

Abstract
Blood–brain barrier disruption represents a key feature in hyperglycaemia-aggravated cerebral damage after an ischaemic stroke. Although the underlying mechanisms remain largely unknown, activation of protein kinase C (PKC) is thought to play a critical role. This study examined whether apoptosis of human brain microvascular endothelial cells (HBMEC) might contribute to hyperglycaemia-evoked barrier damage and assessed the specific role of PKC in this phenomenon. Treatments with hyperglycaemia (25 mM) or phorbol myristate acetate (PMA, a protein kinase C activator, 100 nM) significantly increased NADPH oxidase activity, O2•- generation, proapoptotic protein Bax expression, TUNEL-positive staining and caspase-3/7 activities. Pharmacological inhibition of NADPH oxidase, PKC-a, PKC-ß or PKC-ßI via their specific inhibitors and neutralisation of O2•- by a cell-permeable superoxide dismutase mimetic, MnTBAP normalised all the aforementioned increases induced by hyperglycaemia. Suppression of these PKC isoforms also negated the stimulatory effects of hyperglycaemia on the protein expression of NADPH oxidase membrane-bound components, Nox2 and p22-phox which determine the overall enzymatic activity. Silencing of PKC-ßI gene through use of specific siRNAs abolished the effects of both hyperglycaemia and PMA on endothelial cell NADPH oxidase activity, O2•- production and apoptosis and consequently improved the integrity and function of an in vitro model of human cerebral barrier comprising HBMEC, astrocytes and pericytes. Hyperglycaemia-mediated apoptosis of HBMEC contributes to cerebral barrier dysfunction and is modulated by sequential activations of PKC-ßI and NADPH oxidase.
Keywords: Apoptosis, Endothelial cell, Hyperglycaemia, Protein kinase C, NADPH oxidase
Highlights
-
•
Hyperglycemia disrupts cerebral barrier via protein kinase C-ßI-evoked apoptosis.
-
•
Protein kinase C-ßI promotes oxidative stress through NADPH oxidase activation.
-
•
NADPH oxidase regulates apoptotic pathway by elevating caspase-3/7 activity.
-
•
Like hyperglycemia, induction of protein kinase C perse perturbs barrier function.
-
•
Silencing of protein kinase C-ßI protects cerebral barrier from both pathologies.
Graphical abstract
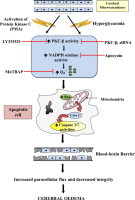
Introduction
Diabetes and acute stress hyperglycaemia increase the rates of mortality and morbidity in patients with ischaemic stroke by exacerbating the breakdown of blood–brain barrier (BBB) [1]. Endothelial cells cover the entire inner surface of all blood vessels and help form the BBB in the central nervous system to prevent the entry of circulating molecules into the brain parenchyma. Anomalies in programmed death (apoptosis) of cells which make up the BBB, especially that of brain microvascular endothelial cells (BMEC), may represent a key phenomenon in hyperglycaemia-evoked BBB disruption. Indeed, the apoptotic effect of hyperglycaemia on endothelial cells derived from large arteries and venous system e.g. aorta and umbilical vein is well-documented [2,3]. Although the mechanisms involved remain unclear, hyperglycaemia is known to elicit cellular injury by increasing the quantities of pro-apoptotic proteins (Bad, Bax and Bid) and possibly by decreasing those of anti-apoptotic bcl-2 proteins (Bcl-2 and bcl-XL) [4]. Sufficient elevations in Bax to Bcl-2 ratio in turn trigger the conversion of inactive procaspase-3 to caspase-3, the major executor of apoptotic process [3].
Hyperglycaemia can also induce apoptosis through stimulation of oxidative stress characterised by the exaggerated synthesis or release of free radicals, in particular superoxide anion (O2•-) [5,6]. Higher concentrations of O2•- may mediate the rates of endothelial cell apoptosis through activation of a variety of redox-sensitive mechanisms including mitochondrial dysfunction, Bax protein overexpression and p38 mitogen-activated protein kinase (p38MAPK) [3,4,7]. As the main source of O2•- in vasculature, NADPH oxidase is anticipated to be intimately involved in BMEC survival/death [8,9].
Hyperglycaemia-mediated activation of protein kinase C (PKC) is regarded as a key step in peripheral and cerebral vasculopathies where activation of different PKC isoforms, particularly PKC-ß is coupled to diabetes-induced endothelial dysfunction and neurovascular complications [10,11]. Besides PKC-ß, activations of other PKC isoforms have also been shown to induce apoptosis through utilisation of different downstream effectors. For instance, stimulation of p38aMAPK by PKC-d and NADPH oxidase-mediated O2•- production by PKC-ßII have been shown to account for hyperglycaemia- and TNF-a-induced apoptosis of bovine retinal pericytes and human umbilical vein endothelial cells, respectively [6,12].
In light of the above, the aims of the current study were three-fold. Firstly, it was to investigate whether and to what extent hyperglycaemia could compromise BMEC apoptosis. Secondly, it was to determine if activation of PKC compromised BMEC viability in a comparable manner. Finally, it was to assess whether inhibition of PKC-ßI might prevent cells from undergoing apoptosis and thus protect BBB integrity.
Materials and methods
Cell culture
Human BMEC (HBMEC) (TCS Cellworks Ltd., Buckingham, UK), between passages 4 and 7, were cultured to almost 70% confluence in its specialised media before exposure to normoglycaemia (NG; 5.5 mM d-glucose), hyperglycaemia (HG; 25 mM d-glucose) or d-mannitol (5.5 mM d-glucose + 19.5 mM d-mannitol) for 72 h. The relevance of individual PKC isoforms to hyperglycaemia-mediated particular outcomes was investigated through simultaneous application of specific inhibitors for PKC-a (Ro-32-0432, 1 µM), PKC-ß (LY333531, 0.05 µM) or PKC-ßII (CGP53353, 1 µM) with hyperglycaemia for 72 h. Potential contribution of O2•- (MnTBAP, 2.5 µM), NADPH oxidase (apocynin, 1 mM), nitric oxide synthase (l-NAME, 100 µM), xanthine oxidase (allopurinol, 100 µM), mitochondrial complex I (rotenone, 2 µM) and cyclooxygenase (indomethacin, 50 µM) to hyperglycaemia-related changes was also studied in similar conditions using specific inhibitors indicated in parentheses.
To ascertain the specific role of PKC activation in the outlined parameters, in some experiments HBMEC were treated with a PKC activator, phorbol myristate acetate (PMA, 0.1 µM) for 4 h.
TUNEL staining
This is performed for detection and quantification of apoptosis at a single cell level, based on in situ labelling of fragmented DNA by the DeadEnd colourimetric TUNEL system (Promega, WI, USA). Apoptotic cells, characterised by dark brown staining, were then counted via light microscopy.
