

Abstract
A good number of abstracts and research articles (in total 74) published, so far, for evaluating antioxidant activity of various samples of research interest were gone through where 407 methods were come across, which were repeated from 29 different methods. These were classified as in vitro and in vivo methods. And those are described and discussed below in this review article. In the later part of this review article, frequency of in vitro as well as in vivo methods is analyzed with a bar diagram. Solvents are important for extracting antioxidants from natural sources. Frequency of solvents used for extraction is also portrayed and the results are discussed in this article. As per this review there are 19 in vitro methods and 10 in vivo methods that are being used for the evaluation of antioxidant activity of the sample of interest. DPPH method was found to be used mostly for the in vitro antioxidant activity evaluation purpose while LPO was found as mostly used in vivo antioxidant assay. Ethanol was with the highest frequency as solvent for extraction purpose.
Keywords: Antioxidant, In vivo methods, In vitro methods
1. Introduction
The human body has a complex system of natural enzymatic and non-enzymatic antioxidant defenses which counteract the harmful effects of free radicals and other oxidants. Free radicals are responsible for causing a large number of diseases including cancer (Kinnula and Crapo, 2004), cardiovascular disease (Singh and Jialal, 2006), neural disorders (Sas et al., 2007), Alzheimer’s disease (Smith et al., 2000), mild cognitive impairment (Guidi et al., 2006), Parkinson’s disease (Bolton et al., 2000), alcohol induced liver disease (Arteel, 2003), ulcerative colitis (Ramakrishna et al., 1997), aging (Hyun et al., 2006) and atherosclerosis (Upston et al., 2003). Protection against free radicals can be enhanced by ample intake of dietary antioxidants. Substantial evidence indicates that foods containing antioxidants and possibly in particular the antioxidant nutrients may be of major importance in disease prevention. There is, however, a growing consensus among scientists that a combination of antioxidants, rather than single entities, may be more effective over the long term. Antioxidants may be of great benefit in improving the quality of life by preventing or postponing the onset of degenerative diseases. In addition, they have a potential for substantial savings in the cost of health care delivery.
Various methods are used to investigate the antioxidant property of samples (diets, plant extracts, commercial antioxidants etc.). The objective of this review article is to accumulate all probable methods that are used to evaluate the antioxidant property of various samples. A compiled description of all available in vitro and in vivo antioxidant models would provide prolific advantages to the researchers of this arena by reducing their time for literature review and method development. Two review articles have been published earlier (Chanda and Dave, 2009 and Badarinath et al., 2010) on in vitro evaluation of antioxidant activity. In this article, attempts have been taken to include in vivo too and to analyze the frequency of the use of different methods.
2. Methods
Internet browsing from Google Scholar database was used to identify and to download abstracts and research papers related to antioxidant activity study using suitable keywords (antioxidant + plant extract + in vitro + in vivo) in the month of August 2011. In the first thirty-four pages, a total of three hundred and forty articles appeared and those were subjected to preliminary screening. The basis of the selection of the articles was (i) antioxidant activity of plant extracts and (ii) description of antioxidant test procedures. A total of seventy-four papers and abstracts were identified and reviewed for in vivo and in vitro methods related to antioxidant evaluation. Solvents used for extraction purpose are also reviewed from the downloaded scientific records.
3. In vitro methods
Antioxidant activity should not be concluded based on a single antioxidant test model. And in practice several in vitro test procedures are carried out for evaluating antioxidant activities with the samples of interest. Another aspect is that antioxidant test models vary in different respects. Therefore, it is difficult to compare fully one method to other one. To some extent comparison among different in vitro methods has been done by Badarinath et al. (2010), while we discussed the methods in terms of grouping in the present manuscript. Researcher has to critically verify methods of analysis before adopting that one for his/her research purpose. Generally in vitro antioxidant tests using free radical traps are relatively straightforward to perform. Among free radical scavenging methods, DPPH method is furthermore rapid, simple (i.e. not involved with many steps and reagents) and inexpensive in comparison to other test models. On the other hand ABTS decolorization assay is applicable for both hydrophilic and lipophilic antioxidants. In this article all in vitro methods are described and it is important to note that one may optimize logically the respective method to serve his/her experimental objective as no one method is absolute in nature rather than an example.
3.1. DPPH scavenging activity
The molecule 1, 1-diphenyl-2-picrylhydrazyl (α,α-diphenyl-β-picrylhydrazyl; DPPH) is characterized as a stable free radical by virtue of the delocalisation of the spare electron over the molecule as a whole, so that the molecule does not dimerize, as would be the case with most other free radicals. The delocalization of electron also gives rise to the deep violet color, characterized by an absorption band in ethanol solution centered at about 517 nm. When a solution of DPPH is mixed with that of a substrate (AH) that can donate a hydrogen atom, then this gives rise to the reduced form with the loss of this violet color.
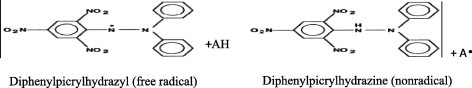
In order to evaluate the antioxidant potential through free radical scavenging by the test samples, the change in optical density of DPPH radicals is monitored. According to Manzocco et al., 1998 the sample extract (0.2 mL) is diluted with methanol and 2 mL of DPPH solution (0.5 mM) is added. After 30 min, the absorbance is measured at 517 nm. The percentage of the DPPH radical scavenging is calculated using the equation as given below:
where Abr is the absorbance before reaction and Aar is the absorbance after reaction has taken place.
