

Abstract
Retinal tissue receives its supply of oxygen from two sources – the retinal and choroidal circulations. Decreases in retinal blood flow occur in the early stages of diabetes, with the eventual development of hypoxia thought to contribute to pathological neovascularization. Oxygen consumption in the retina has been found to decrease in diabetes, possibly due to either a reduction in neuronal metabolism or to cell death. Diabetes also enhances the rate of conversion of oxygen to superoxide in the retina, with experimental evidence suggesting that mitochondrial superoxide not only drives the overall production of reactive oxygen species, but also initiates several pathways leading to retinopathy, including the increased activity of the polyol and hexosamine pathways, increased production of advanced glycation end products and expression of their receptors, and activation of protein kinase C.
Keywords: Diabetes, Retina, Oxygen, Oxidative stress, Superoxide
Highlights
-
•
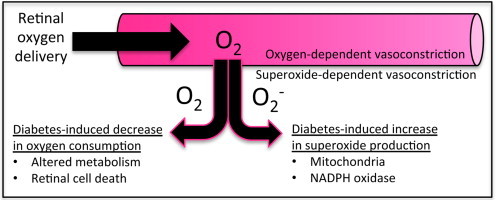
Diabetes alters oxygen delivery and consumption in the retina.
-
•
Conversion of oxygen to superoxide increases in the diabetic retina.
-
•
An initial production of mitochondrial superoxide generates further ROS.
-
•
ROS have been found to mediate deleterious pathways in the diabetic retina.
Graphical Abstract
Oxygen supply to the retina
The oxygen demand of the retina is high, with the retina among the most metabolically active tissues in the body. The retina is in a constant state of activity, converting light into neural signals, and with the activity continuing (and oxygen consumption even increasing) in the dark. The cells of the retina require a constant supply of oxygen; however, a dense vascular network anterior to the photoreceptors would interfere with light transmission to these light-sensing cells. To circumvent this potential interference, the outer one-third to one-half of the retina receives almost all of its oxygen via the choroidal microcirculation, which is just posterior to the retinal tissue (see Fig. 1). The choroidal circulation branches from the posterior ciliary arteries, which in turn derive from the ophthalmic artery. The inner one-half to two-thirds of the retina receives its requirement of oxygen from the retinal circulation, which is a separate branch (via the central retinal artery) of the ophthalmic circulation.
Fig. 1.
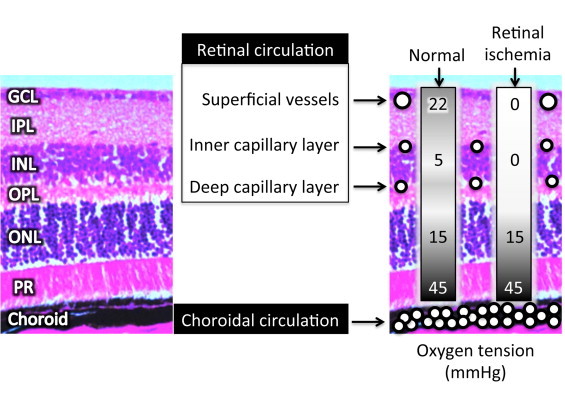
Shown to the left in the figure are the distinct layers of the retina, as seen in a hematoxylin–eosin stained cross-section of a mouse. The layers are the ganglion cell layer (GCL), the inner plexiform layer (IPL), the inner nuclear layer (INL), the outer plexiform layer (OPL), the outer nuclear layer (ONL), and the photoreceptors (PR). The retina receives its oxygen from two different circulations, the retinal circulation that feeds the inner retina, and the choroidal circulation that feeds the outer retina, with cross-sectional depictions of these blood vessels superimposed on the picture at the right. Values of retinal oxygen tension are shown in the boxes to the right, with the values demonstrating the importance of the retinal circulation to the inner retina as described further in the text.
The retinal and choroidal circulations differ in their vascular density, autoregulatory control, and their arterio-venous oxygen differences, but both circulations are critical for supplying sufficient oxygen to the entire retina. Using microelectrodes, Yu et al. [1,2] have measured oxygen partial pressures in the retinas of mice and rats, and have found a very sharp decline from the retinal tissue near the choroid (~45 mmHg; see Fig. 1), with oxygen tension dropping to ~10 to 15 mmHg just anterior to the photoreceptors, consistent with the high metabolic rate of these neurons. Similar profiles have been obtained for pigs and cats [3]. The high consumption of oxygen in the outer retina prevents the choroidal circulation from providing sufficient oxygen to the inner retina in most mammals, excluding rabbits and guinea pigs. The contribution of the retinal microcirculation to the oxygenation of the inner retina was demonstrated by Yu et al. [2], with experiments in which the retinal circulation of rats was occluded by laser (retinal ischemia in Fig. 1), and measurements of oxygen tension were close to 0 mmHg throughout the inner retina. This experimental protocol additionally allowed the researchers to model the oxygen consumption of the inner retina, since the presence of flowing vessels in the tissue interferes with such analysis. Their analysis indicated that oxygen consumption in the inner retina is substantial, and of the same magnitude as that of the outer retina.
The choroidal circulation is much more dense than the retinal circulation (Fig. 1), with the sparse nature of the latter important for light transmission through the retina as mentioned previously. However, the limited retinal circulation has been speculated to contribute to the vulnerability of the retina to vascular disease [4].
Utilization of oxygen in the retina
As mentioned, oxygen profiles have helped to identify the sites of oxygen usage in the retina, with three dominant layers of consumption identified in the rat as the inner segment of the photoreceptors (where the mitochondria are localized), the outer plexiform layer (OPL in Fig. 1) and the deeper region of the inner plexiform layer (IPL) [4]. Accordingly, these three layers of the retina are also where retinal cross-sections stain most intensely for activity of cytochrome oxidase [5], the enzyme in the final step of the mitochondrial respiratory chain in which oxygen is consumed in the production of ATP. Much of the oxygen may be used for synaptic transmission [2], Na–K ATPase activity, phototransduction [6], and processes that require substantial amounts of energy such as the maintenance of the dark current and the generation of guanosine triphosphate [3].