Caspase-3/7 assay
In view of the roles of the caspase family members in mammalian cell apoptosis, the activities of caspase-3/7 were also measured in HBMEC exposed to different experimental conditions using the Apo-ONE homogeneous caspase-3/7 assay kit (Promega). Briefly, onto HBMEC grown in 96-well opaque black plates, 100 µl of diluted caspase-3/7 profluorescent substrate were added. The mixture was incubated at room temperature for 2 h and the enzymatic activities were assessed using a fluorescent plate reader.
Establishment of an in vitro model of human BBB
A triple-culture model of human BBB was set up. For this, human astrocytes were seeded onto the outer surface of Transwell inserts (12 mm diameter, 0.4 µm pore size; Corning Costar, High Wycombe, UK) directed upside down. On the following day, the inserts were inverted the correct way and placed into 12-well culture plates containing human pericytes. HBMEC were then seeded onto the inner side of the inserts before culturing all cell layers to confluence.
Analyses of the BBB characteristics
The integrity and function of the BBB were evaluated as previously described by transendothelial electrical resistance and the flux of high (Evan's Blue-labelled albumin, 67 kDa) and low (sodium fluorescein, 376 Da) molecular weight permeability markers, respectively [13].
Western blotting
Equal amounts of total cellular proteins (~40 µg) were run on 10–15% sodium dodecyl sulphate-polyacrylamide gels before transferring onto polyvinylidene fluoride membranes and successively incubating with a combination of ß-actin (reference protein, Sigma Aldrich, Poole, UK) and anti-Bax (Cell Signalling Technology, Danvers, MA, USA), Nox2, PKC-ßI (Santa Cruz, Dallas, Texas, USA) or p22-phox (Abcam, Cambridge, UK) primary antibodies and appropriate infrared dye-conjugated secondary antibodies (Li-Cor Biosci, Cambridge, UK). Blots were analysed by densitometry using the Li-Cor Odyssey infrared imaging system.
Measurements of O2- production and NADPH oxidase activity
The level of O2•- production was measured by cytochrome C reduction as previously described [8,14]. In brief, cell pellets were sonicated in cold lysis buffer containing 20 mM HEPES buffer (pH 7.2), 1 mM EGTA, 210 mM mannitol and 70 mM sucrose. Equal amounts of homogenate (50 µl) were then incubated with 50 µM cytochrome C for 60 min at 37 °C. O2•- generation was measured as the reduction of cytochrome C and monitored as the change in absorbance at 550 nm using a FLUOstar Omega plate reader (BMG, Aylesbury, UK).
NAD(P)H oxidase activity was measured by the lucigenin chemiluminescence assay. HBMEC homogenates (50 µl) were incubated at 37 °C with assay buffer (50 mM potassium phosphate buffer (pH 7.0), 1 mM EGTA, 150 mM sucrose, and 5 µM lucigenin) containing the specific inhibitors of enzymes that are known to generate reactive oxygen species (ROS), namely nitric oxide synthase (l-NAME, 100 µM), xanthine oxidase (allopurinol, 100 µM), mitochondrial complex I (rotenone, 50 µM) and cyclooxygenase (indomethacin, 50 µM). After 15 min NADPH (100 µM; Sigma Aldrich, Poole, UK) was added to initiate the reaction. The reaction was monitored every minute for 4 h and the rate of reaction calculated. Buffer blanks were also run for both assays and subtracted from the data.
Small interfering RNA knockdown
Semi-confluent HBMEC were transfected for 24 h with DharmaFECT small interfering RNA (siRNA) transfection reagent 4 containing 50 nM of ON-TARGET plus SMART pool human siRNA against PKC-ßI (Thermo Scientific Dharmacon, Lafayette, CO, USA). HBMEC transfected with non-targeting pool of siRNA served as controls. After exposure to different experimental conditions, HBMEC were harvested for different assays.
Statistical analysis
Data are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism 6.0 statistical software package. Data were analysed by nonparametric Mann–Whitney U test or one-way ANOVA followed by Dunnett's post-hoc analyses, where appropriate. P < 0.05 was considered significant.
Results
Hyperglycaemia evokes oxidative stress in endothelial cells via activation of NADPH oxidase
Exposure of HBMEC to hyperglycaemia led to a significant increase in total O2•- generation which was normalised by specific inhibition of NADPH oxidase, PKC-a, PKC-ß and PKC-ßII but not those of nitric oxide synthase, mitochondrial complex I, cyclooxygenase or xanthine oxidase. Treatment of HBMEC with a potent free radical scavenger, MnTBAP almost completely abolished the hyperglycaemia-evoked O2•- generation. Inhibition of the aforementioned PKC isoforms negated hyperglycaemia-mediated increases observed in protein expressions of NADPH oxidase membrane-bound components, namely p22-phox and Nox2 which determine enzymatic activity and stability as a whole (Fig. 1A–C).
Fig. 1.
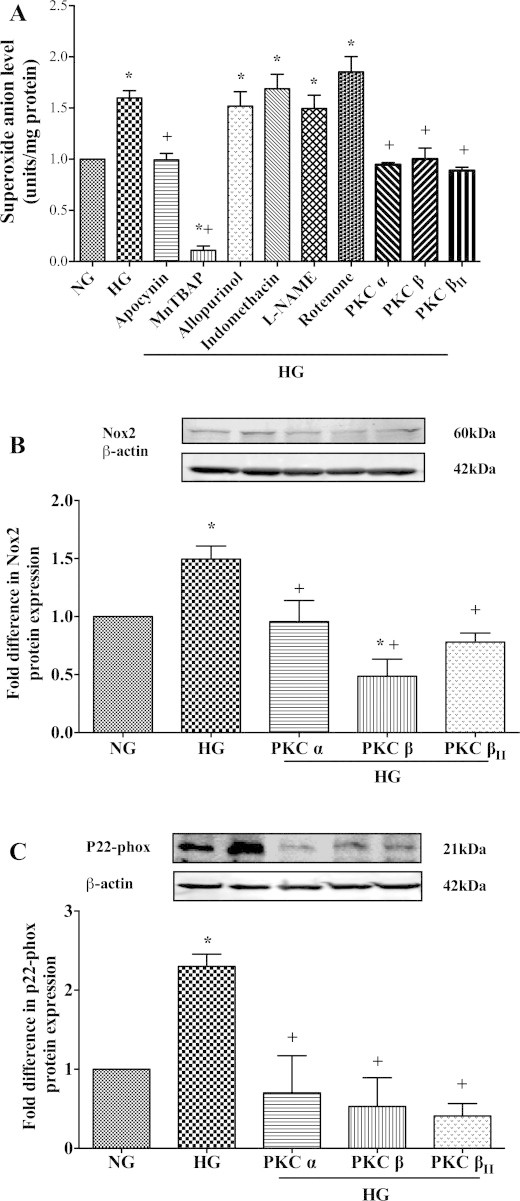
(A) Effects of specific inhibitors targeting the main free radical-generating enzymes and protein kinase C (PKC)-a, PKC-ß and PKC-ßII isoforms on O2•- production (n = 3) in human brain microvascular endothelial cells (HBMEC) subjected to hyperglycaemia (HG; 25 mM d-glucose) for 72 h. Effects of inhibitors for PKC-a, PKC-ß and PKC-ßII on gp91-phox(n = 3) (B) and p22-phox (n = 3) (C) protein expressions in HBMEC exposed to HG. Data are expressed as mean ± SEM. *P < 0.05 versus normoglycaemia (NG), +P < 0.05 versus HG.