3.2. Hydrogen peroxide scavenging (H2O2) assay
Human beings are exposed to H2O2 indirectly via the environment nearly about 0.28 mg/kg/day with intake mostly from leaf crops. Hydrogen peroxide may enter into the human body through inhalation of vapor or mist and through eye or skin contact. H2O2 is rapidly decomposed into oxygen and water and this may produce hydroxyl radicals (OH•) that can initiate lipid peroxidation and cause DNA damage in the body.
The ability of plant extracts to scavenge hydrogen peroxide can be estimated according to the method of Ruch et al. (1989). A solution of hydrogen peroxide (40 mM) is prepared in phosphate buffer (50 mM pH 7.4). The concentration of hydrogen peroxide is determined by absorption at 230 nm using a spectrophotometer. Extract (20–60 μg/mL) in distilled water is added to hydrogen peroxide and absorbance at 230 nm is determined after 10 min against a blank solution containing phosphate buffer without hydrogen peroxide. The percentage of hydrogen peroxide scavenging is calculated as follows:
where Ai is the absorbance of control and At is the absorbance of test.
3.3. Nitric oxide scavenging activity
NO• is generated in biological tissues by specific nitric oxide synthases, which metabolizes arginine to citrulline with the formation of NO• via a five electron oxidative reaction (David, 1999; Ghafourifar and Cadenas, 2005; Marletta, 1989; Moncada et al., 1989; and Virginia et al., 2003). The compound sodium nitroprusside is known to decompose in aqueous solution at physiological pH (7.2) producing NO•. Under aerobic conditions, NO• reacts with oxygen to produce stable products (nitrate and nitrite), the quantities of which can be determined using Griess reagent (Marcocci et al., 1994). Two (2) mL of 10 mM sodium nitroprusside dissolved in 0.5 mL phosphate buffer saline (pH 7.4) is mixed with 0.5 mL of sample at various concentrations (0.2–0.8 mg/mL). The mixture is then incubated at 25 °C. After 150 min of incubation, 0.5 mL of the incubated solution is withdrawn and mixed with 0.5 mL of Griess reagent [(1.0 mL sulfanilic acid reagent (0.33% in 20% glacial acetic acid at room temperature for 5 min with 1 mL of naphthylethylenediamine dichloride (0.1% w/v)]. The mixture is then incubated at room temperature for 30 min and its absorbance pouring into a cuvette is measured at 546 nm. The amount of nitric oxide radical inhibition is calculated following this equation:
where A0 is the absorbance before reaction and A1 is the absorbance after reaction has taken place with Griess reagent.
3.4. Peroxynitrite radical scavenging activity
Peroxynitrite (ONOO•) is a cytotoxicant with strong oxidizing properties toward various cellular constituents, including sulfhydryls, lipids, amino acids and nucleotides and can cause cell death, lipid peroxidation, carcinogenesis and aging. It is generated in vivo by endothelial cells, Kupffer cells, neutrophils and macrophages. Peroxynitrite radical is a relatively stable species compared with other free radicals but once protonated gives highly reactive peroxynitrous acid (ONOOH), decomposing with a very short half-life (1.9 s) at 37 °C to form various cytotoxicants and that can induce the oxidation of thiol (−SH) groups on proteins, nitration of tyrosine, lipid peroxidation and also nitrosation reactions, affecting cell metabolism and signal transduction. It can ultimately contribute to cellular and tissue injury with DNA strand breakage and apoptotic cell death, e.g. in thymocytes, cortical cells and HL-60 leukemia cells. Its excessive formation may also be involved in several human diseases such as Alzheimer’s disease, rheumatoid arthritis, cancer and atherosclerosis. Due to the lack of endogenous enzymes responsible for ONOO• inactivation, developing specific ONOO• scavengers is of considerable importance. The method described by Kooy et al., 1994 involves the use of a stock solution of dihydroxyrhodamine 123 (DHR 123, 5 mM) in dimethylformamide that is purged with nitrogen and stored at −80 °C. Working solution with DHR 123 (final concentration 5 μM) is diluted from the stock solution and is placed on ice in the dark immediately prior to the experiment. Buffer solution, 50 mM sodium phosphate (pH 7.4), containing 90 mM sodium chloride and 5 mM potassium chloride with 100 μM diethylenetriaminepentaacetic acid (DTPA) are purged with nitrogen and placed on ice before use. Scavenging activity of ONOO• by the oxidation of DHR 123 is measured on a microplate fluorescence spectrophotometer with excitation and emission wavelengths of 485 nm and 530 nm at room temperature, respectively. The background and final fluorescent intensities are measured 5 min after treatment without 3-morpholino- sydnonimine (SIN-1) or authentic (ONOO•). Oxidation of DHR 123 by decomposition of SIN-1 gradually increased whereas authentic ONOO• rapidly oxidized DHR 123 with its final fluorescent intensity being stable over time.
3.5. Trolox equivalent antioxidant capacity (TEAC) method/ABTS radical cation decolorization assay
This method, uses a diode-array spectrophotometer to measure the loss of color when an antioxidant is added to the blue–green chromophore ABTS· + (2,2-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid)). The antioxidant reduces ABTS·+ to ABTS and decolorize it. ABTS·+ is a stable radical not found in the human body. Antioxidant activity can be measured as described by Seeram et al. (2006). ABTS radical cations are prepared by adding solid manganese dioxide (80 mg) to a 5 mM aqueous stock solution of ABTS (20 mL using a 75 mM Na/K buffer of pH 7). Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid), a water-soluble analog of vitamin E, can be used as an antioxidant standard. A standard calibration curve is constructed for Trolox at 0, 50, 100, 150, 200, 250, 300, and 350 μM concentrations. Samples are diluted appropriately according to antioxidant activity in Na/K buffer pH, 7. Diluted samples are mixed with 200 μL of ABTS•+ radical cation solution in 96-well plates, and absorbance is read (at 750 nm) after 5 min in a microplate reader. TEAC values can be calculated from the Trolox standard curve and expressed as Trolox equivalents (in mM).