Oxygen consumption increases in the dark, with this increase resulting in almost complete tissue anoxia in certain areas of the cat retina [7]. However, according to the data of Yu and Cringle [8], the development of anoxia in the dark is not a general property of mammalian retinas, in as much as oxygen tension in the outer retina does not decrease below 5 mmHg in dark-adapted rats, despite an approximate 50% increase in oxygen consumption. Yu and Cringle explained the lack of dark-induced anoxia in the rat retina as being due to a significant increase in oxygen delivery. In animals in which darkness induces hypoxia, it could be speculated that the hypoxia and/or the subsequent reoxygenation could induce an oxidative stress; however, it is not clear whether experimental evidence has supported this possibility.
Regulation of retinal blood flow by oxygen
Oxygen concentrations help regulate retinal blood flow. Not only does hypoxia stimulate increased perfusion and potentially angiogenesis, but the reverse also is true, whereby hyperoxia decreases retinal blood flow. The latter phenomenon has been demonstrated by Zhu et al. [9] in experiments in which newborn pigs breathed 100% oxygen, causing retinal blood flow to decrease by ~40% within 5–10?min. This response was attenuated by treatments aimed at several endogenous vasoconstrictors including thromboxane, endothelin, and 20-HETE (20-hydroxyeicosatetraeonic acid), but not by the antioxidant combination of superoxide dismutase and catalase. In comparison, diabetes-induced decreases in retinal blood flow rate in rodents (decreases of ~33%) have been found to be attenuated not only by targeting thromboxane [10–13], endothelin [14], 20-HETE [15], and angiotensin II [11], but also by scavenging superoxide with tempol [16], with the latter implicating reactive oxygen species (ROS) in retinal blood flow control during diabetes. Whether or not oxygen levels regulate these vasoconstrictor pathways in the diabetic retina has yet to be clarified.
Altered metabolism in the diabetic retina
Diabetes-induced decreases in retinal oxygen consumption have been reported for rabbits [17,18] and cats [19], but with conflicting reports of arterio-venous oxygen differences and consumption in the diabetic rat retina [20–22]. However, Obrosova et al. [23] have studied retinal metabolism in streptozotocin (STZ)-induced diabetes in both rats and also mice, at a time point of 6 weeks. They measured several metabolites in the retina including pyruvate, lactate, glutamate, a-ketoglutarate, ammonia, and free NAD+/NADH. In rats, each of these were decreased significantly by ~30% to 50%, but only two of the metabolites were decreased in mice (pyruvate by 10%, and ammonia by 15%). In humans, arterio-venous oxygen differences have been reported to decrease throughout all stages of retinopathy [24]. In as much as early decreases in retinal blood flow have been found in diabetic patients [25], it is likely that oxygen consumption rates simultaneously decrease as is the case in several experimental models of the disease. Any decrease in retinal oxygen consumption, whether transient or sustained, could result in a decrease in retinal blood flow, with Small et al. [26] and Rimmer and Linsenmeier [27] speculating that this mechanism could be operative in the diabetic retina.
Neuronal cell death may contribute to the decrease in metabolism reported in the diabetic retina. STZ-induced diabetes in the rat has been found in one study to produce retinal cell death in the time frame of 1–12 months [28], and in another study, the number of apoptotic photoreceptors in the STZ rat continued to increase throughout the period from 4–24 weeks of hyperglycemia [29]. The increase in neuronal cell death in the retina also has been reported in mice injected with STZ [30,31], and in the Ins2Akita diabetic mouse [31]. Cell death contributes to the phenomenon of retinal thinning, which is found for both rodent [30,32] and human [33–36] diabetes.
Altered oxygen levels in the diabetic retina
The development of diabetic retinopathy is hypothesized, in part, to be a function of retinal hypoxia leading to the uncontrolled growth of new retinal blood vessels [37]. Interestingly, investigators using oxygen microelectrodes have found no signs of retinal hypoxia within the first year of experimentally induced diabetes in cats and dogs [38–40]. Our own lab has studied diabetic mice and rats, and despite finding 20–45% decreases in retinal blood flow rates [10–16], we have not seen significant signs of hypoxia (measured with the probe pimonidazole, and by immunostaining for the hypoxia-inducible factor HIF-1a) early in the progression of hyperglycemia (1–6 months) [41–43]. In fact, we have detected a possible phase of increased retinal oxygen in rats at a time point of 3 months diabetes [41], with this surprising finding also observed in the inner retina by Lau and Linsenmeier [44] at the same time point, using a more direct measurement with oxygen microelectrodes. However, other groups have found significant increases in HIF-1a and/or pimonidazole staining [45–48], and therefore, more studies may be required to resolve the time course of either hypoxia or hyperoxia early in the progression of experimental and human diabetes.
Even though the most direct measure of retinal oxygen, with microelectrodes, has not found hypoxia during the early weeks to months in experimental diabetes, Linsenmeier et al. [19] used this technique to reveal significant hypoxia at a much longer duration (6–7 years of diabetes) in cats, with inner retinal oxygen tension decreased by almost 50% compared to controls.
