

Abstract
Plaque instability associated with acute coronary syndromes results in part from apoptosis and senescence of cells within the atherosclerotic (AS) lesion. Increased cellular oxidative stress has been proposed to contribute to plaque progression and changes in composition, leading to plaque instability. Our objective was to examine the role of NADPH oxidase in smooth muscle cell (SMC) phenotypes associated with an unstable plaque. Aortae were isolated from pre-lesion (8 weeks of age) and post-lesion (35 weeks of age) hypercholesterolemic mice (ApoE-/-/LDLR-/-, AS), and age-matched normal C57BL/6J mice. We observed an age-dependent increase in reactive oxygen species (ROS) in aorta from AS mice, with evidence for elevated ROS prior to lesion development. Whereas macrophage infiltration was restricted to the lesion, oxidized lipids extended beyond the plaque and into the vessel wall. Consistent with these findings, we observed dynamic changes in the expression of NADPH oxidases in AS vessels. Specifically, Nox1 expression was increased early and decreased with lesion progression, while induction of Nox4 was a late event. Nox2 and p22phox were elevated throughout lesion development. Similar to observations in aortae, SMCs isolated from the lesion of AS aortae had decreased Nox1 and increased Nox4 levels as compared to SMCs from normal mice. AS SMCs demonstrated increased generation of ROS, cell cycle arrest, evidence of senescence, and increased susceptibility to apoptosis. Overexpression of Nox4 in normal SMCs recapitulated the phenotypes of the AS SMCs. We conclude that increased expression of Nox4 in AS may drive SMC phenotypes that lead to the plaque instability and rupture responsible for myocardial infarction and stroke.
Keywords: Atherosclerosis, Smooth muscle, Reactive oxygen species, NADPH oxidase, Oxidative stress
Highlights
-
•
Increases in ROS and Nox1 expression precede atherosclerotic lesion development.
-
•
Development of atherosclerotic plaque is associated with expression of Nox4.
-
•
SMCs derived from atherosclerotic vessels have elevated Nox4 expression and ROS.
-
•
Nox4 contributes to the senescent phenotype and apoptosis in plaque SMCs.
-
•
Increased Nox4 may play a role in plaque rupture and myocardial infarction.
Graphical abstract
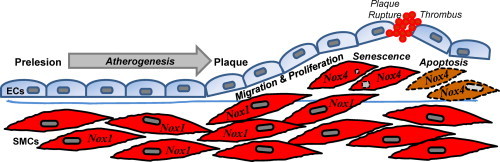
Introduction
Plaque rupture is the primary inciting event in the acute onset of myocardial infarction and stroke. Plaque instability is a multifactorial process that includes increased macrophage content and redox-dependent activation of matrix-degrading enzymes that lead to thinning of the fibrous cap. As a primary source of the supportive extracellular matrix, loss of smooth muscle cells (SMCs) also contributes to plaque instability and rupture. A better understanding of the complex mechanisms that promote plaque rupture may lead to preventative strategies.
Vascular oxidative stress is a mechanistic link between macrophage infiltration, matrix metalloproteinase activation and SMC apoptosis in plaque instability. Although there are multiple potential enzymatic sources of reactive oxygen species (ROS) in vascular cells, including xanthine oxidase, lipoxygenase, nitric oxide synthases, and mitochondrial oxidases [1,2], NADPH oxidases are the primary source of intracellular ROS in atherosclerosis [3]. Studies using mice deficient in catalytic subunits Nox1 or Nox2 or the cytosolic subunit p47phox revealed roles for NADPH oxidase in atherogenesis [[4], [5], [6], [7]]. However, Nox homolog-specific changes in gene expression occur at different times in disease development. In atherosclerotic human coronary arteries, Nox4 expression progressively increases from stage I to stage IV disease [8]. Nox1 expression is increased early after wire injury, whereas Nox4 is increased late [9]. The absence of good mouse models has limited new mechanistic insights into plaque rupture.
SMCs contained within the lesion are phenotypically distinct from medial contractile SMCs [10,11]. In this study, our objective was to examine the contribution of NADPH oxidase in plaque-derived SMCs. Our data identify a relationship between Nox4 expression and lesion progression, ROS levels, apoptosis, and cell cycle arrest. These results implicate Nox4 in senescence and apoptosis of plaque-derived SMCs, suggesting an integral role for Nox4 in plaque stability.
Methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the University of Iowa Animal Care and Use Committee (ACURF Protocol number: 0208216).
Animals
Studies were performed in normal (C57BL/6J) and hypercholesterolemic (ApoE and LDL receptor deficient) mice fed a normal chow diet. Double homozygous ApoE and LDL-receptor deficient mice are >10 generation hybrids (C57BL/6J background) originally obtained from Jackson Laboratory. Mice were studied at 8 weeks (prelesion) and 24–35 weeks (established lesions) of age.
Cell culture
Vascular smooth muscle cells were isolated from the media of normal (C57Bl/6J) aorta or atherosclerotic intima of ApoE/LDL receptor-deficient aorta with minor modification of methods previously described [10,12]. Multiple aorta from mice 24–35 weeks of age were pooled for isolation of SMCs. In brief, animals were euthanized with pentobarbital (150 mg/kg i.p.) and aortae were removed under sterile conditions, cleaned of adventitia, opened longitudinally, and incubated in DMEM supplemented with fungizone (2.5 µg/mL), penicillin G (10,000 U/dL), and streptomycin (10,000 U/dL). After 4 h, the tissue was transferred to phosphate buffered saline (PBS) containing type 1 collagenase (2 mg/mL) at 37 °C for 20 min. The control aorta segment was then scraped to remove remaining endothelium. In atherosclerotic aorta, the intima was microdissected from the medial layer. Tissue was cut into 1-mm strips and incubated in collagenase (2 mg/mL), elastase (0.5 mg/mL), and bovine serum albumin (1 mg/mL) for 40 min. Cell suspensions were centrifuged, the pellet resuspended in DMEM containing 10% fetal bovine serum (FBS), and cells plated on collagen-treated surfaces. After 24 h, medium was removed, and cells were rinsed with DMEM to remove debris. Growth media consisting of DMEM containing 10% FBS was changed twice weekly and cells were used for experiments at 90–100% confluence. All studies were performed on cells from the fourth to eighth passages. For each passage, cells were identified as SMCs by positive immunohistochemical staining for smooth muscle a-actin. Studies were performed in four different isolations of AS and normal SMCs. Human coronary artery SMCs (HCASMCs) were purchased from ATCC and cultured in Vascular Cell Basal Media (ATCC) supplemented with Vascular Smooth Muscle Growth Kit components (ATCC).