3.6. Total radical-trapping antioxidant parameter (TRAP) method
This method is based on the protection provided by antioxidants on the fluorescence decay of R-phycoerythrin (R-PE) during a controlled peroxidation reaction. The fluorescence of R-Phycoerythrin is quenched by ABAP (2,2′-azo–bis(2-amidino-propane)hydrochloride) as a radical generator. This quenching reaction is measured in the presence of antioxidants. The antioxidative potential is evaluated by measuring the decay in decoloration. According to Ghiselli et al. (1995) 120 μL of diluted sample is added to 2.4 mL of phosphate buffer (pH 7.4), 375 μL of bidistilled water, 30 μL of diluted R-PE and 75 μL of ABAP; the reaction kinetics at 38 °C is recorded for 45 min by a luminescence spectrometer. TRAP values are calculated from the length of the lag-phase due to the sample compared with standard.
3.7. Ferric reducing-antioxidant power (FRAP) assay
This method measures the ability of antioxidants to reduce ferric iron. It is based on the reduction of the complex of ferric iron and 2,3,5-triphenyl-1,3,4-triaza-2-azoniacyclopenta-1,4-diene chloride (TPTZ) to the ferrous form at low pH. This reduction is monitored by measuring the change in absorption at 593 nm, using a diode-array spectrophotometer. Antioxidant assay can be conducted by the method developed by Benzie and Strain (1999). Three milliliter of prepared FRAP reagent is mixed with 100 μL of diluted sample; the absorbance at 593 nm is recorded after a 30 min incubation at 37 °C. FRAP values can be obtained by comparing the absorption change in the test mixture with those obtained from increasing concentrations of Fe3+ and expressed as mM of Fe2+ equivalents per kg (solid food) or per L (beverages) of sample.
3.8. Superoxide radical scavenging activity (SOD)
Although superoxide anion is a weak oxidant, it ultimately produces powerful and dangerous hydroxyl radicals as well as singlet oxygen, both of which contribute to oxidative stress (Meyer and Isaksen, 1995). The superoxide anion scavenging activity can be measured as described by Robak and Gryglewski (1988). The superoxide anion radicals are generated in 3.0 mL of Tris–HCl buffer (16 mM, pH 8.0), containing 0.5 mL of nitroblue tetrazolium (NBT) (0.3 mM), 0.5 mL NADH (0.936 mM) solution, 1.0 mL extract and 0.5 mL Tris–HCl buffer (16 mM, pH 8.0). The reaction is initiated by adding 0.5 mL phenazine methosulfate (PMS) solution (0.12 mM) to the mixture, incubated at 25 °C for 5 min and then the absorbance is measured at 560 nm against a blank sample.
3.9. Hydroxyl radical scavenging activity
Hydroxyl radical is one of the potent reactive oxygen species in the biological system that reacts with polyunsaturated fatty acid moieties of cell membrane phospholipids and causes damage to cell. The scavenging ability of hydroxyl radicals is measured by the method of Kunchandy and Rao (1990). The reaction mixture (1.0 mL) consist of 100 μL of 2-deoxy-Dribose (28 mM in 20 mM KH2PO4-KOH buffer, pH 7.4), 500 μL of the extract, 200 μL EDTA (1.04 mM) and 200 μM FeCl3 (1:1 v/v), 100 μL of H2O2 (1.0 mM) and 100 μL ascorbic acid (1.0 mM) which is incubated at 37 °C for 1 h. One milliliter of thiobarbituric acid (1%) and 1.0 mL of trichloroacetic acid (2.8%) are added and incubated at 100 °C for 20 min. After cooling, absorbance is measured at 532 nm, against a blank sample.
3.10. Hydroxyl radical averting capacity (HORAC) method
The HORAC assay described by Ou et al. (2002) measures the metal-chelating activity of antioxidants in the conditions of Fenton-like reactions employing a Co (II) complex and hence the protecting ability against formation of hydroxyl radical. Hydrogen peroxide solution of 0.55 M is prepared in distilled water and 4.6 mM Co (II) is prepared by dissolving 15.7 mg of CoF2·4H2O and 20 mg of picolinic acid in 20 mL of distilled water. Fluorescein – 170 μL (60 nM, final concentration) and 10 μL of sample are incubated at 37 °C for 10 min. directly in the plate reader. After incubation 10 μL H2O2 (27.5 mM, final concentration) and 10 μL of Co (II) (230 μM final concentration) solutions are added subsequently. The initial fluorescence is measured after which the readings are taken every min. after shaking. For the blank sample, phosphate buffer solution is used. 100, 200, 600, 800 and 1000 μM standard antioxidant solutions (in phosphate buffer 75 mM, pH 7.4) are used for building the standard curve. The final HORAC values are calculated using a regression equation between the standard antioxidant concentration and the net area under the curve. One HORAC unit is assigned to the net protection area provided by 1 μM standard antioxidant and the activity of the sample is expressed as μM standard antioxidant equivalents per gram of fresh weight of the samples. Gallic acid can be used as standard antioxidant.