Evidence for increased superoxide in experimental diabetic retinopathy
Superoxide can be generated from oxygen by a number of different enzymes, including mitochondrial complex Q, NADPH oxidase, xanthine oxidase, p450 mono-oxygenase, uncoupled nitric oxide synthase (NOS), cyclooxygenase, and lipoxygenase. The overproduction of superoxide via one or more of these pathways has been implicated in the development of diabetic retinopathy. Many reports have been made of increased retinal superoxide production in rodent models of diabetic retinopathy. In one of these studies, using the STZ rat model, retinal superoxide production was found to be increased following 2 months of hyperglycemia, with the increase in superoxide shown to be produced primarily from the mitochondria, with small amounts being produced from NADPH oxidase and NOS [49]. In an STZ mouse model, superoxide increased in the photoreceptors following 2 months of hyperglycemia: the increase in superoxide occurred in both light-adapted and dark-adapted diabetic eyes [50].
The activity of the enzyme responsible for converting superoxide into oxygen and hydrogen peroxide, superoxide dismutase (SOD), has been reported to decrease in STZ-induced diabetic rats following 6 weeks [51], 2 months [52], and 11 months [53] of hyperglycemia. However, it should be mentioned that an increase in SOD activity (measured as an inhibition of xanthine oxidase) in STZ-induced diabetic rats has also been reported [54]. An increase in SOD would have the potential of decreasing levels of superoxide due to the conversion to hydrogen peroxide (H2O2), which in turn would be efficiently converted to H2O in the presence of adequate catalase and/or reaction with glutathione (GSH) in the presence of glutathione peroxidase (GSH-Px). Catalase has been found to slightly increase in the diabetic rat retina, with no change in GSH but a decrease in GSH-Px [23].
Role of reactive oxygen species (ROS) in the diabetic retina
According to the “unified hypothesis” of diabetes-induced pathology [55–57], superoxide overproduction is central to the initiation of parallel deleterious signaling pathways, including the increased activity of the polyol and hexosamine pathways, increased production of advanced glycation end products (AGEs) and expression of their receptors, and activation of protein kinase C. As shown in Fig. 2, the overproduction of superoxide due to hyperglycemia could involve the mitochondrial electron transport chain, due to the increased metabolism of glucose generating pyruvate that enters Kreb’s cycle. The electron donors (NADH, FADH2) that are produced enter the mitochondrial electron transport chain, resulting in the extrusion of protons and the crossing of a critical threshold of mitochondrial membrane voltage. Once this threshold is exceeded, electron transfer (in complex III) is blocked, causing coenzyme Q to donate electrons to O2, generating superoxide. Additional reactive oxidants are formed when superoxide dismutase converts O2- to hydrogen peroxide (H2O2), which can be reduced to the highly reactive hydroxide anion (OH-). Superoxide also can react with nitric oxide to form the highly reactive (and protein modifying) anion peroxynitrite (ONOO-).
Fig. 2.
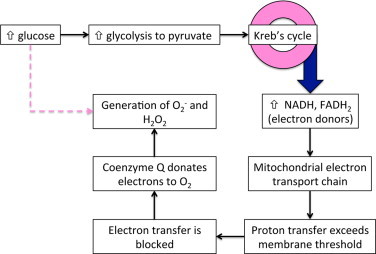
Hyperglycemia induces superoxide and ROS generation. One pathway of this generation includes the mitochondrial electron transport chain, where excessive production of electrons induces their donation to oxygen, creating the superoxide radical. Other hyperglycemia-induced pathways leading to ROS (indicated by the dashed line) also may contribute to oxidative stress.
Evidence for the role of mitochondrial superoxide as the initial reactive oxidant produced by hyperglycemia has been demonstrated with in vitro experiments in which the mitochondrial voltage gradient was collapsed by overexpressing the uncoupling protein (UCP-1) that helps regulate the production of superoxide. This manipulation, and also overexpressing MnSOD (found almost exclusively in the mitochondria) were both able to prevent hyperglycemia-induced production of ROS in vitro [58]. Overexpression of MnSOD also has been performed in vivo, in diabetic mice, with the retinas protected from oxidative damage to proteins and DNA [59].
Although the mitochondria may be the initial producer of superoxide due to hyperglycemia, this process in turn may activate other superoxide-generating pathways in diabetic tissue such as redox changes, uncoupled NOS, and NADPH oxidase [56]. The potential importance of NADPH oxidase (specifically, NOX2) was found in experiments in which blocking the effects of the enzyme was able to completely eliminate the diabetes-induced increase in ROS production, as measured by dichlorofluorescein (DCF) intensity in cross-sections of the diabetic mouse retina [60]. In these experiments, DCF intensity was no greater than controls in either NOX2-deficient mice or in mice treated with the NOX inhibitor apocynin. This may appear to differ from the findings of Nishikawa et al. [58], who found evidence that mitochondrial superoxide is responsible for hyperglycemia-induced increases in ROS generation. However, a link between these two mechanisms could exist, as suggested by Al-Shabrawey et al. [60], who proposed that mitochondria-derived ROS may trigger protein kinase C to activate NOX, and with other studies [61-63] providing evidence that NOX can sustain the ROS formation initiated by the mitochondria.
The evidence for increased retinal oxidative stress in the diabetic retina has prompted experimental studies of antioxidant therapies in animal models of diabetic retinopathy. As reviewed by Madsen-Bouterse and Kowluru [64], administration of lipoic acid to rats protects retinal capillary cells from apoptosis and attenuates retinal mitochondrial dysfunction [53,55]. Vitamins C and E given to diabetic rats decrease the number of acellular capillaries (that is, lost pericytes and endothelial cells) in the retinal microvasculature [65], with the number of acellular capillaries also decreased by overexpression of MnSOD in diabetic mice, which reduces oxidative stress [66]. The same benefits in diabetic rats are seen via administration of green tea extract [67] and other nutritional supplements [68]. Whether these treatments will be effective in human diabetic retinopathy have yet to be determined: diet recall in diabetic patients has not demonstrated any strong evidence for protection by the antioxidants ascorbic acid, a-tocopherol, and vitamins C and E [69,70], but a comprehensive literature review of the effects of vitamin C and SOD on human diabetic retinopathy indicates that no research to date has adequately addressed the potential for clinical benefits [71].