Real-time quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from SMC using TRI reagent (Molecular Research Center, Inc.) and treated with DNase I (Ambion) to remove contaminating DNA. First-strand cDNA synthesis was performed using total RNA, random hexamer, dNTP, and superscript II reverse transcriptase (Invitrogen). Relative levels of p22phox, Nox1, Nox2 and Nox4 mRNA were quantified with real-time, quantitative PCR using the QuantiTect SYBR green I PCR kit (Qiagen) in a PerkinElmer 7000 real-time thermocycler as previously described [13]. Primer sequences are as follows: Nox1 forward 5'-CATCCAGTCTCCAAACATGACAG-3', reverse 5'-TGCAACTCCCCTTATGGTCATC-3'; Nox2 forward 5'- CAGGAACCTCACTTTCCATAAGATG-3'; reverse 5'-AACGTTGAAGAGATGTGCAATTGT-3'; Nox4 forward 5'-CTGGTCTGACGGGTGTCTGCATGGTG-3', reverse 5'-CTCCGCACAATAAAGGCACAAAGGTCCAG-3'; p22phox forward 5'-AGATCGAGTGGGCCATGTGGGCCAACGAAC-3', reverse 5'-CTTGGGTTTAGGCTCAATGGGAGTCCACTGCTCA-3'. Data were normalized to 18S rRNA (Alternate 18S Internal Standards, Ambion).
ROS measurements
Superoxide levels in vessels
Aortic segments (3 mm) were obtained from the aortic arch, rinsed in cold PBS, flash frozen in OCT, sectioned (30 µm), and incubated in dihydroethidium (DHE, 10 µM, Molecular Probes) for 30 min at 37 °C as previously described [12]. DHE fluorescence in aortic sections was detected with a Zeiss LSM 510 META confocal microscope (excitation, 514 nm; emission, 575 nm long pass filter). Where indicated, sections were preincubated with polyethylene glycolated superoxide dismutase (PEG-SOD, 500 units/mL) prior to DHE.
Intracellular superoxide and H2O2 in cells
Intracellular superoxide and H2O2 levels were detected in SMCs with dihydroethidium (DHE) or 5-(and-6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA), respectively as described previously [13]. Briefly, cells were treated with DHE (10 µM) or CM-H2DCFDA (20 µM) for 30 min at 37 °C. DHE fluorescence intensity was visualized with Zeiss LSM 510 META laser confocal microscope and quantitated with NIH ImageJ software. In each experiment, 7–8 randomly picked fields per sample (29–54 cells in each field) were visualized and captured. 2',7'-dichlorofluorescein intensity was determined on a Becton Dickinson FACScan (BD Biosciences) and analyzed with the FlowJo software package (Tree Star, Inc.). Data were normalized to cell number. Because both DHE and dichlorofluorescein fluorescence is known to have limitations regarding specificity for detection of superoxide and peroxides, respectively [14], we performed the additional measurements of ROS.
Extracellular H2O2 in cells
Cells were incubated with Amplex Red (50 µM) and HRP (1 U/mL) for 1 h, after which the fluorescence intensity of the media was determined (excitation and emission wavelengths of 545 and 590 nm, respectively) and normalized to cell number as previously described [15].
NADPH-stimulated superoxide
Membrane-enriched fractions were isolated as previously described [16]. Briefly, cells were lysed by sonication in ice-cold protease inhibitor buffer, centrifuged at 500g for 10 min at 4 °C to remove cell debris, and then the supernatant centrifuged at 100,000g for 60 min at 4 °C. The pellet was suspended in protease inhibitor buffer and protein concentration measured. NADPH oxidase activity of 20 µg protein was measured by lucigenin-enhanced chemiluminescence (5 × 10-6 M) after addition of NADPH (10-4 M). Duplicate samples were also incubated with catalase (250 units/mL) and the catalase-inhibitable chemiluminescence was measured in an FB12 luminometer (Zylux Corp) and RLU/sec normalized to protein. Where indicated, cells were treated with 0.1 mM diphenyleneiodonium (DPI) prior to lysis.
Immunohistochemistry
Oxidized phospholipids were assessed in aortic sections using IgM E06 (1:400 dilution) followed by appropriate secondary antibody and avidin-biotin-AP complex as previously described [17]. E06 binds the phosphocholine head group of oxidized but not native phospholipids. For macrophage immunostaining, sections were blocked with serum and incubated overnight at 4 °C with mouse macrophage antigen (MOMA)-2 (Accurate Chemicals, 1:100), then with biotinylated immunoglobulin followed by avidin–biotin–AP complex and visualized with Vector Red alkaline phosphatase substrate (Vector Laboratories). No staining was seen when serial sections were treated with nonimmune sera instead of primary antibody.
Measurement of antioxidant activities
Superoxide dismutase (SOD) activities were measured as described previously [18]. One unit of SOD activity was defined as the amount of protein that yields 50% of maximal inhibition of nitroblue tetrazolium reduction by superoxide. Catalase activity was determined by monitoring the disappearance of H2O2 and expressed in ? units/mg protein [19].
ß-Galactosidase
ß-Galactosidase activity was measured in extracts from SMCs cultured under normal growth conditions using the Galacto-Light Plus Chemiluminescence reporter assay kit (#BL100P, Applied Biosystems) per the manufacturer's instructions. Data are expressed as relative light units (RLU) per min and normalized to protein concentration.
Cell growth
SMCs at passage 4 were plated at equivalent densities (104 cells/dish) in 6-well plate and cultured in standard growth medium with 10% FBS for 6 days. Cell counts were performed 6 h after plating (day 0) to ensure equivalent efficiency for both SMC. At intervals after plating, cells were trypsinized, and cell numbers were determined using a Coulter Counter. Cell proliferation was also examined as described previously by measuring incorporation of [3H]-leucine [10]. Briefly, cells were serum-starved in 0.5% FBS for 72 h, and then were either maintained in 0.1% FBS or cultured in standard growth medium with 10% FBS supplemented with [3H]-leucine for 24 h. Data are expressed at counts per minute (CPM).