3.11. Oxygen radical absorbance capacity (ORAC) Method
ORAC is an exciting and revolutionary new test tube analysis that can be utilized to test “Antioxidant Power” of foods and other chemical substances. This test can be done using either β-phycoerythrin (β-PE) or fluorescein as target molecule. While reviewing, β-PE is not encountered in the publications later than 2005. Thus it seems fluorescein is replacing the β-PE as target molecule in ORAC assay. The test is performed using Trolox (a water-soluble analog of Vitamin E) as a standard to determine the Trolox Equivalent (TE). The ORAC value is then calculated from the Trolox Equivalent and expressed as ORAC units or value. The higher the ORAC value, the greater the “Antioxidant Power”.
This assay is based on generation of free radical using AAPH (2,2-azobis 2-amidopropane dihydrochloride) and measurement of decrease in fluorescence in the presence of free radical scavengers. Prior et al. (2003) have reported an automated ORAC assay. In this assay β-phycoerythrin (β-PE) was used as target free radical damage, AAPH as a peroxy radical generator and Trolox as a standard control. After addition of AAPH to the test solution, the fluorescence is recorded and the antioxidant activity is expressed as trolox equivalent (Cao et al., 1993; Frei et al., 1990).
The assay can be carried out according to Prior et al. (2003) in 96-well polypropylene fluorescence plates with a final volume of 200 μL. Assays are conducted at pH 7.0 with Trolox (6.25, 12.5, 25, and 50 μmol/L for lipophilic assays; 12.5, 25, 50 and 100 μmol/L hydrophilic assays) as the standard and 75 mM/L phosphate buffer as the blank. After the addition of AAPH, the plate is placed immediately in a multilabel counter preheated to 37 °C. The plate is shaken in an orbital manner for 10 s and the fluorescence is read at 1 min intervals for 35 min at the excitation wavelength of 485 nm and emission wavelength of 520 nm. Area-under-the-curve is calculated for each sample using Wallac Workout 1.5 software. Final computation of results is made by taking the difference of areas-under-the-decay curves between blank and sample and/or standard (Trolox) and expressing this in μM of Trolox equivalents (TE) per g dry weight of sample (μM TE/g).
3.12. Reducing power method (RP)
This method is based on the principle of increase in the absorbance of the reaction mixtures. Increase in the absorbance indicates an increase in the antioxidant activity. In this method, antioxidant compound forms a colored complex with potassium ferricyanide, trichloro acetic acid and ferric chloride, which is measured at 700 nm. Increase in absorbance of the reaction mixture indicates the reducing power of the samples (Jayaprakash et al., 2001). In the method described by Oyaizu (1986) 2.5 mL of 0.2 M phosphate buffer (pH 6.6) and 2.5 mL of K3Fe (CN)6 (1% w/v) are added to 1.0 mL of sample dissolved in distilled water. The resulting mixture is incubated at 50 °C for 20 min, followed by the addition of 2.5 mL of Trichloro acetic acid (10% w/v). The mixture is centrifuged at 3000 rpm for 10 min to collect the upper layer of the solution (2.5 mL), mixed with distilled water (2.5 mL) and 0.5 mL of FeCl3 (0.1%, w/v). The absorbance is then measured at 700 nm against blank sample.
3.13. Phosphomolybdenum method
Total antioxidant capacity assay is a spectroscopic method for the quantitative determination of antioxidant capacity, through the formation of phosphomolybdenum complex. The assay is based on the reduction of Mo (VI) to Mo (V) by the sample analyte and subsequent formation of a green phosphate Mo (V) complex at acidic pH. Total antioxidant capacity can be calculated by the method described by Prieto et al. (1999). 0.1 mL of sample (100 μg) solution is combined with 1 mL of reagent (0.6 M sulfuric acid, 28 mM sodium phosphate and 4 mM ammonium molybdate). The tube is capped and incubated in a boiling water bath at 95 °C for 90 min. After cooling the sample to room temperature, the absorbance of the aqueous solution is measured at 695 nm against blank in UV spectrophotometer. A typical blank solution contained 1 mL of reagent solution and the appropriate volume of the same solvent used for the sample and it is incubated under same conditions as rest of the sample. For samples of unknown composition, antioxidant capacity can be expressed as equivalents of α-tocopherol.
3.14. Ferric thiocyanate (FTC) method
This method can be exploited to determine the antioxidant activity as illustrated by Kikuzaki et al. (1991). A mixture of 4 mg of sample (final concentration of 0.02% w/v) in 4 mL ethanol, 4.1 mL of 2.51% linoleic acid in ethanol, 8.0 mL of 0.02 M phosphate buffer (pH 7.0) and 3.9 mL of distilled water contained in screw cap vial is placed in an oven at 40 °C in the dark. 0.1 mL of the reaction mixture is transferred to a test tube and, to it; 9.7 mL of 75% (v/v) aqueous ethanol, followed by 0.1 mL of 30% aqueous ammonium thiocyanate and 0.1 mL of 0.02 M ferrous chloride in 3.5% hydrochloric acid are added. Three minutes after the addition of ferrous chloride to the reaction mixture, the absorbance of the resulting mixture (red color) is measured at 500 nm every 24 h until the absorbance of the control reached its maximum. Standard antioxidant (final concentration of 0.02% w/v) is used as positive control, and the mixture without the sample is used as the negative control.
3.15. Thiobarbituric acid (TBA) method
TBA method, described by Ottolenghi (1959) is as follows: The final sample concentration of 0.02% w/v was used in this method. Two mL of 20% trichloroacetic acid and 2 mL of 0.67% of thiobarbituric acid were added to 1 mL of sample solution. The mixture was placed in a boiling water bath for 10 min and then centrifuged after cooling at 3000 rpm for 20 min. The absorbance activity of the supernatant was measured at 552 nm and recorded after it has reached its maximum.