Connection between mitochondrial superoxide and parallel pathways of damage
Diabetes causes a decrease in the activity of the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and with this decrease, glycolytic intermediates upstream of GAPDH increase, which increases flux into the parallel pathways mentioned earlier, that is, the polyol and hexosamine pathways, production of advanced glycation end products (AGEs) and expression of their receptors, and activation of protein kinase C [56]. GAPDH activity is inhibited by hyperglycemia in vivo in a mechanism whereby superoxide increases levels of poly(ADP-ribose) polymerase (PARP) [72]. The resulting polymers of ADP-ribose accumulate on GAPDH, rendering it inactive. However, this inhibition of GAPDH activity does not occur in hyperglycemia when mitochondrial production of superoxide is inhibited [73].
Regulation of hypoxia inducible factors by ROS
Neovascularization is a characteristic of the later stages of diabetic retinopathy [74,75]. It is thought that the stabilization and activation of hypoxia inducible factor 1a (HIF-1a) contributes to the upregulation of angiogenic factors leading to the neovascularization [76]. Kietzmann and Görlach [77] have reviewed potential mechanisms by which ROS may regulate the expression of HIF-1a, including via prolyl hydroxylase, extracellular-signal-regulated kinase (ERK), p38 mitogen-activated protein kinase, and protein kinase B, with the source of ROS in these mechanisms likely to include those derived by the mitochondria and by NADPH oxidase [78]. ROS-dependent regulation of HIF-1a does not always correlate with the level of hypoxia, and interestingly, hyperoxia can stimulate the mitochondrial production of ROS in endothelial cells, leading to activation of NADPH oxidase [79]. Other studies as well have documented the production of ROS due to hyperoxia [80-83]. However, whether ROS generated by hyperoxia contributes to oxygen-dependent vasoconstriction in the retina is questionable, given the previously mentioned study by Zhu et al. [9] in which SOD/catalase was unable to attenuate hyperoxia-induced decreases in retinal blood flow.
Although ROS have been shown to stabilize and activate HIF-1a, hyperglycemia has been shown to downregulate HIF-1a [84], and the mechanism of downregulation appears to be in part due to binding of HIF-1a by methylglyoxal [85,86], a byproduct of glycolysis. The methylglyoxal-induced downregulation of HIF-1a has been implicated as preventing ischemia-induced vasculogenesis in diabetes [86]; which also could possibly contribute to the presence of ischemic regions in the diabetic retina. However, one or more aspects of this mechanism may be altered or bypassed in certain areas of the retina in the later stages of diabetic retinopathy, given the deleterious angiogenesis that occurs. Further investigation into the mechanisms of HIF-1a stabilization and activation, and the clinical implications of these results is warranted. Fig. 3 summarizes some of the pathways initiated by retinal production of superoxide in diabetes, incorporating the pathways involving PARP-induced decreases in GAPDH, and methylglyoxal- and ROS-mediated changes in HIF-1a.
Fig. 3.
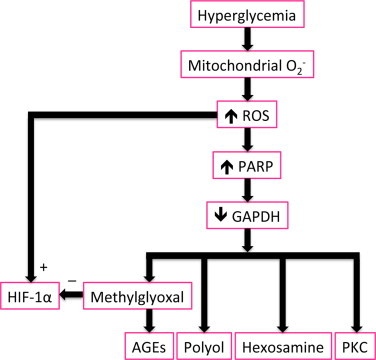
Hyperglycemia-induced ROS production increases levels of poly(ADP-ribose) polymerase (PARP), which inhibits glyceraldehyde 3-phosphate dehydrogenase (GAPDH) activity. The consequences of inhibited GAPDH include an increase in methylglyoxal, increased activity of the polyol and hexosamine pathways, increased production of advanced glycation end products (AGEs) and expression of their receptors, and activation of protein kinase C (PKC). The effects on HIF-1a may be mixed, being stimulated by ROS but inhibited by methylglyoxal.
Summary
Diabetic retinopathy involves alterations in oxygen delivery, oxygen consumption, and superoxide production, with the interdependence of these mechanisms still yet to be determined. Reports of increased retinal oxidative stress in the diabetic retina have led to experimental studies of antioxidant therapies, which have shown promise in animal models of diabetic retinopathy. Such treatments have included agents such as lipoic acid, vitamins C and E, MnSOD, green tea extract, and nutritional antioxidant supplements [64]. To date, less is known concerning the potential long-term effects of targeting diabetes-induced changes in blood flow and oxygen delivery. The degree to which therapies directed at oxygen delivery, usage, and production of superoxide will attenuate human diabetic retinopathy awaits further investigation.
Acknowledgements
Funding through NIHEY017599 (NRH).