Hyperglycaemia promotes endothelial cell apoptosis through NADPH oxidase and PKC activations
Hyperglycaemia significantly increased TUNEL-positive staining (DNA fragmentation), caspase-3/7 activities and Bax protein expression in HBMEC which appeared to be independent of hyperglycaemia-mediated increases in osmolality. Inhibition of NADPH oxidase, PKC-a, PKC-ß and PKC-ßII and neutralisation of O2•- effectively suppressed all the increases observed in the apoptotic parameters. However, reductions in DNA fragmentation rates stayed significantly higher in MnTBAP-treated cells compared to controls (Fig. 2A–E).
Fig. 2.
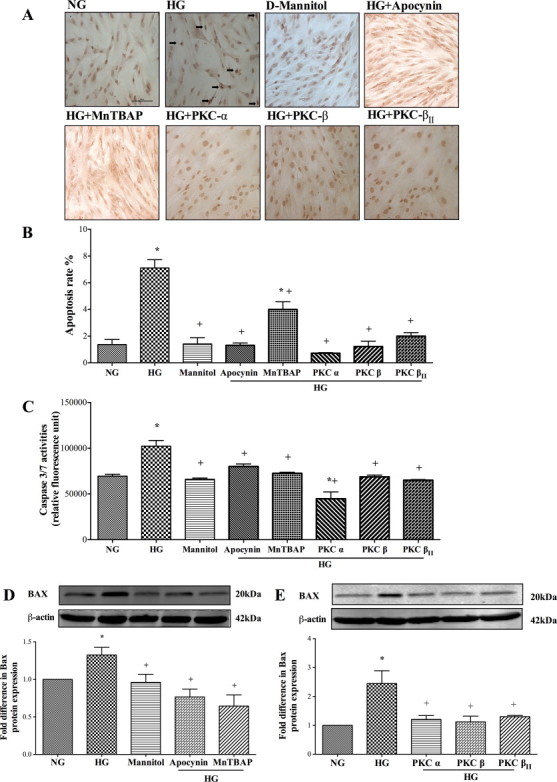
Effects of inhibitors for NADPH oxidase (apocynin), superoxide anion (MnTBAP) and protein kinase C-a (PKC-a), PKC-ß and PKC-ßII on (A) TUNEL staining (Bar, 100 µm), (B) DNA fragmentation rates (n = 3), (C) caspase-3/7 activities (n = 3), (D, E) Bax protein expressions (n = 4 and 3) in human brain microvascular endothelial cells (HBMEC) exposed to hyperglycaemia (HG; 25 mM d-glucose) for 72 h. Data are expressed as mean ± SEM. *P < 0.05 versus normoglycaemia (NG), +P < 0.05 versus HG.
Induction of PKC promotes endothelial cell apoptosis
To determine whether elevations in PKC activity during a hyperglycaemic insult may constitute the main cause of apoptosis, in some experiments the PKC activity was increased by exposing HBMEC to increasing concentrations of PMA (0.1–1 µM) for 2–4 h. As these revealed time-dependent increases in total PKC activity, in subsequent studies the cells were treated with 0.1 µM of PMA for 4 h which produced significant increases in PKC-ßI activity and all apoptotic parameters i.e. DNA fragmentation rates, caspase-3/7 activities and Bax protein expression (Fig. 3A–E). Silencing of PKC-ßI gene dramatically diminished its protein expression and attenuated both hyperglycaemia- and PMA-evoked increases observed in NADPH oxidase activity and O2•- production (Fig. 4A–E).
Fig. 3.
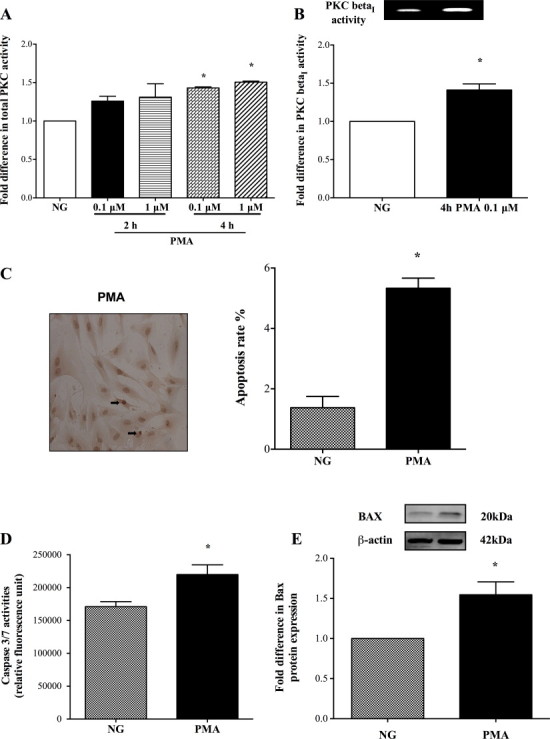
Effect of phorbol myristate acetate (PMA; 0.1–1 µM) on total protein kinase C (PKC) (n = 4) (A) and PKC-ßI(n = 4) (B) activities in human brain microvascular endothelial cells (HBMEC). Effect of PMA (4 h, 100 nM) on HBMEC (C) DNA fragmentation (n = 3), (D) caspase-3/7 activities (n = 4) and (E) Bax protein expression (n = 4). Data are expressed as mean ± SEM. *P < 0.05 versus normoglycaemia (NG).
Fig. 4.
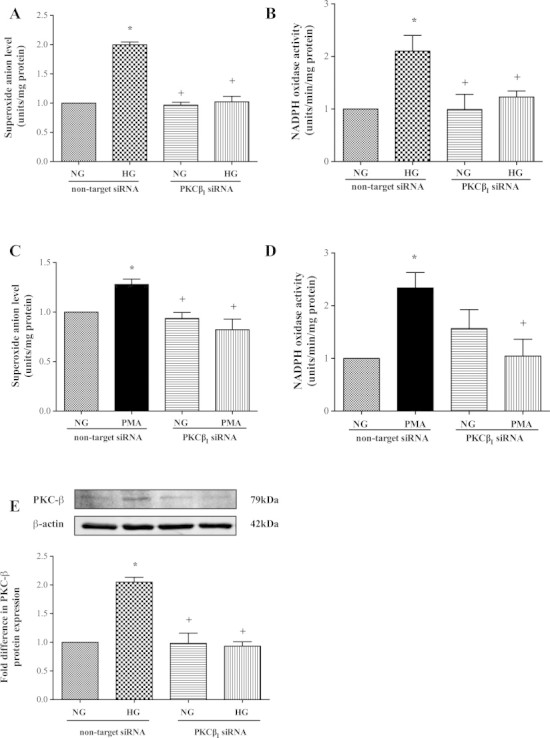
Knockdown of protein kinase C (PKC)-ßI gene normalises the effects of hyperglycaemia (HG) (A, B) (n = 3 and 4) and PMA (C, D) (both n = 3) on superoxide anion generation and NADPH oxidase activity and reduces PKC-ßI protein expression (n = 3) (E) in human brain microvascular endothelial cells. Data are expressed as mean ± SEM. *P < 0.05 versus normoglycaemia (NG), +P < 0.05 versus HG or PMA (4 h, 100 nM).