3.16. DMPD (N,N-dimethyl-p-phenylene diamine dihydrochloride) method
DMPD radical cation decolorization method has been developed for the measurement of the antioxidant activity in food and biological samples. This assay is based on the reduction of buffered solution of colored DMPD in acetate buffer and ferric chloride. The procedure involves measurement of decrease in absorbance of DMPD in the presence of scavengers at its absorption maximum of 505 nm. The activity was expressed as percentage reduction of DMPD.
Fogliano et al. (1999) obtained the radical by mixing 1 mL of DMPD solution (200 mM), 0.4 mL of ferric chloride (III) (0.05 M), and 100 mL of sodium acetate buffer solution at 0.1 M, modifying the pH to 5.25. The reactive mixture has to be kept in darkness, under refrigeration, and at a low temperature (4–5 °C) .The reaction takes place when 50 μL of the sample (a dilution of 1:10 in water) is added to 950 μL of the DMPD·+ solution. Absorbance is measured after 10 min of continuous stirring, which is the time taken to reach constant decolorization values. The results are quantified in mM Trolox on the relevant calibration curve.
3.17. β-carotene linoleic acid method/conjugated diene assay
This is one of the rapid method to screen antioxidants, which is mainly based on the principle that linoleic acid, which is an unsaturated fatty acid, gets oxidized by “Reactive Oxygen Species” (ROS) produced by oxygenated water. The products formed will initiate the β-carotene oxidation, which will lead to discoloration. Antioxidants decrease the extent of discoloration, which is measured at 434 nm and the activity is measured.
The method as described by Kabouche et al. (2007): β-carotene (0.5 mg) in 1 mL of chloroform is added to 25 μL of linoleic acid and 200 mg of tween-80 emulsified mixture. Chloroform is evaporated at 40 °C, 100 mL of distilled water saturated with oxygen is slowly added to the residue and the solution is vigorously agitated to form a stable emulsion. Four mL of this mixture is added into the test tubes containing 200 μL of sample prepared in methanol at final concentrations (25, 50, 100, 200 and 400 μg/mL). As soon as the emulsified solution is added to the tubes, zero time absorbance is measured at 470 nm. The tubes are incubated for 2 h at 50 °C. Vitamin C can be used as standard. Antioxidant activity is calculated as percentage of inhibition (I%) relative to the control using the following equation:
where As was initial absorbance, As120 was the absorbance of the sample at 120 min, Ac was initial absorbance of negative control and Ac120 was the absorbance of the negative control at 120 min.
3.18. Xanthine oxidase method
The xanthine oxidase activity with xanthine as sub-substrate can be measured spectrophotometrically, by the method of Noro et al. (1983). The extract (500 μL of 0.1 mg/mL) and allopurinol (100 μg/mL) (in methanol) are mixed with 1.3 mL phosphate buffer (0.05 M, pH 7.5) and 0.2 mL of 0.2 units/mL xanthine oxidase solution. After 10 min of incubation at room temperature (25 °C), 1.5 mL of 0.15 M xanthine substrate solution is added to this mixture. The mixture is again incubated for 30 min at room temperature (25 °C) and then the absorbance is measured at 293 nm using a spectrophotometer against blank (0.5 mL methanol, 1.3 mL phosphate buffer, 0.2 mL xanthine oxidase). The solution of 0.5 mL methanol, 1.3 mL phosphate buffer, 0.2 mL xanthine oxidase and 1.5 mL xanthine substrate is used as a control. Percentage of inhibition is calculated using the formula:
where As and Ac are the absorbance values of the test sample and control, respectively.
3.19. Cupric ion reducing antioxidant capacity (CUPRAC) method
The chromogenic oxidizing reagent of the developed CUPRAC method, that is, bis(neocuproine)copper(II) chloride [Cu(II)-Nc], reacts with polyphenols [Ar(OH)n] in the manner.
where the liberated protons may be buffered with the relatively concentrated ammonium acetate buffer solution. In this reaction, the reactive Ar-OH groups of polyphenols are oxidized to the corresponding quinones and Cu (II)-Nc is reduced to the highly colored Cu (I)-Nc chelate showing maximum absorption at 450 nm.
According to Apak et al. (2008), 1 mL of 10−2 M of CuCl2, 1 mL of 7.5 × 10−3 M neocuproine and 1 M NH4CH3COO solution are added into the glass test tube. Then, 400 μL of freshly prepared standard solution is added and diluted to the final volume of 4.1 mL with deionized water. This procedure is repeated for 400 μL, 300 μL, 200 μL, 100 μL and 50 μL additions of freshly prepared solutions of the sample. The prepared solutions are mixed and incubated at room temperature for 30 min. The absorbance at 450 nm is determined against a reagent blank by spectrometer. The calculation of antioxidant capacity of compounds as Trolox equivalents (TEAC values) by the CUPRAC method has been reported.
3.20. Metal chelating activity
Ferrozine can form a complex with a red color by forming chelates with Fe2+. This reaction is restricted in the presence of other chelating agents and results in a decrease of the red color of the ferrozine-Fe2+ complexes. Measurement of the color reduction determines the chelating activity to compete with ferrozine for the ferrous ions (Soler-Rivas et al., 2000). The chelation of ferrous ions is estimated using the method of Dinis et al. (1994). 0.1 mL of the extract is added to a solution of 0.5 mL ferrous chloride (0.2 mM). The reaction is started by the addition of 0.2 mL of ferrozine (5 mM) and incubated at room temperature for 10 min and then the absorbance is measured at 562 nm. EDTA or citric acid (Dinis et al., 1994) can be used as a positive control.