References
- 1.Yu D.Y., Cringle S.J. Oxygen distribution in the mouse retina. Investigative Ophthalmology and Visual Science. 2006;47:1109–1112. doi: 10.1167/iovs.05-1118. 16505048 [DOI] [PubMed] [Google Scholar]
- 2.Yu D.Y., Cringle S.J., Yu P.K., Su E.N. Intraretinal oxygen distribution and consumption during retinal artery occlusion and graded hyperoxic ventilation in the rat. Investigative Ophthalmology and Visual Science. 2007;48:2290–2296. doi: 10.1167/iovs.06-1197. 17460293 [DOI] [PubMed] [Google Scholar]
- 3.Wangsa-Wirawan N.D., Linsenmeier R.A. Retinal oxygen: fundamental and clinical aspects. Archives of Ophthalmology. 2003;121:547–557. doi: 10.1001/archopht.121.4.547. 12695252 [DOI] [PubMed] [Google Scholar]
- 4.Yu D.Y., Cringle S.J. Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Progress in Retinal and Eye Research. 2001;20:175–208. doi: 10.1016/s1350-9462(00)00027-6. 11173251 [DOI] [PubMed] [Google Scholar]
- 5.Kur J., Newman E.A., Chan-Ling T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Progress in Retinal and Eye Research. 2012;31:377–406. doi: 10.1016/j.preteyeres.2012.04.004. 22580107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ames A., 3rd, Li Y.Y., Heher E.C., Kimble C.R. Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1992;12:840–853. doi: 10.1523/JNEUROSCI.12-03-00840.1992. 1312136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linsenmeier R.A. Effects of light and darkness on oxygen distribution and consumption in the cat retina. Journal of General Physiology. 1986;88:521–542. doi: 10.1085/jgp.88.4.521. 3783124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu D.Y., Cringle S.J. Outer retinal anoxia during dark adaptation is not a general property of mammalian retinas. Comparative Biochemistry and Physiology. Part A, Molecular and Integrative Physiology. 2002;132:47–52. doi: 10.1016/s1095-6433(01)00528-1. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Y., Park T.S., Gidday J.M. Mechanisms of hyperoxia-induced reductions in retinal blood flow in newborn pig. Experimental Eye Research. 1998;67:357–369. doi: 10.1006/exer.1998.0535. 9778417 [DOI] [PubMed] [Google Scholar]
- 10.Wright W.S., Harris N.R. Ozagrel attenuates early streptozotocin-induced constriction of arterioles in the mouse retina. Experimental Eye Research. 2008;86:528–536. doi: 10.1016/j.exer.2007.12.012. 18262522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee S., Harris N.R. Losartan and ozagrel reverse retinal arteriolar constriction in non-obese diabetic mice. Microcirculation. 2008;15:379–387. doi: 10.1080/10739680701829802. 18574741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S., Morgan G.A., Harris N.R. Ozagrel reverses streptozotocin-induced constriction of arterioles in rat retina. Microvascular Research. 2008;76:217–223. doi: 10.1016/j.mvr.2008.07.005. 18718478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright W.S., Messina J.E., Harris N.R. Attenuation of diabetes-induced retinal vasoconstriction by a thromboxane receptor antagonist. Experimental Eye Research. 2009;88:106–112. doi: 10.1016/j.exer.2008.10.008. 18996116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z., Yadav A.S., Leskova W., Harris N.R. Attenuation of streptozotocin-induced microvascular changes in the mouse retina with the endothelin receptor A antagonist atrasentan. Experimental Eye Research. 2010;91:670–675. doi: 10.1016/j.exer.2010.08.008. 20727883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z., Yadav A.S., Leskova W., Harris N.R. Inhibition of 20-HETE attenuates diabetes-induced decreases in retinal hemodynamics. Experimental Eye Research. 2011;93:108–113. doi: 10.1016/j.exer.2011.05.011. 21658386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yadav A.S., Harris N.R. Effect of tempol on diabetes-induced decreases in retinal blood flow in the mouse. Current Eye Research. 2011;36:456–461. doi: 10.3109/02713683.2011.556300. 21501080 [DOI] [PubMed] [Google Scholar]
- 17.Illing E.K., Gray C.H. Retinal metabolism in diabetes; the metabolism of retinae of normal and alloxandiabetic rabbits. Journal of Endocrinology. 1951;7:242–247. doi: 10.1677/joe.0.0070242. 14861355 [DOI] [PubMed] [Google Scholar]
- 18.Sutherland F.S., Stefansson E., Hatchell D.L., Reiser H. Retinal oxygen consumption in vitro. The effect of diabetes mellitus, oxygen and glucose. Acta Ophthalmologica. 1990;68:715–720. doi: 10.1111/j.1755-3768.1990.tb01701.x. [DOI] [PubMed] [Google Scholar]
- 19.Linsenmeier R.A., Braun R.D., McRipley M.A. Retinal hypoxia in long-term diabetic cats. Investigative Ophthalmology and Visual Science. 1998;39:1647–1657. 9699554 [PubMed] [Google Scholar]
- 20.Blair N.P., Wanek J.M., Mori M., Shahidi M. Abnormal retinal vascular oxygen tension response to light flicker in diabetic rats. Investigative Ophthalmology and Visual Science. 2009;50:5444–5448. doi: 10.1167/iovs.09-3465. 19553624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wanek J., Teng P.Y., Blair N.P., Shahidi M. Inner retinal oxygen delivery and metabolism in streptozotocin diabetic rats. Investigative Ophthalmology and Visual Science. 2014;55:1588–1593. doi: 10.1167/iovs.13-13537. 24550355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Roetth A. Metabolism of the alloxan diabetic rat retina. Transactions of the American Ophthalmological Society. 1963;61:429–458. 16693624 [PMC free article] [PubMed] [Google Scholar]