Cell cycle analysis
Normally cycling cells were incubated with 10 µM 5-bromo-2'-deoxyuridine (BrdU) for 30 min, trypsinized, and fixed 70% ice-cold ethanol. Fixed cells were incubated in 0.2 mg/mL pepsin prepared in 2N HCl for 30 min, and then neutralized with 0.3 M borax. Cells were next incubated in anti-BrdU primary antibody (BD Biosciences) followed by FITC-conjugated secondary antibody. Cells were then treated with 1 mg/mL RNase A for 30 min then incubated with 17.5 µg/mL propidium iodide for 60 min. Cells were then analyzed on a FACScan (Becton Dickinson) using the CELLQUEST flow cytometric analysis software (BD Biosciences) and ModFit (Verity) was used to analyze cell cycle alterations.
Western blot analysis
Cells were lysed in lysis buffer containing 62.5 mM Tris, pH 6.8, 2% SDS, 10% glycerol, and 50 mM diethylenetriaminepentaacetic acid (DTT). After sonication, protein concentration was determined and 50 µg protein was subjected to SDS-PAGE, transferred to nitrocellulose membranes, blocked with 5% milk, and incubated with primary antibodies (1:1000 anti-cyclin D1: #2926 Cell Signaling; 1:500 mouse monoclonal anti-retinoblastoma protein: BD Biosciences; 1:10,000 mouse monoclonal anti-ß-actin: Sigma) and HRP-conjugated secondary antibodies. Bands were visualized using SuperSignal West Pico Chemiluminescent Substrate from Pierce.
TUNEL staining
Mice were euthanized by intraperitoneal (IP) injection of pentobarbital (50 mg/kg) and aorta removed and dissected free of loosely-adhering tissue. A section of the thoracic aorta was snap-frozen aorta and sectioned (8 µm thick). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed on aortic sections using the Apop-Tag In Situ Apoptosis Detection kit (Millipore) according to the manufacturer's instructions.
Apoptosis
Apoptosis was assessed using the Vybrant Apoptosis Assay kit (V-13242, Molecular Probes) per manufacturer’s instructions. Briefly, cells were centrifuged and resuspended in annexin-binding buffer (140 mM NaCl, 2.5 mM CaCl2, 10 mM HEPES, pH 7.4), stained with FITC-conjugated annexin V and propidium iodide, and incubated for 15 min at room temperature. Cells were then analyzed on a FACScan (Becton Dickinson) using the CELLQUEST flow cytometric analysis software (BD Biosciences). Cells in the lower right dot plot quadrant (PS-positive and PI-negative) were reported as apoptotic, whereas cells in the upper right dot plot quadrant (PS-positive and PI-positive) were scored as necrotic. For normal (C57Bl6/J) primary aortic SMCs infected with Ad-Nox4 and treated with H2O2, 48 h after infection cells were serum-starved in the presence or absence of 50 µM H2O2 for 24 h prior to assessment of apoptosis as described. All other apoptosis measurements were obtained using cells cultured in serum-containing normal growth media.
Cell viability
Cells were treated with increasing concentrations of H2O2 for 24 h in serum-free media, and then cell viability determined using the CellTiter 96 AQueous One Solution 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) Assay (Promega) per the manufacturer's instructions.
Nox4 knockdown
Nox4 siRNAs were generated from gene specific double-stranded RNA (dsRNA) and transfected into AS SMC using Dicer siRNA Generation Kit (Gene Therapy Systems) according to the manufacturer's instructions. In brief, a plasmid containing murine Nox4 cDNA (kindly provided by Dr. Kathy Griendling, Emory University) was used as template for PCR. Primer sequences: forward primer: 5'- GCGTAATACGACTCACTATAGGGAGAATGCATTTGGCTGTCCCTAAACG-3'; reverse primer: 5'-GCGTAATACGACTCACTATAGGGAGATAAAACAA ACCACCTGAAACATGC-3'. The PCR product was transcribed to dsRNA then digested with recombinant Dicer enzyme to generate siRNA duplexes. After purification and quantification, plaque-derived SMC were transfected with 200 nM siRNA using GeneSilencer siRNA transfection reagent. GFP siRNA served as negative control.
Nox4 overexpression
Senescence experiments
To generate pCMV-Nox4, full-length human Nox4 cDNA (kindly provided by J. David Lambeth, Emory University) was cloned into pCMV-Script after excising Nox4 cDNA from pcDNA3 by restriction digestion with KpnI and NotI, blunting, and insertion into pCMVscript by blunt end ligation. HCASMCs were transfected with either pCMV-Script empty vector or pCMV-Script containing Nox4 cDNA using Lipofectamine (Invitrogen). Experiments were performed 48 h after transfection.
Apoptosis experiments
The University of Iowa Gene Transfer Vector Core directionally cloned Nox4 cDNA from pCMV-Nox4 into the replication deficient recombinant adenoviral vector containing CMV, Ad5CMV, to generate Ad-Nox4. The vector was sequenced for confirmation. All adenoviruses were generated by the University of Iowa Gene Transfer Vector Core. Normal (C57Bl6/J) primary aortic SMCs were infected with Ad-Nox4 or Ad-GFP at multiplicity of infection (MOI) of 300 in serum-free media in the presence of cationic polymer poly-l-lysine (250 molecules/virus particle) as previously described [20]. After 6 h, fresh 10% FBS-containing medium was added, and experiments were conducted 48 h after infection.
Statistical analysis
Results are expressed as mean ± SEM. Statistical comparisons were performed by one-way or two-way analysis of variance (ANOVA) with appropriate post-hoc analysis. For analysis of Nox mRNA levels in vessels, the one sample t-test was used. For cell viability assays in the presence of H2O2, analysis was performed with two-way repeated measures ANOVA with Bonferroni post-test. A p value of <0.05 was considered significant.
Results
Increased superoxide production precedes lesion formation in atherosclerotic aortae
Using the ApoE-/-/LDLR-/- mice as a model of atherogenesis, we first evaluated the time-dependent increase in superoxide levels in aorta of AS mice as compared to normal C57BL/6J mice. As previously reported, superoxide levels were increased in aorta after the development of atherosclerotic plaques (Fig. 1A, AS 35 weeks). Interestingly, the increase in superoxide levels preceded development of lesions (AS 8 weeks). This early increase in cellular superoxide appeared primarily along the endothelial layer and within the adventitia. Pre-incubation of AS 35 weeks aortic sections with antioxidant superoxide dismutase (PEG-SOD) normalized superoxide levels to those observed in C57BL/6J mice (Fig. 1A). Thus, an early indicator in atherogenesis is an increase in superoxide levels, which precede the development of atherosclerotic lesions.