4. In vivo models
For all in vivo methods the samples that are to be tested are usually administered to the testing animals (mice, rats, etc.) at a definite dosage regimen as described by the respective method.
After a specified period of time, the animals are usually sacrificed and blood or tissues are used for the assay.
4.1. Ferric reducing ability of plasma
It is one of the most rapid test and very useful for routine analysis (Umesh et al., 2010). The antioxidative activity is estimated by measuring the increase in absorbance caused by the formation of ferrous ions from FRAP reagent containing TPTZ (2,4,6-tripyridyl-s-triazine) and FeCl2·6H2O. The absorbance is measured spectrophotometrically at 593 nm.
The method illustrated by Benzie and Strain (1996) involves the use of blood samples that are collected from the rat retro-orbital venous plexus into heparinized glass tubes at 0, 7 and 14 days of treatment. Three mL of freshly prepared and warm (37 °C) FRAP reagent [1 mL (10 mM) of 2,4,6 tripyridyl-s-triazine (TPTZ) solution in 40 mM HCl, 1 mL 20 mM FeCl2·6H2O, 10 mL of 0.3 M acetate buffer (pH 3.6)] is mixed with 0.375 mL distilled water and 0.025 mL of test samples. The absorbance of developed color in organic layer is measured at 593 nm. The temperature is maintained at 37 °C. The readings at 180 s are selected for the calculation of FRAP values.
4.2. Reduced glutathione (GSH) estimation
GSH is an intra-cellular reductant and plays major role in catalysis, metabolism and transport. It protects cells against free radicals, peroxides and other toxic compounds (Sapakal et al., 2008) Deficiency of GSH in the lens leads to cataract formation. Glutathione also plays an important role in the kidney and takes part in a transport system involved in the reabsorption of amino acids. The method illustrated by Ellman (1959) can be used for determination of antioxidant activity. The tissue homogenate (in 0.1 M phosphate buffer pH 7.4) is taken and added with equal volume of 20% trichloroacetic acid (TCA) containing 1 mM EDTA to precipitate the tissue proteins. The mixture is allowed to stand for 5 min prior to centrifugation for 10 min at 2000 rpm. The supernatant (200 μL) is then transferred to a new set of test tubes and added with 1.8 mL of the Ellman’s reagent (5,5′-dithiobis-2-nitrobenzoic acid (0.1 mM) prepared in 0.3 M phosphate buffer with 1% of sodium citrate solution). Then all the test tubes are made up to the volume of 2 mL. After completion of the total reaction, solutions are measured at 412 nm against blank. Absorbance values were compared with a standard curve generated from known GSH.
4.3. Glutathione peroxidase (GSHPx) estimation
GSHPX is a seleno-enzyme two third of which (in liver) is present in the cytosol and one third in the mitochondria. It catalyzes the reaction of hydroperoxides with reduced glutathione to form glutathione disulfide (GSSG) and the reduction product of hydroperoxide. GSHPx is found throughout the tissues, being present as four different isoenzymes, cellular glutathione peroxidase, extracellular glutathione peroxidase, phospholipid hydroperoxide glutathione peroxidase and gastrointestinal glutathione peroxidase. GSHPx measurement is considered in particular with patients who are under oxidative stress for any reason; low activity of this enzyme is one of the early consequences of a disturbance of the prooxidant/antioxidant balance (Paglia and Valentin, 1967; Yang et al., 1984).
According to Wood (1970), Cytosolic GPx is assayed via a 3-mL cuvette containing 2.0 mL of 75 mM/L phosphate buffer, pH 7.0. The following solutions are then added: 50 μL of 60 mM/L glutathione reductase solution (30 U/mL), 50 μL of 0.12 M/L NaN3, 0.10 of 0.15 mM/L Na2EDTA, 100 μL of 3.0 mM/L NADPH, and 100 μL of cytosolic fraction obtained after centrifugation at 20,000 g for 25 min. Water is added to make a total volume of 2.9 mL. The reaction is started by the addition of 100 μL of 7.5 mM/L H2O2, and the conversion of NADPH to NADP is monitored by a continuous recording of the change of absorbance at 340 nm at 1 min interval for 5 min. Enzyme activity of GSHPx was expressed in terms of mg of proteins.
4.4. Glutathione-S-transferase (GSt)
Glutathione-S-transferase is thought to play a physiological role in initiating the detoxication of potential alkylating agents, including pharmacologically active compounds. These enzymes catalyze the reaction of such compounds with the -SH group of glutathione, thereby neutralizing their electrophilic sites and rendering the products more water-soluble. The method can be used as described by Jocelyn (1972). The reaction mixture (1 mL) consisted of 0.1 N potassium phosphate (pH 6.5), 1 nM/L GSt, 1 M/L l-chloro-2, 4-dinitrobenzene as substrate and a suitable amount of cytosol (6 mg protein/mL). The reaction mixture is incubated at 37 °C for 5 min and the reaction is initiated by the addition of the substrate. The increase in absorbance at 340 nm was measured spectrophotometrically.
