Anticancer Research. 2005;25(2):709–713. [PubMed] [Google Scholar]
- 96.Mohamed MM, El-Ghonaimy EA, Nouh MA, Schneider RJ, Sloane BF, El-Shinawi M. Cytokines secreted by macrophages isolated from tumor microenvironment of inflammatory breast cancer patients possess chemotactic properties. International Journal of Biochemistry and Cell Biology. 2014;46:138–147. doi: 10.1016/j.biocel.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chakraborty G, Jain S, Kundu GC. Osteopontin promotes vascular endothelial growth factor-dependent breast tumor growth and angiogenesis via autocrine and paracrine mechanisms. Cancer Research. 2008;68(1):152–161. doi: 10.1158/0008-5472.CAN-07-2126. [DOI] [PubMed] [Google Scholar]
- 98.Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biology. 2000;2(10):737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Storgard CM, Stupack DG, Jonczyk A, Goodman SL, Fox RI, Cheresh DA. Decreased angiogenesis and arthritic disease in rabbits treated with an αvβ3 antagonist. Journal of Clinical Investigation. 1999;103(1):47–54. doi: 10.1172/JCI3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nakahara H, Song J, Sugimoto M, et al. Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis & Rheumatism. 2003;48(6):1521–1529. doi: 10.1002/art.11143. [DOI] [PubMed] [Google Scholar]
- 101.Woods JM, Mogollon A, Amin MA, Martinez RJ, Koch AE. The role of COX-2 in angiogenesis and rheumatoid arthritis. Experimental and Molecular Pathology. 2003;74(3):282–290. doi: 10.1016/s0014-4800(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 102.Scott BB, Zaratin PF, Colombo A, Hansbury MJ, Winkler JD, Jackson JR. Constitutive expression of angiopoietin-1 and -2 and modulation of their expression by inflammatory cytokines in rheumatoid arthritis synovial fibroblasts. Journal of Rheumatology. 2002;29(2):230–239. [PubMed] [Google Scholar]
- 103.Ballara S, Taylor PC, Reusch P, et al. Raised serum vascular endothelial growth factor levels are associated with destructive change in inflammatory arthritis. Arthritis & Rheumatology. 2001;44(9):2055–2064. doi: 10.1002/1529-0131(200109)44:9<2055::AID-ART355>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 104.Marumo T, Schini-Kerth VB, Busse R. Vascular endothelial growth factor activates nuclear factor-κB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes. 1999;48(5):1131–1137. doi: 10.2337/diabetes.48.5.1131. [DOI] [PubMed] [Google Scholar]
- 105.Tchetverikov I, Ronday HK, Van El B, et al. MMP profile in paired serum and synovial fluid samples of patients with rheumatoid arthritis. Annals of the Rheumatic Diseases. 2004;63(7):881–883. doi: 10.1136/ard.2003.013243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Keyszer G, Lambiri I, Nagel R, et al. Circulating levels of matrix metalloproteinases MMP-3 and MMP-1, tissue inhibitor of metalloproteinases 1 (TIMP-1), and MMP-1/TIMP-1 complex in rheumatic disease. Correlation with clinical activity of rheumatoid arthritis versus other surrogate markers. Journal of Rheumatology. 1999;26(2):251–258. [PubMed] [Google Scholar]
- 107.Haywood L, McWilliams DF, Pearson CI, et al. Inflammation and angiogenesis in osteoarthritis. Arthritis & Rheumatism. 2003;48(8):2173–2177. doi: 10.1002/art.11094. [DOI] [PubMed] [Google Scholar]
- 108.Henrotin Y, Lambert C, Richette P. Importance of synovitis in osteoarthritis: evidence for the use of glycosaminoglycans against synovial inflammation. Seminars in Arthritis and Rheumatism. 2014;43(5):579–587. doi: 10.1016/j.semarthrit.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 109.Jackson JR, Minton JA, Ho ML, Wei N, Winkler JD. Expression of vascular endothelial growth factor in synovial fibroblasts is induced by hypoxia and interleukin 1beta. Journal of Rheumatology. 1997;24:1253–1259. [PubMed] [Google Scholar]
- 110.Frater-Schroder M, Muller G, Birchmeier W, Bohlen P. Transforming growth factor-beta inhibits endothelial cell proliferation. Biochemical and Biophysical Research Communications. 1986;137(1):295–302. doi: 10.1016/0006-291x(86)91209-x. [DOI] [PubMed] [Google Scholar]