Fig. 1.
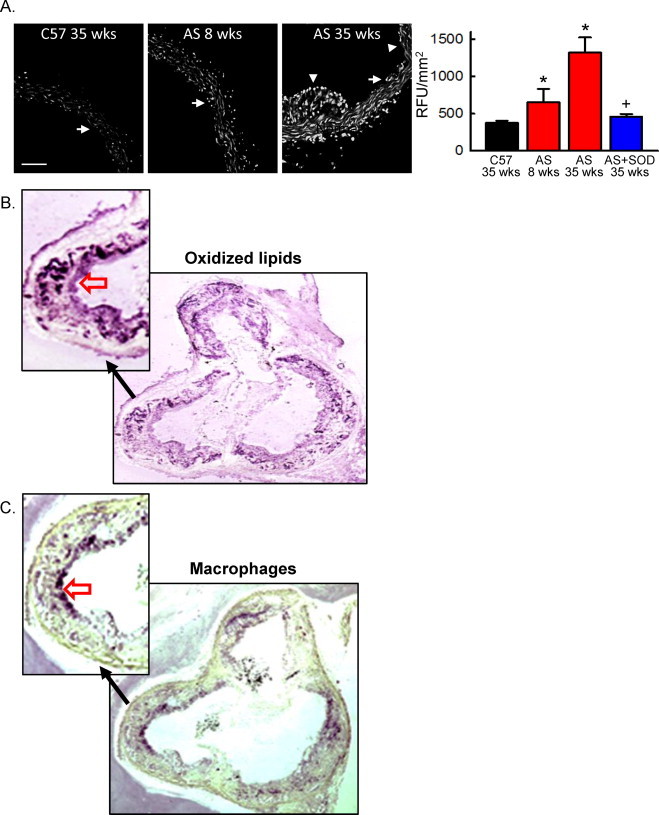
Increased aortic superoxide levels precede lesion development in hypercholesterolemic mice. (A) Confocal fluorescent images of DHE-stained aortic cross-sections from normal (C57) or hypercholesterolemic ApoE-/-LDLR-/- (AS) mice. White arrows denote the endothelial layer; arrowhead denotes plaque. Scale bar = 100 µm. Right panel, summary data of DHE fluorescence in the absence or presence of PEG-SOD (SOD). Data were normalized to vessel area. RFU, relative fluorescent units. n = 4, *p < 0.05 vs. C57 at 35 weeks; +p< 0.05 vs. AS at 35 weeks. (B and C) Oxidized phospholipids (B) and macrophage infiltration (C) was examined by immunohistochemistry in the aortic sinus of AS mice at 35 weeks of age. The open red arrows indicate staining of oxidized lipids (B) and macrophages (C). The no primary controls showed no staining (not shown).
In the aortic sinus, the distribution of oxidized phospholipids extended into the vessel wall (Fig. 1B), whereas macrophage infiltration was in general restricted to the plaque (Fig. 1C). This observation suggests that the increased superoxide is not limited to macrophages but is derived from enzymatic sources within the vessel wall.
Variable NADPH oxidase expression with atherogenesis
NADPH oxidases are the primary source of superoxide in the vessel wall. We next determined the relationship between NADPH oxidase expression and development of atherosclerosis. In the AS aorta prior to lesion development (AS 8 weeks), mRNA levels of p22phox, Nox1 and Nox2 but not Nox4 were increased as compared to aorta from normal C57BL/6J mice (Fig. 2A). By 24 weeks of age, Nox1 levels were no longer increased, whereas Nox2 levels remained elevated (Fig. 2B). In contrast to the pre-lesion, Nox4 levels were also increased by 24 weeks. The dynamic changes in Nox1 and Nox4 expression suggest distinct functional contributions of these subunits in atherosclerosis.
Fig. 2.
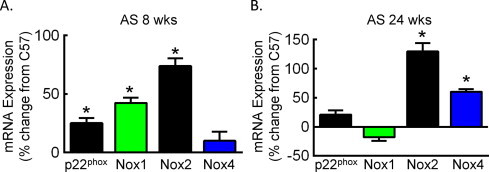
NADPH oxidase subunits are differentially expressed in pre-lesion and post-lesion aorta. Relative mRNA expression as determine by qRT-PCR in aorta from AS mice at 8 (A) and 24 (B) weeks of age. Data were normalized to 18S RNA and quantitated as the percent change in expression in aorta from normal age-matched C57 mice (n = 4). *p < 0.05 vs. C57.
Cultured AS SMCs have increased NADPH oxidase activity
The primary cellular component of the vascular wall is SMCs. In response to activation, SMCs migrate to the subendothelial space and proliferate, contributing to lesion formation. Within the atherosclerotic lesion, SMCs provide extracellular matrix deposition and plaque stability. Based on our observations of dynamic changes in Nox1 and Nox4 in the aorta during atherogenesis, we next focused our studies on SMCs, which express both of these subunits. SMCs were isolated and cultured from aorta of AS mice (>24 weeks of age) or from C57Bl6/J (normal) aorta. Consistent with our observations in AS aorta (24 weeks), Nox1 levels in SMCs derived from AS aorta were ˜1.5-fold lower whereas levels of both Nox4 and p22phox were 3-fold higher as compared to normal SMCs (Fig. 3A). NADPH oxidase activity, as measured by NADPH-stimulated superoxide, was greater in AS SMCs as compared to normal SMCs. Superoxide generation was prevented by the flavoenzyme inhibitor diphenyleneiodonium (DPI, Fig. 3B). We next measured ROS levels and observed significantly higher intracellular superoxide and H2O2 levels as well as extracellular H2O2 in AS SMCs relative to normal SMCs (Fig. 3C–E). Knockdown of Nox4 in AS SMCs decreased intracellular ROS levels by 40% (siNox4: 168+/-16; siGFP: 265+/-17 RFU/cell normalized to normal SMCs, p < 0.05). Antioxidant activity was also elevated in AS SMCs, with a 3.67 ± 0.07-fold increase in Cu2+/Zn+ SOD, a 2.15 ± 0.15-fold increase in Mn2+ SOD, and a 2.87 ± 0.14-fold increase in catalase (n = 4; * p < 0.05 vs. normal SMCs), in accord with increased antioxidative enzyme expression in human atherosclerotic lesions [21]. These data are consistent with the hypothesis that increased cellular ROS in AS SMCs results from increased NADPH oxidase activity, in particular Nox4.