4.5. Superoxide dismutase (SOD) method
This method is well described by Mccord and Fridovich (1969) and can be applied for determination of antioxidant activity of a sample. It is estimated in the erythrocyte lysate prepared from the 5% RBC suspension. To 50 μL of the lysate, 75 mM of Tris–HCl buffer (pH 8.2), 30 mM EDTA and 2 mM of pyrogallol are added. An increase in absorbance is recorded at 420 nm for 3 min by spectrophotometer. One unit of enzyme activity is 50% inhibition of the rate of autooxidation of pyrogallol as determined by change in absorbance/min at 420 nm. The activity of SOD is expressed as units/mg protein.
4.6. Catalase (CAT)
Catalase activity can be determined in erythrocyte lysate using Aebi’s method (Aebi, 1984). Fifty microliter of the lysate is added to a cuvette containing 2 mL of phosphate buffer (pH 7.0) and 1 mL of 30 mM H2O2. Catalase activity is measured at 240 nm for 1 min using spectrophotometer. The molar extinction coefficient of H2O2, 43.6 M cm−1 was used to determine the catalase activity. One unit of activity is equal to 1 mmol of H2O2 degraded per minute and is expressed as units per milligram of protein.
4.7. γ-Glutamyl transpeptidase activity (GGT) assay
According to Singhal et al. (1982), the serum sample is added to a substrate solution containing glycylglycine, MgCl2 and g-Glutamyl-p-nitroanilide in 0.05 M tris (free base), pH 8.2. The mixture is incubated at 37 °C for 1 min and the absorbance read at 405 nm at 1 m interval for 5 m. The activity of GGT is calculated from the absorbance values.
4.8. Glutathione reductase (GR) assay
The ubiquitous tripeptide glutathione (GSH), which is the most abundant low molecular weight thiol in almost all cells, is involved in a wide range of enzymatic reactions. A major function of GSH is to serve as a reductant in oxidation–reduction processes; a function resulting in the formation of glutathione disulfide (GSSG). A heat labile system capable of reducing GSSG was discovered in liver. The enzyme directly involved in reduction of GSSG.
The method illustrated by Kakkar et al. (1984) is as follows: Livers (about 400 g) are obtained from killed rats (200–250 g). The livers are cut into small pieces and homogenized in 9 mL of 0.25 M ice-cold sucrose per g of rat liver in a blender. The homogenate is centrifuged for 45 min at 14,000 rpm. The pellets are suspended in a small volume of 0.25 M sucrose and centrifuged. The supernatants are combined with the previous centrifugate. The pooled material is adjusted to pH 5.5 with cold 0.2 M acetic acid and centrifuged again for 45 min at 14,000 rpm. The rate of oxidation of NADPH by GSSG at 30 °C is used as a standard measure of enzymatic activity. The reaction system of 1 mL contained: 1.0 mM GSSG, 0.1 mM NADPH, 0.5 mM EDTA, 0.10 M sodium phosphate buffer (pH 7.6), and a suitable amount of the glutathione reductase sample to give a change in absorbance of 0.05–0.03/min. The oxidation of 1 μM of NADPH/min under these conditions is used as a unit of glutathione reductase activity. The specific activity is expressed as units per mg of protein.
4.9. Lipid peroxidation (LPO) assay
LPO is an autocatalytic process, which is a common consequence of cell death. This process may cause peroxidative tissue damage in inflammation, cancer and toxicity of xenobiotics and aging. Malondialdehyde (MDA) is one of the end products in the lipid peroxidation process. Malondialdehyde (MDA) is formed during oxidative degeneration as a product of free oxygen radicals, which is accepted as an indicator of lipid peroxidation.
This method described by Ohkawa et al. (1979) is as follows: The tissues are homogenized in 0.1 M buffer pH 7.4 with a Teflon-glass homogenizer. LPO in this homogenate is determined by measuring the amounts of malondialdehyde (MDA) produced primarily. Tissue homogenate (0.2 mL), 0.2 mL of 8.1% sodium dodecyl sulfate (SDS), 1.5 mL of 20% acetic acid and 1.5 mL of 8% TBA are added. The volume of the mixture is made up to 4 mL with distilled water and then heated at 95 °C on a water bath for 60 min using glass balls as condenser. After incubation the tubes are cooled to room temperature and final volume was made to 5 mL in each tube. Five mL of butanol: pyridine (15:1) mixture is added and the contents are vortexed thoroughly for 2 min. After centrifugation at 3000 rpm for 10 min, the upper organic layer is taken and its OD is taken at 532 nm against an appropriate blank without the sample. The levels of lipid peroxides can be expressed as n moles of thiobarbituric acid reactive substances (TBARS)/mg protein using an extinction coefficient of 1.56 × 105 ML cm−1.
4.10. LDL assay
The isolated LDL is washed and dialyzed against 150 mmol/L NaCl and 1 mmol/L Na2EDTA (pH 7.4) at 4 °C. The LDL is then sterilized by filtration (0.45 μM), kept under nitrogen in the dark at 4 °C. LDL (100 μg of protein/mL) is incubated for 10 min at room temperature with samples. Then, 5 μmol/L of CuSO4 is added, and the tubes are incubated for 2 h at 37 °C. Cu2+-induced oxidation is terminated by the addition of butylated hydroxytoluene (BHT, 10 μM). At the end of the incubation, the extent of LDL oxidation is determined by measuring the generated amount of lipid peroxides and also by the thiobarbituric acid reactive substances (TBARS) assay at 532 nm, using malondialdehyde (MDA) for the standard curve as described by Buege and Aust, 1978; El-Saadani et al., 1989.