- 23.Obrosova I.G., Drel V.R., Kumagai A.K., Szabo C., Pacher P., Stevens M.J. Early diabetes-induced biochemical changes in the retina: comparison of rat and mouse models. Diabetologia. 2006;49:2525–2533. doi: 10.1007/s00125-006-0356-7. 16896942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammer M., Vilser W., Riemer T. Diabetic patients with retinopathy show increased retinal venous oxygen saturation. Graefe’s Archive for Clinical and Experimental Ophthalmology (Albrecht von Graefes Archiv für Klinische und Experimentelle Ophthalmologie) 2009;247:1025–1030. doi: 10.1007/s00417-009-1078-6. 19404666 [DOI] [PubMed] [Google Scholar]
- 25.Clermont A.C., Aiello L.P., Mori F., Aiello L.M., Bursell S.E. Vascular endothelial growth factor and severity of nonproliferative diabetic retinopathy mediate retinal hemodynamics in vivo: a potential role for vascular endothelial growth factor in the progression of nonproliferative diabetic retinopathy. American Journal of Ophthalmology. 1997;124:433–446. doi: 10.1016/s0002-9394(14)70860-8. 9323935 [DOI] [PubMed] [Google Scholar]
- 26.Small K.W., Stefansson E., Hatchell D.L. Retinal blood flow in normal and diabetic dogs. Investigative Ophthalmology and Visual Science. 1987;28:672–675. 3557871 [PubMed] [Google Scholar]
- 27.Rimmer T., Linsenmeier R.A. Resistance of diabetic rat electroretinogram to hypoxemia. Investigative Ophthalmology and Visual Science. 1993;34:3246–3252. 8225859 [PubMed] [Google Scholar]
- 28.Barber A.J., Lieth E., Khin S.A., Antonetti D.A., Buchanan A.G., Gardner T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. Journal of Clinical Investigation. 1998;102:783–791. doi: 10.1172/JCI2425. 9710447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park S.H., Park J.W., Park S.J. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia. 2003;46:1260–1268. doi: 10.1007/s00125-003-1177-6. 12898017 [DOI] [PubMed] [Google Scholar]
- 30.Martin P.M., Roon P., Van Ells T.K., Ganapathy V., Smith S.B. Death of retinal neurons in streptozotocin-induced diabetic mice. Investigative Ophthalmology and Visual Science. 2004;45:3330–3336. doi: 10.1167/iovs.04-0247. 15326158 [DOI] [PubMed] [Google Scholar]
- 31.Losiewicz M.K., Fort P.E. Diabetes impairs the neuroprotective properties of retinal alpha-crystallins. Investigative Ophthalmology and Visual Science. 2011;52:5034–5042. doi: 10.1167/iovs.10-6931. 21467180 [DOI] [PubMed] [Google Scholar]
- 32.Zhang J., Wu Y., Jin Y. Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Investigative Ophthalmology and Visual Science. 2008;49:732–742. doi: 10.1167/iovs.07-0721. 18235022 [DOI] [PubMed] [Google Scholar]
- 33.Biallosterski C., van Velthoven M.E., Michels R.P., Schlingemann R.O., DeVries J.H., Verbraak F.D. Decreased optical coherence tomography-measured pericentral retinal thickness in patients with diabetes mellitus type 1 with minimal diabetic retinopathy. British Journal of Ophthalmology. 2007;91:1135–1138. doi: 10.1136/bjo.2006.111534. 17383994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bronson-Castain K.W., Bearse M.A., Jr., Neuville J. Adolescents with Type 2 diabetes: early indications of focal retinal neuropathy, retinal thinning, and venular dilation. Retina. 2009;29:618–626. doi: 10.1097/IAE.0b013e31819a988b. 19262432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nilsson M., von Wendt G., Wanger P., Martin L. Early detection of macular changes in patients with diabetes using rarebit fovea test and optical coherence tomography. British Journal of Ophthalmology. 2007;91:1596–1598. doi: 10.1136/bjo.2007.124461. 17591672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Dijk H.W., Kok P.H., Garvin M. Selective loss of inner retinal layer thickness in type 1 diabetic patients with minimal diabetic retinopathy. Investigative Ophthalmology and Visual Science. 2009;50:3404–3409. doi: 10.1167/iovs.08-3143. 19151397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arden G.B., Sivaprasad S. The pathogenesis of early retinal changes of diabetic retinopathy. Documenta Ophthalmologica. Advances in Ophthalmology. 2012;124:15–26. doi: 10.1007/s10633-011-9305-y. 22302291 [DOI] [PubMed] [Google Scholar]
- 38.Ernest J.T., Goldstick T.K., Engerman R.L. Hyperglycemia impairs retinal oxygen autoregulation in normal and diabetic dogs. Investigative Ophthalmology and Visual Science. 1983;24:985–989. 6862799 [PubMed] [Google Scholar]
- 39.Stefansson E., Hatchell D.L., Fisher B.L., Sutherland F.S., Machemer R. Panretinal photocoagulation and retinal oxygenation in normal and diabetic cats. American Journal of Ophthalmology. 1986;101:657–664. doi: 10.1016/0002-9394(86)90765-8. 3717248 [DOI] [PubMed] [Google Scholar]
- 40.Stefansson E., Peterson J.I., Wang Y.H. Intraocular oxygen tension measured with a fiber-optic sensor in normal and diabetic dogs. American Journal of Physiology. 1989;256:H1127–H1133. doi: 10.1152/ajpheart.1989.256.4.H1127. 2705554 [DOI] [PubMed] [Google Scholar]
- 41.Wright W.S., McElhatten R.M., Messina J.E., Harris N.R. Hypoxia and the expression of HIF-1alpha and HIF-2alpha in the retina of streptozotocin-injected mice and rats. Experimental Eye Research. 2010;90:405–412. doi: 10.1016/j.exer.2009.12.002. 20005221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wright W.S., McElhatten R.M., Harris N.R. Increase in retinal hypoxia-inducible factor-2alpha, but not hypoxia, early in the progression of diabetes in the rat. Experimental Eye Research. 2011;93:437–441. doi: 10.1016/j.exer.2011.06.003. 21689648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright W.S., Yadav A.S., McElhatten R.M., Harris N.R. Retinal blood flow abnormalities following six months of hyperglycemia in the Ins2(Akita) mouse. Experimental Eye Research. 2012;98:9–15. doi: 10.1016/j.exer.2012.03.003. 22440813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lau J.C., Linsenmeier R.A. Oxygen consumption and distribution in the diabetic rat retina. Investigative Ophthalmology and Visual Science. 2010;51:5644. E-abstract. [Google Scholar]