Suppression of PKC-ßI prevents apoptosis and protects cerebral barrier
Knockdown of PKC-ßI also effectively suppressed both hyperglycaemia-induced apoptosis as evidenced by radical decreases observed in all apoptotic parameters (Fig. 5A–D). Although silencing of PKC-ßI gene neutralised PMA-mediated increases in apoptosis rate and Bax protein expression, it failed to normalise caspase-3/7 activities (Fig. 6A–D). PKC-ßI knockdown also negated the deleterious effects of both hyperglycaemia and PMA on cerebral barrier integrity and function as proven by marked increases in transendothelial electrical resistance and concurrent decreases in paracellular flux of Evan's Blue-labelled albumin and sodium fluorescein, respectively (Fig. 7A–C).
Fig. 5.
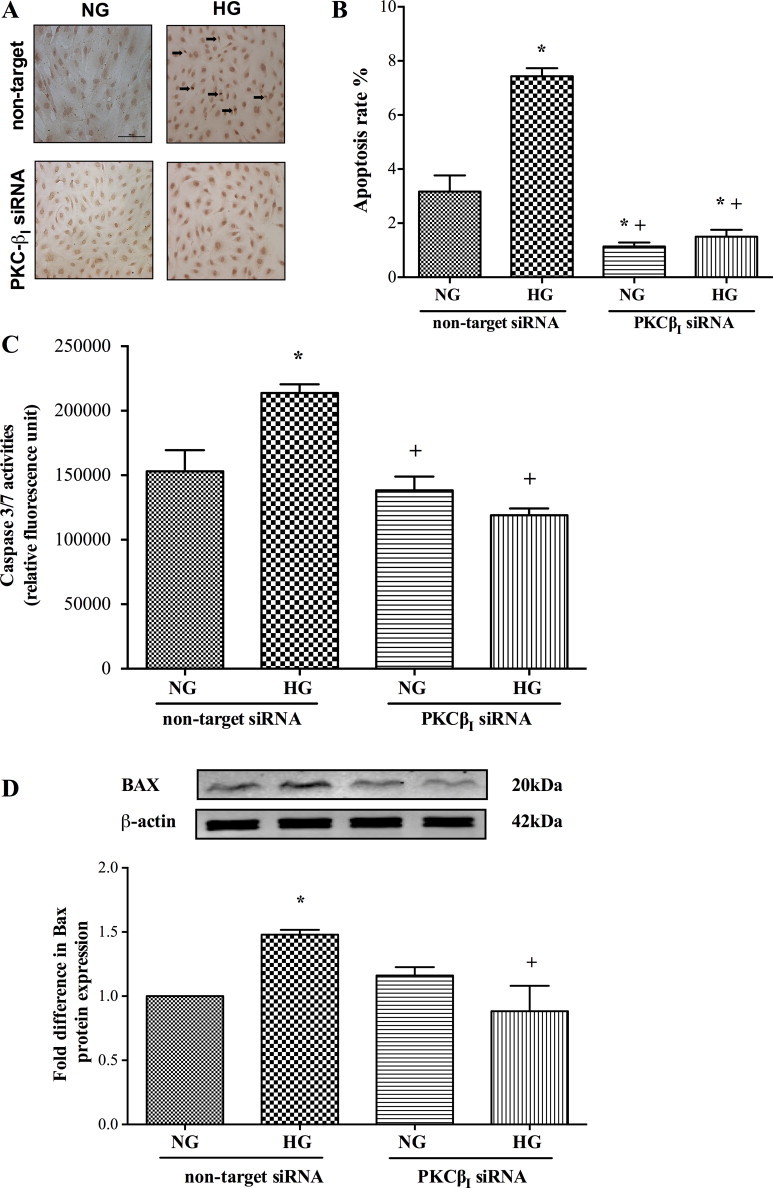
Effect of protein kinase C-ßI (PKC-ßI) gene knockdown on TUNEL staining (Bar, 100 µm) (A), DNA fragmentation rates (n = 3) (B), caspase-3/7 activities (n = 6) (C) and Bax protein expression (n = 3) (D) in human brain microvascular endothelial cells exposed to hyperglycaemia (HG). Data are expressed as mean ± SEM. *P < 0.05 versus normoglycaemia (NG), +P < 0.05 versus HG.
Fig. 6.
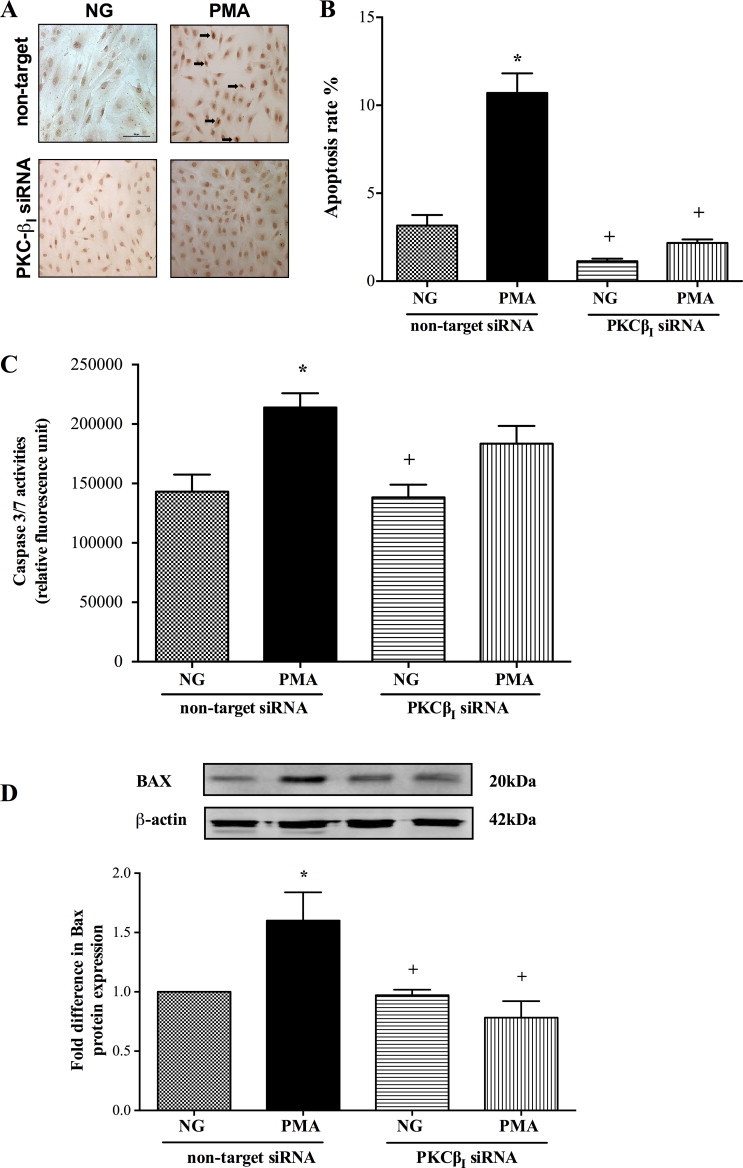
Effect of protein kinase C-ßI (PKC-ßI) gene silencing on TUNEL staining (Bar, 100 µm) (A), DNA fragmentation rates (n = 3) (B), caspase-3/7 activities (n = 6) (C) and Bax protein expression (n = 3) (D) in human brain microvascular endothelial cells exposed to PMA (100 nM) for 4 h. Data are expressed as mean ± SEM. *P < 0.05 versus normoglycaemia (NG), +P < 0.05 versus PMA.