Fig. 3.
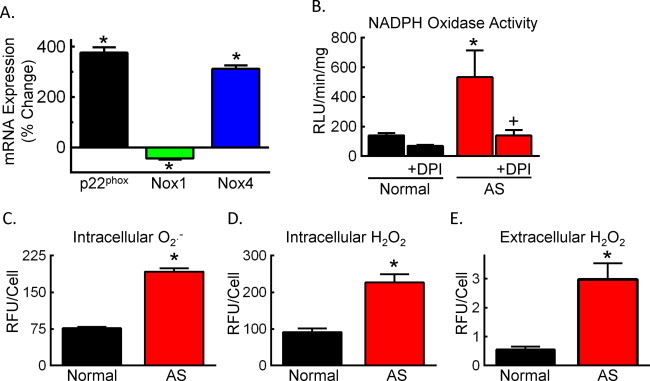
Isolated AS SMCs have increased NADPH oxidase expression and activity. (A) Percent change in mRNA expression in AS SMCs relative normal C57 SMCs. (B) NADPH oxidase activity in normal C57 (normal) or AS SMCs as indicated by the NADPH-stimulated lucigenin-enhanced chemiluminescence in the absence or presence of 100 µM DPI. Data were normalized to protein concentration. RLU, relative light units. (C and D) Intracellular superoxide (O2-•, C) and H2O2 (D) levels as determined by flow cytometry after DHE or DCFH staining, respectively. (E) Extracellular H2O2 levels as determined by Amplex Red. n = 4–10 Experiments. *p < 0.05 vs. normal SMCs; +p< 0.05 vs. untreated AS SMCs.
Nox4 mediates a senescent phenotype in AS SMCs
Redox-dependent activation of SMCs by NADPH oxidases promotes cell growth [3]. Therefore, we compared growth in normal and AS SMCs and found that, over 6 days of culture in complete media, the number of AS SMCs was dramatically lower than normal SMCs (Fig. 4A). Confirming a decrease in DNA synthesis, [3H]-thymidine incorporation was impaired in AS SMCs as compared to normal SMCs (Fig. 4B). H2O2 has been shown to induce replicative senescence as demonstrated by an increase in senescence-associated ß-galactosidase activity and cell cycle arrest [22,23]. Analysis of ß-galactosidase activity revealed a 1.9-fold increase in AS SMCs relative to normal SMCs (574+/-39 vs. 306+/-11 RLU/min/mg, respectively, n = 4, p < 0.01). In addition, we observed an increased percentage of AS SMCs in G0/G1 (Fig. 4C and D), consistent with previous reports that atherosclerotic plaque-derived SMCs have a senescent phenotype [11]. The AS SMC arrest in G0/G1 corresponded with decreases in cyclin D1and retinoblastoma protein (Rb, Fig. 4E). To evaluate the contribution of Nox4 to the senescent phenotype in AS SMCs, we attempted to restore cell cycle progression by knocking down Nox4. Despite a 40% reduction in intracellular H2O2 levels, the knockdown of Nox4 failed to release cells from G0/G1 arrest (data not shown). This observation is consistent with senescence representing terminal differentiation, and the loss of proliferative potential is irreversible [24]. Therefore, we next determined the potential for Nox4 to induce a senescent-like phenotype by overexpressing Nox4 in normal SMCs. We confirmed that overexpression of Nox4 increased cellular H2O2 by greater than 2-fold (Fig. 4F). Nox4 overexpression induced an increase in the percentage of cells in G0/G1 and a decrease in S phase (Fig. 4G), indicative of cell cycle arrest in G0/G1. These data provide evidence for a role for Nox4 in the senescent phenotype in AS SMCs.
Fig. 4.
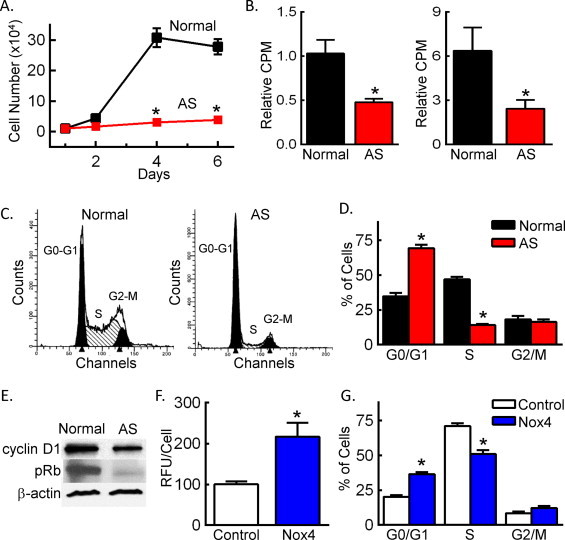
Overexpression of Nox4 in normal SMCs mimics the senescent phenotype observed in AS SMCs. (A) Cell growth in 10% FBS as determined by cell number. *p < 0.05 vs. normal SMCs. (B) Cell growth in 0.1% (left panel) or 10% FBS (right panel) as determined by [3H]-leucine uptake. Data are reported as counts per minute (CPM). *p < 0.05 vs. normal SMCs. (C) Representative cell cycle profiles. (D) Quantitation of cell cycle distribution. *p < 0.05 vs. normal SMCs in same cell cycle phase. (E) Expression of G0/G1 cell cycle markers by Western blotting. (F) H2O2 levels as determined by DCFH staining and flow cytometry in HCASMCs transfected with either control plasmid (control) or plasmid containing Nox4 cDNA (Nox4). *p < 0.05 vs. control. (G) Quantitation of cell cycle distribution in HCASMCs transfected with control or Nox4 plasmid. *p < 0.05 vs. control in same cell cycle phase. n = 3–5 Independent experiments.