- 111.Lehmann W, Edgar CM, Wang K, et al. Tumor necrosis factor alpha (TNF-α) coordinately regulates the expression of specific matrix metalloproteinases (MMPS) and angiogenic factors during fracture healing. Bone. 2005;36(2):300–310. doi: 10.1016/j.bone.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 112.Koch AE, Burrows JC, Haines GK, Carlos TM, Harlan JM, Leibovich SJ. Immunolocalization of endothelial and leukocyte adhesion molecules in human rheumatoid and osteoarthritic synovial tissues. Laboratory Investigation. 1991;64(3):313–320. [PubMed] [Google Scholar]
- 113.Nickoloff BJ, Mitra RS, Varani J, Dixit VM, Polverini PJ. Aberrant production of interleukin-8 and thrombospondin-1 by psoriatic keratinocytes mediates angiogenesis. American Journal of Pathology. 1994;144(4):820–828. [PMC free article] [PubMed] [Google Scholar]
- 114.Kumari S, Bonnet MC, Ulvmar MH, et al. Tumor necrosis factor receptor signaling in keratinocytes triggers interleukin-24-dependent psoriasis-like skin inflammation in mice. Immunity. 2013;39(5):899–911. doi: 10.1016/j.immuni.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 115.Singh TP, Schön MP, Wallbrecht K, Gruber-Wackernagel A, Wang XJ, Wolf P. Involvement of IL-9 in Th17-associated inflammation and angiogenesis of psoriasis. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0051752.e51752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bhushan M, McLaughlin B, Weiss JB, Griffiths CEM. Levels of endothelial cell stimulating angiogenesis factor and vascular endothelial growth factor are elevated in psoriasis. British Journal of Dermatology. 1999;141(6):1054–1060. doi: 10.1046/j.1365-2133.1999.03205.x. [DOI] [PubMed] [Google Scholar]
- 117.Simonetti O, Lucarini G, Goteri G, et al. VEGF is likely a key factor in the link between inflammation and angiogenesis in psoriasis: results of an immunohistochemical study. International Journal of Immunopathology and Pharmacology. 2006;19(4):751–760. doi: 10.1177/039463200601900405. [DOI] [PubMed] [Google Scholar]
- 118.Nofal A, Al-Makhzangy I, Attwa E, Nassar A, Abdalmoati A. Vascular endothelial growth factor in psoriasis: an indicator of disease severity and control. Journal of the European Academy of Dermatology and Venereology. 2009;23(7):803–806. doi: 10.1111/j.1468-3083.2009.03181.x. [DOI] [PubMed] [Google Scholar]
- 119.Flisiak I, Myśliwiec H, Chodynicka B. Effect of psoriasis treatment on plasma concentrations of metalloproteinase-1 and tissue inhibitor of metalloproteinases-1. Journal of the European Academy of Dermatology and Venereology. 2005;19(4):418–421. doi: 10.1111/j.1468-3083.2005.01199.x. [DOI] [PubMed] [Google Scholar]
- 120.Suomela S, Kariniemi A-L, Impola U, et al. Matrix metalloproteinase-19 is expressed by keratinocytes in psoriasis. Acta Dermato-Venereologica. 2003;83(2):108–114. doi: 10.1080/00015550310007445. [DOI] [PubMed] [Google Scholar]
- 121.Cordiali-Fei P, Trento E, D’Agosto G, et al. Decreased levels of metalloproteinase-9 and angiogenic factors in skin lesions of patients with psoriatic arthritis after therapy with anti-TNF-α . Journal of Autoimmune Diseases. 2006;3, article 5 doi: 10.1186/1740-2557-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Funk M, Kriechbaum K, Prager F, et al. Intraocular concentrations of growth factors and cytokines in retinal vein occlusion and the effect of therapy with bevacizumab. Investigative Ophthalmology and Visual Science. 2009;50(3):1025–1032. doi: 10.1167/iovs.08-2510. [DOI] [PubMed] [Google Scholar]
- 123.Funk M, Schmidinger G, Maar N, et al. Angiogenic and inflammatory markers in the intraocular fluid of eyes with diabetic macular edema and influence of therapy with bevacizumab. Retina. 2010;30(9):1412–1419. doi: 10.1097/IAE.0b013e3181e095c0. [DOI] [PubMed] [Google Scholar]
- 124.Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. Journal of Experimental Medicine. 2003;198(3):483–489. doi: 10.1084/jem.20022027. [DOI] [PMC free article] [PubMed] [Google Scholar]