Fig. 7.
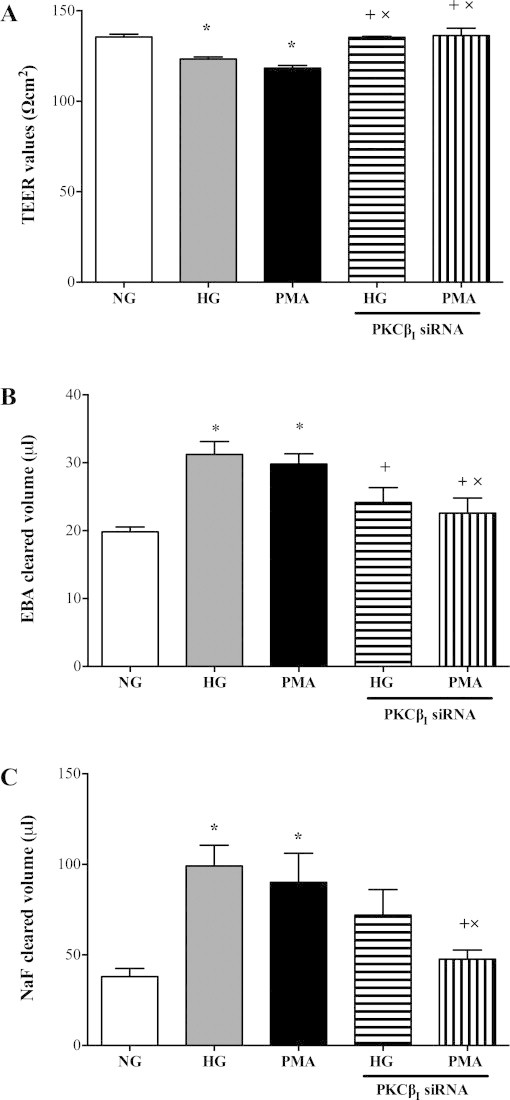
Effects of protein kinase C-ßI (PKC-ßI) gene knockdown on (A) transendothelial electrical resistance (TEER) (n = 3) and paracellular flux of high and low molecular weight permeability markers, namely Evan's Blue-labelled albumin (n = 3) (B) and sodium fluorescein (n = 3) (C) across an in vitro model of human blood–brain barrier composed of human brain microvascular endothelial cells, astrocytes and pericytes in the absence or presence of PMA (100 nM, 4 h) or hyperglycaemia (HG; 25 mM d-glucose). Data are expressed as mean ± SEM from at least three different experiments. *P < 0.05 versus normoglycaemia (NG), +P < 0.05 versus HG, xP < 0.05 versus PMA.
Discussion
Increased prevalence of BBB failure in stroke patients with diabetes bestows a pivotal role on hyperglycaemia in the pathogenesis of this defect. However, the mechanisms involved remain ambiguous and require in depth investigation in order to devise efficacious novel therapies to prevent barrier leakage. Using an in vitro model of human cerebral barrier, we have recently shown that hyperglycaemia compromises the structural and functional capacities of BBB through a mechanism involving concurrent activations of PKC-ß and NADPH oxidase [14]. In light of this, the present study investigated whether putative increases in apoptosis of BMEC, the most prominent cellular component of the cerebral barrier, may also contribute to the hyperglycaemia-evoked BBB dysfunction and is affected by the activities of PKC and/or NADPH oxidase.
Accumulating evidence has displayed hyperglycaemia-mediated apoptosis as one of the key mediators of vascular complications, such as retinopathy, to which induction of NF-?B, c-jun NH2 terminal kinase, shp-1 and PKC-d appear to contribute [12,15]. However, given that endothelial cells derive from different parts of the vascular tree respond to the effects of hyperglycaemia in a different manner, it was important to ascertain whether and to what extent hyperglycaemia alters BMEC viability and reveal the mechanisms involved [16]. To this end, the present study shows that hyperglycaemia evokes significant increases in HBMEC apoptosis as evidenced by increases in DNA fragmentation rates, caspase-3/7 activities and in expression of proapoptotic protein Bax. Marked increases in Bax or Bax/Bcl-2 ratio have previously been linked to hyperglycaemia-mediated apoptosis of human aortic endothelial cells [3]. Hyperglycaemia-evoked translocation of Bax protein into mitochondrial membrane leads to disruption of mitochondrial membrane integrity thereby facilitating the escape of cytochrome C from mitochondria and consequent activation of caspase-9 [3,17]. Caspase-9, in turn, activates several downstream caspases amongst which caspase-3 and caspase-7 were shown to be of particular importance in HBMEC.
Oxidative stress, associated with excessive availability of O2•- may account for hyperglycaemia-evoked apoptosis. Using specific inhibitors of the major prooxidant enzymes, the current study has shown NADPH oxidase as the main source of O2•- in hyperglycaemic endothelial cells. Indeed, specific inhibition of this oxidase protected HBMEC from apoptosis as evidenced by marked decreases in all apoptotic parameters. Interestingly, despite almost completely eradicating the availability of O2•-, MnTBAP, a cell-permeable superoxide dismutase mimetic failed to normalise HG-mediated elevations in DNA fragmentation rates. Taken together, these data ascribe additional benefits to inhibition of vascular NADPH oxidase beyond its O2•--related effects.
NADPH oxidases make up a dedicated family of O2•--forming enzymes. In general, they are activated by coupling of Nox2, the catalytic subunit, with other subunits, p22-phox, p47-phox, p40-phox and p67-phox. Although seven isoforms of Nox have been identified to date, only Nox1, Nox2, Nox4 and Nox5 are known to be expressed in vascular cells [18,19]. In light of our past and present studies proving Nox2-derived O2•- as the key regulator of blood–brain barrier integrity, endothelial function and microvascular endothelial cell growth, we specifically focused on this particular isoform in the current study [20–23]. Discovery of considerably smaller cerebral infarcts in Nox2-deficient mice subjected to middle cerebral artery occlusion further corroborate the correlation between Nox2 availability and cerebral homeostasis [24].