Nox4 contributes to increased susceptibility of AS SMCs to cell death
Apoptosis is increased in multiple cell types in atherosclerotic lesions [25]. In AS vessels, we observed increased TUNEL staining within the lesion relative to the adjacent medial layer (Fig. 5A). In isolated SMCs cultured in full serum, more than 10% of AS SMCs were apoptotic (Fig. 5B). We next examined the effect of oxidative stress on cell viability and found that, with increasing concentrations of exogenous H2O2, AS SMCs had reduced viability as compared to normal SMCs (Fig. 5C). Addition of the flavoenzyme inhibitor DPI improved AS SMCs viability in the presence of H2O2 (Fig. 5D). Furthermore, overexpression of Nox4 in normal SMCs increased apoptosis under normal growth conditions (Fig. 5E) and in response to H2O2 (Fig. 5F). These data suggest that the observed increased in Nox4 expression in AS SMCs contributes to the increased susceptibility to apoptosis.
Fig. 5.
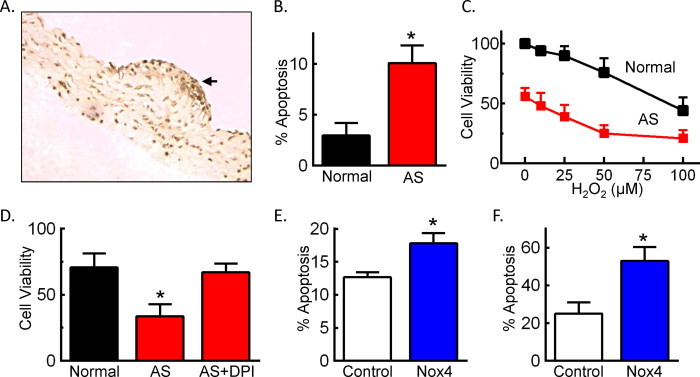
Overexpression of Nox4 in normal SMCs mimics the apoptotic phenotype observed in AS SMCs. (A) Representative TUNEL staining of aortic sections from 24-week old AS mice. Arrow indicates lesion. (B) Percent apoptotic cells as determined by propidium iodide and annexin V staining of normal or AS SMCs cultured in 10% FBS (n = 7–10). (C) Cell viability as determined by MTS assay in the absence or presence of increasing concentrations of H2O2 in 0.5% FBS for 24 h. Data were quantitated relative to normal SMCs without H2O2 (n = 4). *p < 0.05 normal vs. AS by two-way repeated measures ANOVA with Bonferroni post-test. (D) Cell viability as measured by MTS assay in serum-starved cells treated with 50 µM H2O2 in the presence or absence of 10 µM DPI for 24 h. Data were quantitated relative to normal SMCs without H2O2 (n = 4). (E) Percent apoptotic cells as determined by annexin V/propidium iodide staining of normal SMCs infected with either Ad-GFP (control) or Ad-Nox4 (Nox4) for 48 h (n = 7). (F) Percent apoptotic cells after infection of normal SMCs with either Ad-GFP (control) or Ad-Nox4 (Nox4) and treatment 48 h later with 50 µM H2O2 in 0.5% FBS for 24 h (n = 5–6). *p < 0.05 vs. normal SMCs (B–D) or control adenovirus (E and F).
Discussion
Previous studies from patients with established coronary artery disease demonstrate differential expression of NADPH oxidase subunits [8,26]. Our objective was to evaluate the changes in NADPH oxidases with progression of atherosclerosis. Studies in AS mice demonstrated a prelesional increase in Nox1, whereas development of plaque was associated with increased expression of Nox4. SMCs isolated from AS aorta showed a decrease in growth, G0/G1 arrest with corresponding alterations in cell cycle regulators, and increased levels of the senescence marker ß-galactosidase. Furthermore, AS SMCs had decreased viability and increased susceptibility to apoptosis. Similar phenotypes were observed with overexpression of Nox4 in normal SMCs. Together, our data implicate Nox4 in SMC growth inhibition, apoptosis, and senescence, processes that are known to contribute to plaque destabilization and myocardial infarction.
A number of studies have evaluated the role of NADPH oxidases in atherogenesis. Early evidence for increased NADPH oxidase in lesions came from examination of p22phox expression in human coronary arteries from patients with or without coronary artery disease [27]. Subsequent studies demonstrated that Nox2 but not Nox1 is also increased in advanced human atherosclerotic lesions [8,26]. In non-human primates on an atherogenic diet, both Nox1 and Nox2 levels are increased and return to baseline with normal diet [28]. Genetic deficiency of p47phox, Nox1, and Nox2 are all associated with protection against lesion development in ApoE-/- AS mice [[4], [5], [6], [7]]. However, a limitation of these genetic mouse models is they are designed to primarily examine the role of NADPH oxidase in the initiation and early progression of atherogenesis. A strength of our study is that we define the temporal changes in NADPH oxidases and identify a role for SMC Nox4 in the later stages of atherosclerosis, in particular the plaque phenotypes of apoptosis and senescence. Administration of a dual Nox1/Nox4 pharmacologic inhibitor (GenKyoTex) reduces lesion area, ROS generation, and markers of oxidative stress in AS mice [29]. Consistent with the role of Nox4 in plaque phenotypes rather than plaque formation, it has been recently demonstrated that deficiency of Nox4 on an ApoE-/- model does not impact lesion area [30].
Recent reports suggest a protective effect for Nox4-derived H2O2 in the vasculature [[31], [32], [33]]. Whereas these studies have primarily focused on the role of Nox4 in endothelial cells, it is unclear whether increased expression of Nox4 in SMCs is protective. Based on our data, Nox4 mediates changes in cell growth, apoptosis, and senescence of SMCs. Together, these observations imply that Nox4-derived H2O2 is necessary for signaling adaptive survival pathways; however, increased Nox4 expression in SMCs can lead to oxidative stress and be detrimental. Consistent with this interpretation, others have shown using a model of ischemic stroke that the absence of Nox4 is neuroprotective [34].
We observed an increase in Nox1 expression prior to the development of overt plaque, which is consistent with Nox1 activation in the early response to vascular injury [4,9]. Nox2 levels remained elevated throughout the disease continuum, reflecting the predominant contribution of inflammatory cells in the atherosclerotic lesion [35]. Data regarding changes in Nox4 expression in late stage atherosclerosis in human and non-human primates have been inconclusive [26,28,36]. In our AS murine model, we observed an increase in Nox4 mRNA levels in advanced lesions. Importantly, the relative changes in Nox4 were more pronounced when measured in AS SMCs (Fig. 3A) as compared to the total AS vessel (Fig. 2B). This observation may in part explain the failure of prior studies to show changes in Nox4 since plaque SMCs only represent a small proportion of the total SMCs in an intact vessel. The observed temporal changes in Nox1 and Nox4 expression during atherogenesis suggest distinct functional consequences of the Nox-derived ROS. Whereas Nox1-derived ROS are important in SMC migration and proliferation [10,37], our data suggest the Nox4-derived ROS contribute to SMC growth inhibition, apoptosis, and senescence.