- 45.Li C., Chen P., Zhang J. Enzyme-induced vitreolysis can alleviate the progression of diabetic retinopathy through the HIF-1alpha pathway. Investigative Ophthalmology and Visual Science. 2013;54:4964–4970. doi: 10.1167/iovs.12-11443. 23847321 [DOI] [PubMed] [Google Scholar]
- 46.Ly A., Yee P., Vessey K.A., Phipps J.A., Jobling A.I., Fletcher E.L. Early inner retinal astrocyte dysfunction during diabetes and development of hypoxia, retinal stress, and neuronal functional loss. Investigative Ophthalmology and Visual Science. 2011;52:9316–9326. doi: 10.1167/iovs.11-7879. 22110070 [DOI] [PubMed] [Google Scholar]
- 47.Pouliot M., Talbot S., Senecal J., Dotigny F., Vaucher E., Couture R. Ocular application of the kinin B1 receptor antagonist LF22-0542 inhibits retinal inflammation and oxidative stress in streptozotocin-diabetic rats. PloS One. 2012;7:e33864. doi: 10.1371/journal.pone.0033864. 22470485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reddy V.S., Raghu G., Reddy S.S., Pasupulati A.K., Suryanarayana P., Reddy G.B. Response of small heat shock proteins in diabetic rat retina. Investigative Ophthalmology and Visual Science. 2013;54:7674–7682. doi: 10.1167/iovs.13-12715. 24159092 [DOI] [PubMed] [Google Scholar]
- 49.Du Y., Miller C.M., Kern T.S. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radical Biology and Medicine. 2003;35:1491–1499. doi: 10.1016/j.freeradbiomed.2003.08.018. 14642397 [DOI] [PubMed] [Google Scholar]
- 50.Du Y., Veenstra A., Palczewski K., Kern T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:16586–16591. doi: 10.1073/pnas.1314575110. 24067647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Obrosova I.G., Fathallah L., Greene D.A. Early changes in lipid peroxidation and antioxidative defense in diabetic rat retina: effect of DL-alpha-lipoic acid. European Journal of Pharmacology. 2000;398:139–146. doi: 10.1016/s0014-2999(00)00286-7. 10856458 [DOI] [PubMed] [Google Scholar]
- 52.Kowluru R.A., Kern T.S., Engerman R.L. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. IV. Antioxidant defense system. Free Radical Biology and Medicine. 1997;22:587–592. doi: 10.1016/s0891-5849(96)00347-4. 9013121 [DOI] [PubMed] [Google Scholar]
- 53.Kowluru R.A., Atasi L., Ho Y.S. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Investigative Ophthalmologyand Visual Science. 2006;47:1594–1599. doi: 10.1167/iovs.05-1276. 16565397 [DOI] [PubMed] [Google Scholar]
- 54.Dene B.A., Maritim A.C., Sanders R.A., Watkins J.B., 3rd Effects of antioxidant treatment on normal and diabetic rat retinal enzyme activities. Journal of Ocular Pharmacology and Therapeutics: The Official Journal of the Association for Ocular Pharmacology and Therapeutics. 2005;21:28–35. doi: 10.1089/jop.2005.21.28. 15718825 [DOI] [PubMed] [Google Scholar]
- 55.Brownlee M. The pathobiology of diabetic complications:a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. 15919781 [DOI] [PubMed] [Google Scholar]
- 56.Giacco F., Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. 21030723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishikawa T., Edelstein D., Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney International. 2000;77(Suppl.):S26–S30. doi: 10.1046/j.1523-1755.2000.07705.x. 10997687 [DOI] [PubMed] [Google Scholar]
- 58.Nishikawa T., Edelstein D., Du X.L. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. 10783895 [DOI] [PubMed] [Google Scholar]
- 59.Kowluru R.A., Kowluru V., Xiong Y., Ho Y.S. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radical Biology and Medicine. 2006;41:1191–1196. doi: 10.1016/j.freeradbiomed.2006.01.012. 17015165 [DOI] [PubMed] [Google Scholar]
- 60.Al-Shabrawey M., Rojas M., Sanders T. Role of NADPH oxidase in retinal vascular inflammation. Investigative Ophthalmology and Visual Science. 2008;49:3239–3244. doi: 10.1167/iovs.08-1755. 18378574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kimura S., Zhang G.X., Nishiyama A. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860–866. doi: 10.1161/01.HYP.0000163462.98381.7f. 15824196 [DOI] [PubMed] [Google Scholar]
- 62.Lee S.B., Bae I.H., Bae Y.S., Um H.D. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. Journal of Biological Chemistry. 2006;281:36228–36235. doi: 10.1074/jbc.M606702200. 17015444 [DOI] [PubMed] [Google Scholar]
- 63.Schafer M., Schafer C., Ewald N., Piper H.M., Noll T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circulation Research. 2003;92:1010–1015. doi: 10.1161/01.RES.0000070882.81508.FC. 12690038 [DOI] [PubMed] [Google Scholar]
- 64.Madsen-Bouterse S.A., Kowluru R.A. Oxidative stress and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Reviews in Endocrineand Metabolic Disorders. 2008;9:315–327. doi: 10.1007/s11154-008-9090-4. 18654858 [DOI] [PubMed] [Google Scholar]
- 65.Kowluru R.A., Tang J., Kern T.S. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001;50:1938–1942. doi: 10.2337/diabetes.50.8.1938. 11473058 [DOI] [PubMed] [Google Scholar]