Despite constituting the main Nox isoform in colon epithelial cells, Nox1 is also associated with production of low levels of O2•- in vasculature [25,26]. However, through a complex reaction involving concomitant induction of PKC-, MAPK- and PKA-dependent mechanisms, the vascular pathologies appear to elevate Nox1-mediated release of O2•- [27–29] which in turn may trigger BMEC apoptosis to elicit barrier permeability. In this context, the hyperglycaemia-evoked apoptosis of a murine BMEC line, bEnd3 has recently been attributed to NF-?B-dependent upregulation of p22-phox and Nox1 isoforms. However, negation of apoptosis by agents that inhibit the activity of NADPH oxidase complex, namely apocynin and resveratrol, a polyphenolic antioxidant suggest the involvement of other Nox isoforms, in particular Nox2, in this phenomenon [30].
Unlike other isoforms of Nox, Nox4 predominantly generates H2O2 [31]. As it is mainly implicated in cellular senescence and vascular and renal complications of diabetes, its inhibition, in various models of diabetes, has unsurprisingly led to reduced oxidative stress, inflammation and availability of profibrotic markers and thus protected renovascular function as evidenced by attenuation of albuminuria and glomerular macrophage infiltration [32–36]. Conversely, other studies reported endogenous Nox4 as a pivotal stabiliser of vascular integrity owing to its anti-apoptotic effects which seem to be mediated, in part, by activation of akonitase in various cell types including microvascular endothelial cells exposed to diabetic, ischaemic or inflammatory stress [37–41]. Considering the alleged protective effects exerted by Nox4 itself, it is unlikely that its inhibition can contribute to apocynin-mediated BBB protection observed in this study.
In contrast to other Noxs, Nox5 does not require p22-phox for activation and is regulated in a Ca2+-sensitive manner [42,43]. Although the biological importance of vascular Nox5 is largely unknown, its involvement in endothelial cell proliferation and oxidative stress-mediated atherosclerotic damage imply a putative role for this isoform diabetic cerebral barrier injury [44,45].
Hyperglycaemia-mediated activation of PKC represents a key step in vasculopathies and is documented in organs that display hyperglycaemia-mediated vascular leakage such as heart and retina [46,47]. In keeping with these findings, stimulation of total PKC activity in HBMEC (by PMA) increased the rates of apoptosis in this study and consequently compromised the integrity and function of an in vitro model of human BBB. Time-dependent increases in the expression and activities of specific PKC isoforms have previously been correlated with the severity of diabetic microvascular complications, endothelial barrier breakdown and apoptosis during cardiac ischaemia [48–50]. Naturally, suppression of PKC-a, PKC-ß or PKC-ßII in the current study prevented BMEC apoptosis. This appeared to be modulated in part by curtailment of NADPH oxidase activity deriving from reductions in its p22-phox and Nox2 subunits, membrane-bound components that determine oxidase activity and stability and O2•- bioavailability. Decreased phosphorylation of cytosolic components, notably p47-phox stemming from PKC inhibition may also contribute to the diminished oxidase activity [14].
Increases in PKC-ß and PKC-ßII activities have been shown to account for much of the elevated total PKC activity in hyperglycaemic HBMEC. However, despite abating total PKC activity, the inhibition of PKC-ßII failed to improve BBB integrity and conferred a critical role on PKC-ßI in this process and possibly in apoptosis [14]. Indeed, silencing of PKC-ßI gene in HBMEC supported this notion by radically diminishing the levels of all apoptotic parameters in cells subjected to hyperglycaemia. Concurrent suppressions of NADPH oxidase activity and O2•- release in the same cells reveal that PKC-ßI acts upstream to NADPH oxidase and regulation of oxidative stress is a crucial factor in its barrier-protective effects. Since knockdown of PKC-ßI failed to normalise the PMA-evoked increases observed in caspase-3/7 activities despite preventing DNA fragmentation and Bax expression in an efficacious fashion, it is likely that other PKC isoforms and associated downstream mechanisms possibly involving other Noxs may also contribute to the regulation of endothelial cell apoptosis. In support of this notion, hyperglycaemia- and PMA-induced production of O2•- in human lung microvascular endothelial cells has recently been ascribed to PKC-a-evoked activation of Nox5. Interestingly, while genetic silencing of PKC-e also reduced PMA-stimulated Nox5 activity in these cells, suppression of PKC-d elevated activity [51]. Furthermore, activation of PKC-? by disturbed blood flow has been shown to induce endothelial apoptosis by modulating p53 and thus contribute to the early events in atherosclerosis [52].
It is possible that other mechanisms such as hyperglycaemia-induced activation of matrix metalloproteinases (MMPs) may also be responsible for BMEC apoptosis similar to this reported in diabetic mice retinas [53]. Given that hyperglycaemia substantially increases MMP-2 release in HBMEC through a process involving both NADPH oxidase and PKC-ß, this hypothesis warrants further investigation [14]. Activation of small GTP-binding protein RhoA by high glucose levels may also be implicated in hyperglycaemia-evoked HBMEC apoptosis due to its roles in cytoskeletal reorganisation, proapoptotic Bax expression and pro-survival phosphatidylinositol-3-kinase/Akt/endothelial nitric oxide synthase pathway [54,55].
In conclusion, the present study reveals that hyperglycaemia perturbs BBB integrity, in part, through promotion of HBMEC apoptosis by sequential activations of PKC and NADPH oxidase where targeting of PKC-ßI may be of therapeutic value. Studies with PMA have revealed that activation of PKC per se compromise endothelial cell viability and cerebral barrier integrity in equal measures and therefore justify targeting of its specific isoform(s) as efficacious therapeutic options in negating hyperglycaemic barrier damage. Exploration of growth profile of cells that make up the BBB, notably HBMEC, astrocytes and pericytes in normal versus hyperglycaemic conditions would be beneficial in unravelling the precise contribution of each cell line to hyperglycaemia-mediated BBB hyperpermeability.
Conflicts of interest
B.S. performed the study, collected and analysed the data and wrote the first draft of the manuscript. U.B. designed and supervised the study, interpreted the data and wrote the manuscript. The authors have no conflict of interest to declare in relation to this manuscript.
Sources of funding
This study was supported by a Ph.D. studentship grant to Dr. Bayraktutan.