Since SMCs contribute to the stability of the fibrous cap in part by synthesizing extracellular matrix, apoptosis of SMCs has been proposed as a mechanism for plaque instability and rupture [38]. Treatment of cultured SMCs with 7-ketocholesterol, an oxysterol present in oxidized LDL, induces apoptosis through increased expression of Nox4 [39]. Our findings take this a step further by providing evidence for Nox4-dependent apoptosis in atherosclerosis. Our study was not designed to look for evidence of plaque rupture in vivo in large part because the animal models have poorly mimicked plaque instability and the formation of occlusive thrombi [38].
In addition to apoptosis, senescence is a hallmark of SMCs within an advanced lesion. Characteristic changes in human plaque SMCs include hypo-phosphorylation of Rb, increased expression of cyclin-dependent kinase inhibitors, and decreased expression of cyclins [24]. In addition to cell cycle alterations, senescence is also evidenced by ß-galactosidase expression [24]. We observed many of these changes, specifically hypo-phosphorylation of Rb, decreased expression of cyclin D1, increased expression of ß-galactosidase, and G0/G1 arrest, SMCs derived from a murine model of AS. Furthermore, we found that overexpression of Nox4 in normal SMCs mimicked the cell cycle arrest observed in AS SMCs. These findings are consistent with recent evidence that Nox4 contributes to a senescent phenotype in endothelial cells [40]. The knockdown of Nox4 in AS SMCs was unsuccessful in restoring normal cell cycle progression. However, this not unexpected given that replicative senescence is a terminal phenotype [24]. Whereas our data do not identify the specific mechanism for increased Nox4 expression in the AS SMCs, it has been reported that TGF-ß levels are increased in advanced atherosclerotic lesions [24]. TGF-ß is known to induce Nox4 expression in SMCs [41].
Conclusions
Vascular cells in AS are known to have a senescent phenotype with increased susceptibility to apoptosis, thereby predisposing to plaque rupture with subsequent thrombosis and myocardial infarction. Novel findings from this study show a similar phenotype in SMCs derived from a murine model of AS and implicate the Nox4 NADPH oxidase in these phenotypic changes. These data provide new insight into the enzymatic source of ROS in advanced AS.
Acknowledgements
The authors acknowledge the Flow Cytometry Core Facility, the DNA Core Facility, the Gene Transfer Vector Core Facility, and the Free Radical Core Facility in the University of Iowa Roy J. and Lucille A. Carver College of Medicine. We thank Kristina W. Thiel for assistance in manuscript preparation. This work was supported by the Office of Research and Development, Department of Veterans Affairs [1BX001729 to F.J.M.] and an American Heart Association Postdoctoral Fellowship (0320075Z to S.U.).
References
- 1.Harrison D., Griendling K.K., Landmesser U., Hornig B., Drexler H. Role of oxidative stress in atherosclerosis. American Journal of Cardiology. 2003;91:7A–11A. doi: 10.1016/s0002-9149(02)03144-2. 12645638 [DOI] [PubMed] [Google Scholar]
- 2.Cai H., Harrison D.G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circulation Research. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. 11073878 [DOI] [PubMed] [Google Scholar]
- 3.Lassegue B., San Martin A., Griendling K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circulation Research. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. 22581922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheehan A.L., Carrell S., Johnson B., Stanic B., Banfi B. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis. 2011;216:321–326. doi: 10.1016/j.atherosclerosis.2011.02.028. 21411092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Judkins C.P., Diep H., Broughton B.R., Mast A.E., Hooker E.U. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE-/- mice. American Journal of Physiology: Heart and Circulatory Physiology. 2010;298:H24–H32. doi: 10.1152/ajpheart.00799.2009. 19837950 [DOI] [PubMed] [Google Scholar]
- 6.Barry-Lane P.A., Patterson C., van der Merwe M., Hu Z.Y., Holland S.M. p47phox is required for atherosclerotic lesion progression in ApoE(-/-) mice. Journal of Clinical Investigation. 2001;108:1513–1522. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vendrov A.E., Hakim Z.S., Madamanchi N.R., Rojas M., Madamanchi C. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:2714–2721. doi: 10.1161/ATVBAHA.107.152629. 17823367 [DOI] [PubMed] [Google Scholar]
- 8.Sorescu D., Weiss D., Lassegue B., Szocs K., Vega J.D. Expression of the NAD(P)H oxidase subunits in human coronary arteries. Circulation. 2001;104:192. [Google Scholar]
- 9.Szocs K., Lassegue B., Sorescu D., Hilenski L.L., Valppu L. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:21–27. doi: 10.1161/hq0102.102189. 11788456 [DOI] [PubMed] [Google Scholar]
- 10.Xu S., Shriver A.S., Jagadeesha D.K., Chamseddine A., Szocs K. Increased expression of Nox1 in neointimal smooth muscle cells promotes activation of matrix metalloproteinase-9. Journal of Vascular Research. 2012;49:242–248. doi: 10.1159/000332958. 22433789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matthews C., Gorenne I., Scott S., Figg N., Kirkpatrick P. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circulation Research. 2006;99:156–164. doi: 10.1161/01.RES.0000233315.38086.bc. 16794190 [DOI] [PubMed] [Google Scholar]
- 12.Miller F.J., Jr., Gutterman D.D., Rios C.D., Heistad D.D., Davidson B.L. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circulation Research. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. 9648726 [DOI] [PubMed] [Google Scholar]
- 13.Stanic B., Katsuyama M., Miller F.J., Jr. An oxidized extracellular oxidation-reduction state increases Nox1 expression and proliferation in vascular smooth muscle cells via epidermal growth factor receptor activation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30:2234–2241. doi: 10.1161/ATVBAHA.110.207639. 20814013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radical Biology and Medicine. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. 17761297 [DOI] [PubMed] [Google Scholar]