- 66.Kanwar M., Chan P.S., Kern T.S., Kowluru R.A. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Investigative Ophthalmologyand Visual Science. 2007;48:3805–3811. doi: 10.1167/iovs.06-1280. 17652755 [DOI] [PubMed] [Google Scholar]
- 67.Mustata G.T., Rosca M., Biemel K.M. Paradoxical effects of green tea (Camellia sinensis) and antioxidant vitamins in diabetic rats: improved retinopathy and renal mitochondrial defects but deterioration of collagen matrix glycoxidation and cross-linking. Diabetes. 2005;54:517–526. doi: 10.2337/diabetes.54.2.517. 15677510 [DOI] [PubMed] [Google Scholar]
- 68.Kowluru R.A., Kanwar M., Chan P.S., Zhang J.P. Inhibition of retinopathy and retinal metabolic abnormalities in diabetic rats with AREDS-based micronutrients. Archives of Ophthalmology. 2008;126:1266–1272. doi: 10.1001/archopht.126.9.1266. 18779489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Millen A.E., Gruber M., Klein R., Klein B.E., Palta M., Mares J.A. Relations of serum ascorbic acid and alpha-tocopherol to diabetic retinopathy in the Third National Health and Nutrition Examination Survey. American Journal of Epidemiology. 2003;158:225–233. doi: 10.1093/aje/kwg116. 12882944 [DOI] [PubMed] [Google Scholar]
- 70.Millen A.E., Klein R., Folsom A.R., Stevens J., Palta M., Mares J.A. Relation between intake of vitamins C and E and risk of diabetic retinopathy in the Atherosclerosis Risk in Communities Study. American Journal of Clinical Nutrition. 2004;79:865–873. doi: 10.1093/ajcn/79.5.865. 15113727 [DOI] [PubMed] [Google Scholar]
- 71.Lopes de Jesus C.C., Atallah A.N., Valente O., Moca Trevisani V.F. Vitamin C and superoxide dismutase (SOD) for diabetic retinopathy. Journal of Cochrane Database of Systematic Reviews. 2008:1–15. doi: 10.1002/14651858.CD006695.pub2. CD006695 [DOI] [PubMed] [Google Scholar]
- 72.Du X., Matsumura T., Edelstein D. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. Journal of Clinical Investigation. 2003;112:1049–1057. doi: 10.1172/JCI18127. 14523042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nascimento N.R., Lessa L.M., Kerntopf M.R. Inositols prevent and reverse endothelial dysfunction in diabetic rat and rabbit vasculature metabolically and by scavenging superoxide. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:218–223. doi: 10.1073/pnas.0509779103. 16373499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Engerman R.L. Pathogenesis of diabetic retinopathy. Diabetes. 1989;38:1203–1206. doi: 10.2337/diab.38.10.1203. [Pubmed: 2676655] [DOI] [PubMed] [Google Scholar]
- 75.Cai J., Boulton M. The pathogenesis of diabetic retinopathy: Old concepts and new questions. Eye. 2002;16:242–260. doi: 10.1038/sj.eye.6700133. 12032713 [DOI] [PubMed] [Google Scholar]
- 76.Campochiaro P.A. Ocular neovascularization. Journal of Molecular Medicine. 2013;91:311–321. doi: 10.1007/s00109-013-0993-5. 23329331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kietzmann T., Görlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Seminars in Cell and Developmental Biology. 2005;16:474–486. doi: 10.1016/j.semcdb.2005.03.010. 15905109 [DOI] [PubMed] [Google Scholar]
- 78.Wilkinson-Berka J.L., Rana I., Armani R., Agrotis A. Reactive oxygen species, Nox and angiotensin II in angiogenesis: implications for retinopathy. Clinical Science. 2013;124:597–615. doi: 10.1042/CS20120212. [DOI] [PubMed] [Google Scholar]
- 79.Brueckl C., Kaestle S., Kerem A. Hyperoxia-induced reactive oxygen species formation in pulmonary capillary endothelial cells in situ. American Journal of Respiratory Cell and Molecular Biology. 2006;34:453–463. doi: 10.1165/rcmb.2005-0223OC. 16357365 [DOI] [PubMed] [Google Scholar]
- 80.Mori H., Oikawa M., Tamagami T. Oxidized proteins in astrocytes generated in a hyperbaric atmosphere induce neuronal apoptosis. Journal of Alzheimer’s Disease. 2007;11(2):165–174. doi: 10.3233/jad-2007-11204. 17522441 [DOI] [PubMed] [Google Scholar]
- 81.Gerstner B., DeSilva T.M., Genz K. Hyperoxia causes maturation-dependent cell death in the developing white matter. Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2008;28:1236–1245. doi: 10.1523/JNEUROSCI.3213-07.2008. 18234901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jamieson D., Chance B., Cadenas E., Boveris A. The relation of free radical production to hyperoxia. Annual Review of Physiology. 1986;48:703–719. doi: 10.1146/annurev.ph.48.030186.003415. 3010832 [DOI] [PubMed] [Google Scholar]
- 83.Yusa T., Beckman J.S., Crapo J.D., Freeman B.A. Hyperoxia increases H2O2 production by brain in vivo. Journal of Applied Physiology. 1987;63:353–358. doi: 10.1152/jappl.1987.63.1.353. 3624137 [DOI] [PubMed] [Google Scholar]
- 84.Catrina S.B., Okamoto K., Pereira T., Brismar K., Poellinger L. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes. 2004;53:3226–3232. doi: 10.2337/diabetes.53.12.3226. 15561954 [DOI] [PubMed] [Google Scholar]
- 85.Bento C.F., Fernandes R., Matafome P., Sena C., Seica R., Pereira P. Methylglyoxal-induced imbalance in the ratio of vascular endothelial growth factor to angiopoietin 2 secreted by retinal pigment epithelial cells leads to endothelial dysfunction. Experimental Physiology. 2010;95:955–970. doi: 10.1113/expphysiol.2010.053561. 20562294 [DOI] [PubMed] [Google Scholar]
- 86.Ceradini D.J., Yao D., Grogan R.H. Decreasing intracellular superoxide corrects defective ischemia-induced new vessel formation in diabetic mice. Journal of Biological Chemistry. 2008;283:10930–10938. doi: 10.1074/jbc.M707451200. 18227068 [DOI] [PMC free article] [PubMed] [Google Scholar]