References
- 1.Baird T.A., Parsons M.W., Barber P.A. The influence of diabetes mellitus and hyperglycaemia on stroke incidence and outcome. Journal of Clinical Neuroscience. 2002;9:618–626. doi: 10.1054/jocn.2002.1081. 12604269 [DOI] [PubMed] [Google Scholar]
- 2.Du X.L., Sui G.Z., Stockklauser-Färber K. Induction of apoptosis by high proinsulin and glucose in cultured human umbilical vein endothelial cells is mediated by reactive oxygen species. Diabetologia. 1998;41:249–256. doi: 10.1007/s001250050900. 9541163 [DOI] [PubMed] [Google Scholar]
- 3.Nakagami H., Morishita R., Yamamoto K. Phosphorylation of p38 mitogen-activated protein kinase downstream of bax-caspase-3 pathway leads to cell death induced by high D-glucose in human endothelial cells. Diabetes. 2001;50:1472–1481. doi: 10.2337/diabetes.50.6.1472. 11375350 [DOI] [PubMed] [Google Scholar]
- 4.Yang Z., Mo X., Gong Q. Critical effect of VEGF in the process of endothelial cell apoptosis induced by high glucose. Apoptosis. 2008;13:1331–1343. doi: 10.1007/s10495-008-0257-y. [DOI] [PubMed] [Google Scholar]
- 5.Cifarelli V., Geng X., Styche A., Lakomy R., Trucco M., Luppi P. C-peptide reduces high-glucose-induced apoptosis of endothelial cells and decreases NAD(P)H-oxidase reactive oxygen species generation in human aortic endothelial cells. Diabetologia. 2011;54:2702–2712. doi: 10.1007/s00125-011-2251-0. [DOI] [PubMed] [Google Scholar]
- 6.Deng B., Xie S., Wang J., Xia Z., Nie R. Inhibition of protein kinase C ß2 prevents tumor necrosis factor-a-induced apoptosis and oxidative stress in endothelial cells: the role of NADPH oxidase subunits. Journal of Vascular Research. 2012;49:144–159. doi: 10.1159/000332337. [DOI] [PubMed] [Google Scholar]
- 7.van den Oever I.A., Raterman H.G., Nurmohamed M.T., Simsek S. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediators of Inflammation. 2010;2010:792393. doi: 10.1155/2010/792393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen C.L., Bayraktutan U. Antioxidants attenuate hyperglycaemia-mediated brain endothelial cell dysfunction and blood–brain barrier hyperpermeability. Diabetes, Obesity and Metabolism. 2009;11:480–490. doi: 10.1111/j.1463-1326.2008.00987.x. [DOI] [PubMed] [Google Scholar]
- 9.Bayraktutan U., Draper N., Lang D., Shah A.M. Expression of a functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovascular Research. 1998;38:256–262. doi: 10.1016/s0008-6363(98)00003-0. [DOI] [PubMed] [Google Scholar]
- 10.Beckman J.A., Goldfine A.B., Gordon M.B., Garrett L.A., Creager M.A. Inhibition of protein kinase Cbeta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circulation Research. 2002;90:107–111. doi: 10.1161/hh0102.102359. [DOI] [PubMed] [Google Scholar]
- 11.Cipolla M.J., Huang Q., Sweet J.G. Inhibition of protein kinase Cß reverses increased blood–brain barrier permeability during hyperglycemic stroke and prevents edema formation in vivo. Stroke. 2011;42:3252–3257. doi: 10.1161/STROKEAHA.111.623991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geraldes P., Hiraoka-Yamamoto J., Matsumoto M. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nature Medicine. 2009;15:1298–1306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen C., Srivastava K., Bayraktutan U. Small GTPase RhoA and its effector Rho kinase mediate oxygen glucose deprivation-evoked in vitro cerebral barrier dysfunction. Stroke. 2010;41:2056–2063. doi: 10.1161/STROKEAHA.109.574939. [DOI] [PubMed] [Google Scholar]
- 14.Shao B., Bayraktutan U. Hyperglycaemia promotes cerebral barrier dysfunction through activation of protein kinase C-ß. Diabetes, Obesity and Metabolism. 2013;15:993–999. doi: 10.1111/dom.12120. [DOI] [PubMed] [Google Scholar]
- 15.Ho F.M., Lin W.W., Chen B.C. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-?B and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cellular Signalling. 2006;18:391–399. doi: 10.1016/j.cellsig.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Kapitulnik J., Benaim C., Sasson S. Endothelial cells derived from the blood–brain barrier and islets of Langerhans differ in their response to the effects of bilirubin on oxidative stress under hyperglycemic conditions. Frontiers in Pharmacology. 2012;3:131. doi: 10.3389/fphar.2012.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 18.Leto T.L., Morand S., Hurt D., Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxidants and Redox Signaling. 2009;11:2607–2619. doi: 10.1089/ars.2009.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petry A., Weitnauer M., Görlach A. Receptor activation of NADPH oxidases. Antioxidants and Redox Signaling. 2010;13:467–487. doi: 10.1089/ars.2009.3026. [DOI] [PubMed] [Google Scholar]
- 20.Rakkar K., Srivastava K., Bayraktutan U. Attenuation of urokinase activity during experimental ischaemia protects the cerebral barrier from damage through regulation of matrix metalloproteinase-2 and NAD(P)H oxidase. European Journal of Neuroscience. 2014 doi: 10.1111/ejn.12552. [DOI] [PubMed] [Google Scholar]
- 21.Bayraktutan U. Nitric oxide synthase and NAD(P)H oxidase modulate coronary endothelial cell growth. Journal of Molecular and Cellular Cardiology. 2004;36:277–286. doi: 10.1016/j.yjmcc.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Bayraktutan U. Coronary microvascular endothelial cell growth regulates expression of the gene encoding p22-phox. Free Radical Biology and Medicine. 2005;39:1342–1352. doi: 10.1016/j.freeradbiomed.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 23.Demirci B., McKeown P.P., Bayraktutan U.B. The bimodal regulation of vascular function by superoxide anion: role of endothelium. BMB Reports. 2008;41:223–229. doi: 10.5483/bmbrep.2008.41.3.223. [DOI] [PubMed] [Google Scholar]
- 24.Chen H., Song Y.S., Chan P.H. Inhibition of NADPH oxidase is neuroprotective after ischemia–reperfusion. Journal of Cerebral Blood Flow and Metabolism. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nisimoto Y., Tsubouchi R., Diebold B.A. Activation of NADPH oxidase 1 in tumour colon epithelial cells. Biochemical Journal. 2008;415:57–65. doi: 10.1042/BJ20080300. [DOI] [PubMed] [Google Scholar]
- 26.Muzaffar S., Shukla N., Bond M. Exogenous hydrogen sulfide inhibits superoxide formation, NOX-1 expression and Rac1 activity in human vascular smooth muscle cells. Journal of Vascular Research. 2008;45:521–528. doi: 10.1159/000129686. [DOI] [PubMed] [Google Scholar]
- 27.Kroviarski Y., Debbabi M., Bachoual R. Phosphorylation of NADPH oxidase activator 1 (NOXA1) on serine 282 by MAP kinases and on serine 172 by protein kinase C and protein kinase A prevents NOX1 hyperactivation. FASEB Journal. 2010;24:2077–2092. doi: 10.1096/fj.09-147629. [DOI] [PubMed] [Google Scholar]