- 15.Takapoo M., Chamseddine A.H., Bhalla R.C., Miller F.J., Jr. Glutathione peroxidase-deficient smooth muscle cells cause paracrine activation of normal smooth muscle cells via cyclophilin A. Vascular Pharmacology. 2011;55:143–148. doi: 10.1016/j.vph.2011.07.002. 21782974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chamseddine A.H., Miller F.J., Jr. gp91phox contributes to NADPH oxidase activity in aortic fibroblasts but not smooth muscle cells. American Journal of Physiology: Heart and Circulatory Physiology. 2003;285:H2284–H2289. doi: 10.1152/ajpheart.00459.2003. 12855428 [DOI] [PubMed] [Google Scholar]
- 17.Tsimikas S., Aikawa M., Miller F.J., Jr., Miller E.R., Torzewski M. Increased plasma oxidized phospholipid:Apolipoprotein B-100 ratio with concomitant depletion of oxidized phospholipids from atherosclerotic lesions after dietary lipid-lowering: a potential biomarker of early atherosclerosis regression. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:175–181. doi: 10.1161/01.ATV.0000251501.86410.03. 17082490 [DOI] [PubMed] [Google Scholar]
- 18.Spitz D.R., Oberley L.W. An assay for superoxide dismutase activity in mammalian tissue homogenates. Analytical Biochemistry. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. 2547324 [DOI] [PubMed] [Google Scholar]
- 19.Spitz D.R., Elwell J.H., Sun Y., Oberley L.W., Oberley T.D. Oxygen toxicity in control and H2O2-resistant Chinese hamster fibroblast cell lines. Archives of Biochemistry and Biophysics. 1990;279:249–260. doi: 10.1016/0003-9861(90)90489-l. 2350176 [DOI] [PubMed] [Google Scholar]
- 20.Fasbender A., Zabner J., Chillon M., Moninger T.O., Puga A.P. Complexes of adenovirus with polycationic polymers and cationic lipids increase the efficiency of gene transfer in vitro and in vivo. Journal of Biological Chemistry. 1997;272:6479–6489. doi: 10.1074/jbc.272.10.6479. 9045673 [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi S., Inoue N., Azumi H., Seno T., Hirata K. Expressional changes of the vascular antioxidant system in atherosclerotic coronary arteries. Journal of Atherosclerosis and Thrombosis. 2002;9:184–190. doi: 10.5551/jat.9.184. 12226550 [DOI] [PubMed] [Google Scholar]
- 22.Chen Q., Ames B.N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:4130–4134. doi: 10.1073/pnas.91.10.4130. 8183882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dimri G.P., Lee X., Basile G., Acosta M., Scott G. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. 7568133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J.C., Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circulation Research. 2012;111:245–259. doi: 10.1161/CIRCRESAHA.111.261388. 22773427 [DOI] [PubMed] [Google Scholar]
- 25.Bennett M.R., Evan G.I., Schwartz S.M. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. Journal of Clinical Investigation. 1995;95:2266–2274. doi: 10.1172/JCI117917. 7738191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guzik T.J., Sadowski J., Guzik B., Jopek A., Kapelak B. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:333–339. doi: 10.1161/01.ATV.0000196651.64776.51. 16293794 [DOI] [PubMed] [Google Scholar]
- 27.Azumi H., Inoue N., Takeshita S., Rikitake Y., Kawashima S. Expression of NADH/NADPH oxidase p22phox in human coronary arteries. Circulation. 1999;100:1494–1498. doi: 10.1161/01.cir.100.14.1494. 10510050 [DOI] [PubMed] [Google Scholar]
- 28.Stanic B., Pandey D., Fulton D.J., Miller F.J., Jr. Increased epidermal growth factor-like ligands are associated with elevated vascular nicotinamide adenine dinucleotide phosphate oxidase in a primate model of atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:2452–2460. doi: 10.1161/ATVBAHA.112.256107. 22879585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vendrov A.E., Madamanchi N.R., Niu X.L., Molnar K.C., Runge M. NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. Journal of Biological Chemistry. 2010;285:26545–26557. doi: 10.1074/jbc.M110.143917. 20558727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gray S.P., Di Marco E., Okabe J., Szyndralewiez C., Heitz F. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127:1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. 23564668 [DOI] [PubMed] [Google Scholar]
- 31.Craige S.M., Chen K., Pei Y., Li C., Huang X. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. 2011;124:731–740. doi: 10.1161/CIRCULATIONAHA.111.030775. 21788590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ray R., Murdoch C.E., Wang M., Santos C.X., Zhang M. Endothelial nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31:1368–1376. doi: 10.1161/ATVBAHA.110.219238. 21415386 [DOI] [PubMed] [Google Scholar]
- 33.Schroder K., Zhang M., Benkhoff S., Mieth A., Pliquett R. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circulation Research. 2012;110:1217–1225. doi: 10.1161/CIRCRESAHA.112.267054. 22456182 [DOI] [PubMed] [Google Scholar]
- 34.Kleinschnitz C., Grund H., Wingler K., Armitage M.E., Jones E. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biology. 2010;8 doi: 10.1371/journal.pbio.1000479. 20877715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hathaway C.A., Heistad D.D., Piegors D.J., Miller F.J., Jr. Regression of atherosclerosis in monkeys reduces vascular superoxide levels. Circulation Research. 2002;90:277–283. doi: 10.1161/hh0302.104724. 11861415 [DOI] [PubMed] [Google Scholar]
- 36.Sorescu D., Weiss D., Lassegue B., Clempus R.E., Szocs K. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. 11914250 [DOI] [PubMed] [Google Scholar]
- 37.Jagadeesha D.K., Takapoo M., Banfi B., Bhalla R.C., Miller F.J., Jr. Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovascular Research. 2012;93:406–413. doi: 10.1093/cvr/cvr308. 22102727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz S.M., Virmani R., Rosenfeld M.E. The good smooth muscle cells in atherosclerosis. Current Atherosclerosis Reports. 2000;2:422–429. doi: 10.1007/s11883-000-0081-5. 11122774 [DOI] [PubMed] [Google Scholar]
- 39.Pedruzzi E., Guichard C., Ollivier V., Driss F., Fay M. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Molecular and Cellular Biology. 2004;24:10703–10717. doi: 10.1128/MCB.24.24.10703-10717.2004. 15572675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koziel R., Pircher H., Kratochwil M., Lener B., Hermann M. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochemical Journal. 2013;452:231–239. doi: 10.1042/BJ20121778. 23514110 [DOI] [PubMed] [Google Scholar]
- 41.Cucoranu I., Clempus R., Dikalova A., Phelan P.J., Ariyan S. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circulation Research. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. 16179589 [DOI] [PubMed] [Google Scholar]