- 28.Dutta S., Rittinger K. Regulation of NOXO1 activity through reversible interactions with p22phox and NOXA1. PLoS ONE. 2010;5:e10478. doi: 10.1371/journal.pone.0010478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivera J., Sobey C.G., Walduck A.K., Drummond G.R. Nox isoforms in vascular pathophysiology: insights from transgenic and knockout mouse models. Redox Report. 2010;15:50–63. doi: 10.1179/174329210X12650506623401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen F., Qian L.H., Deng B., Liu Z.M., Zhao Y., Le Y.Y. Resveratrol protects vascular endothelial cells from high glucose-induced apoptosis through inhibition of NADPH oxidase activation-driven oxidative stress. CNS Neuroscience and Therapeutics. 2013;19:675–681. doi: 10.1111/cns.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu R.F., Ma Z., Liu Z., Terada L.S. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Molecular and Cellular Biology. 2010;30:3553–3568. doi: 10.1128/MCB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lener B., Koziel R., Pircher H. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochemical Journal. 2009;423:363–374. doi: 10.1042/BJ20090666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schilder Y.D., Heiss E.H., Schachner D. NADPH oxidases 1 and 4 mediate cellular senescence induced by resveratrol in human endothelial cells. Free Radical Biology and Medicine. 2009;46:1598–1606. doi: 10.1016/j.freeradbiomed.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 34.Jha J.C., Gray S.P., Barit D. Genetic targeting or pharmacologic inhibition of NADPH oxidase Nox4 provides renoprotection in long-term diabetic nephropathy. Journal of the American Society of Nephrology. 2014 doi: 10.1681/ASN.2013070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manea A., Tanase L.I., Raicu M., Simionescu M. JAK/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30:105–112. doi: 10.1161/ATVBAHA.109.193896. [DOI] [PubMed] [Google Scholar]
- 36.Schröder K. Isoform specific functions of Nox protein-derived reactive oxygen species in the vasculature. Current Opinion in Pharmacology. 2010;10:122–126. doi: 10.1016/j.coph.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 37.Datla S.R., Peshavariya H., Dusting G.J., Mahadev K., Goldstein B.J., Jiang F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:2319–2324. doi: 10.1161/ATVBAHA.107.149450. [DOI] [PubMed] [Google Scholar]
- 38.Basuroy S., Tcheranova D., Bhattacharya S., Leffler C.W., Parfenova H. Nox4 NADPH oxidase-derived reactive oxygen species, via endogenous carbon monoxide, promote survival of brain endothelial cells during TNF-a-induced apoptosis. American Journal of Physiology—Cell Physiology. 2011;300:C256–C265. doi: 10.1152/ajpcell.00272.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edderkaoui M., Nitsche C., Zheng L., Pandol S.J., Gukovsky I., Gukovskaya A.S. NADPH oxidase activation in pancreatic cancer cells is mediated through Akt-dependent up-regulation of p22phox. Journal of Biological Chemistry. 2011;286:7779–7787. doi: 10.1074/jbc.M110.200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee J.K., Edderkaoui M., Truong P. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology. 2007;133:1637–1648. doi: 10.1053/j.gastro.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 41.Wang J., Hong Z., Zeng C., Yu Q., Wang H. NADPH oxidase 4 promotes cardiac microvascular angiogenesis after hypoxia/reoxygenation in vitro. Free Radical Biology and Medicine. 2014;69:278–288. doi: 10.1016/j.freeradbiomed.2014.01.027. [DOI] [PubMed] [Google Scholar]
- 42.Serrander L., Jaquet V., Bedard K. NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie. 2007;89:1159–1167. doi: 10.1016/j.biochi.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Jagnandan D., Church J.E., Banfi B., Stuehr D.J., Marrero M.B., Fulton D.J. Novel mechanism of activation of NADPH oxidase5. Calcium sensitization via phosphorylation. Journal of Biological Bhemistry. 2007;282:6494–6507. doi: 10.1074/jbc.M608966200. [DOI] [PubMed] [Google Scholar]
- 44.Jay D.B., Papaharalambus C.A., Seidel-Rogol B., Dikalova A.E., Lassègue B., Griendling K.K. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radical Biology and Medicine. 2008;45:329–335. doi: 10.1016/j.freeradbiomed.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schulz E., Münzel T. NOX5, a new "radical" player in human atherosclerosis? Journal of the American College of Cardiology. 2008:1810–1812. doi: 10.1016/j.jacc.2008.08.040. [DOI] [PubMed] [Google Scholar]
- 46.Inoguchi T., Battan R., Handler E., Sportsman J.R., Heath W., King G.L. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proceedings of the National Academy of Sciences of the USA. 1992;89:11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shiba T., Inoguchi T., Sportsman J.R., Heath W.F., Bursell S., King G.L. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. American Journal of Physiology. 1993;265:E783–E793. doi: 10.1152/ajpendo.1993.265.5.E783. [DOI] [PubMed] [Google Scholar]
- 48.Wei L., Sun D., Yin Z. A PKC-beta inhibitor protects against cardiac microvascular ischemia reperfusion injury in diabetic rats. Apoptosis. 2010;15:488–498. doi: 10.1007/s10495-009-0439-2. [DOI] [PubMed] [Google Scholar]
- 49.Aiello L.P., Clermont A., Arora V., Davis M.D., Sheetz M.J., Bursell S.E. Inhibition of PKC beta by oral administration of ruboxistaurin is well tolerated and ameliorates diabetes-induced retinal hemodynamic abnormalities in patients. Investigative Ophthalmology and Visual Science. 2006;47:86–92. doi: 10.1167/iovs.05-0757. [DOI] [PubMed] [Google Scholar]
- 50.Kelly D.J., Zhang Y., Hepper C. Protein kinase C beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes. 2003;52:512–518. doi: 10.2337/diabetes.52.2.512. [DOI] [PubMed] [Google Scholar]
- 51.Chen F., Yu Y., Haigh S. Regulation of NADPH oxidase 5 by protein kinase C isoforms. PLoS ONE. 2014;9:e88405. doi: 10.1371/journal.pone.0088405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heo K.S., Lee H., Nigro P. PKC? mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. Journal of Cell Biology. 2011;193:867–884. doi: 10.1083/jcb.201010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kowluru R.A., Mohammad G., dos Santos J.M., Zhong Q. Abrogation of MMP-9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes. 2011;60:3023–3033. doi: 10.2337/db11-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srivastava K., Shao B., Bayraktutan U. PKC-ß exacerbates in vitro brain barrier damage in hyperglycemic settings via regulation of RhoA/Rho-kinase/MLC2 pathway. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:1928–1936. doi: 10.1038/jcbfm.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van der Heijden M., Versteilen A.M., Sipkema P., van Nieuw Amerongen G.P., Musters R.J., Groeneveld A.B. Rho-kinase-dependent F-actin rearrangement is involved in the inhibition of PI3-kinase/Akt during ischemia–reperfusion-induced endothelial cell apoptosis. Apoptosis. 2008;13:404–412. doi: 10.1007/s10495-007-0173-6. [DOI] [PMC free article] [PubMed] [Google Scholar]