5. Results and discussion
In addition to compilation of various methods related to evaluation of antioxidant activity, it was our interest to see the frequency of each method of a given number of citations being used. The results of the said frequency analysis for in vitro and in vivo methods are shown in Figs. 1 and 2, respectively. It is clear from Fig. 1 that four in vitro methods were most frequently used and these were in order of decreasing frequency: DPPH > Hydroxyl radical scavenging > SOD > β-carotene linolate. Out of all the in vitro methods, DPPH is the most easy, simple and reasonably costly method and hence it might have been used mostly for the antioxidant activity evaluation of a sample. If one looks into Fig. 2, it immediately appears that the frequency of use is higher for LPO assay and it was followed by CAT and GSHPx. Lipid is a major component of cell membrane and thus its peroxidation almost directly co-relates peroxidative damage of cell in vivo and hence it might have been found to have the highest frequency in vivo antioxidant activity assay.
Figure 1.
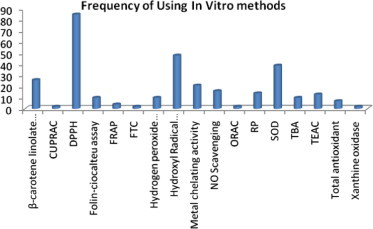
Frequency of commonly used In vitro methods.
Figure 2.
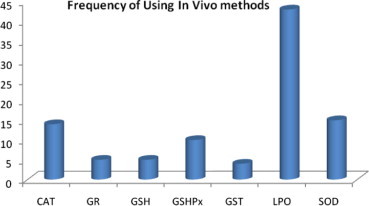
Frequency of commonly used In vivo methods.
Fig. 3 represents the frequency of use of solvent for the extraction of a material to evaluate its antioxidant property. It is evident from the figure that four solvents are prominently being used for the extraction purpose in relation to the stated experiment. These solvents are ethanol, water, methanol and aqueous ethanol. Ethanol, methanol and water have good polarity and hence are used favorably to extract polar compounds such as phenolic compounds and flavonoids which are believed to be effective antioxidants. Ethanol being organic and nontoxic might have the highest frequency of use for extraction purpose. Water needs a different step of freeze drying to remove it from the extract after extraction. Toxicity of methanol limits its use in some extraction and subsequent experiment. Non-polar solvents such as ether and low polarity solvents such as chloroform, ester, acetone etc. have been used in specific cases and their availability also limits their use in the experiment and hence their frequency of use was found very low.
Figure 3.
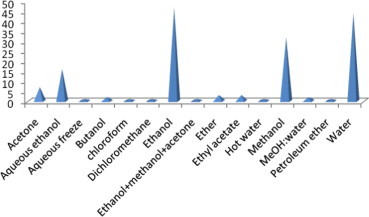
Commonly used extracting solvents for antioxidant study.
6. Conclusion
This review article is focused on in vitro and in vivo methods of antioxidant evaluation. It was prepared based on plenty literature search. Presently, 19 in vitro and 10 in vivo methods are being used for antioxidant evaluation purpose. DPPH method is the most frequently used one for in vitro antioxidant activity evaluation while LPO was found as the mostly used in vivo antioxidant assay. Ethanol extract was found with the highest frequency for antioxidant study. This article will be a comprehensive ready reference for those who are interested on antioxidant study.
Footnotes
Peer review under responsibility of King Saud University

Contributor Information
Md. Nur Alam, Email: nalamju@gmail.com.
Md. Rafiquzzaman, Email: zrafiq18@yahoo.com.
References
- Aebi H. Catalase. Methods Enzymol. 1984;105:121–126. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- Apak R., Güçlü K., Özyürek M., Karademir S.E. Mechanism of antioxidant capacity assays and the CUPRAC (cupric ion reducing antioxidant capacity) assay. Microchim. Acta. 2008;160:413–419. [Google Scholar]
- Arteel G.E. Oxidants and antioxidants in alcohol induced liver disease. Gastroenterol. 2003;124:778–790. doi: 10.1053/gast.2003.50087. [DOI] [PubMed] [Google Scholar]
- Badarinath A.V., RAo K.M., Chetty C.M.S., Ramkanth V., Rajan T.V.S., Gnanaprakash K. A review on in-vitro antioxidant methods: comparisons, correlations and considerations. Int. J. PharmTech Res. 2010;2(2):1276–1285. [Google Scholar]
- Benzie I.F.F., Strain J.J. The ferric reducing ability of plasma (FRAP) as a measure of ‘antioxidant power’: The FRAP assay. Anal. Biochem. 1996;239:70–76. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- Benzie I.F.F., Strain J.J. Ferric reducing antioxidant power assay: direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol. 1999;299:15–27. doi: 10.1016/s0076-6879(99)99005-5. [DOI] [PubMed] [Google Scholar]
- Bolton J.L., Trush M.A., Penning T.M., Dryhurst G., Monks T.J. Role of quinones in toxicology. Chem. Res. Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- Buege J.A., Aust S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–310. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- Cao G., Alessio H.M., Culter R.G. Oxygen radical absorbance capacity assay for antioxidant free radicals. Biol. Med. 1993;14:303–311. doi: 10.1016/0891-5849(93)90027-r. [DOI] [PubMed] [Google Scholar]
- Chanda S., Dave R. In vitro models for antioxidant activity evaluation and some medicinal plants possessing antioxidant properties: an overview. Afr. J. Microbiol. Res. 2009;3(13):981–996. [Google Scholar]
- David S.B. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic. Res. 1999;31(6):577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- Dinis T.C.P., Madeira V.M.C., Almeida L.M. Action of phenolic derivatives (acetaminophen, salicylate and 5-aminosalicylate) as inhibitors of membrane lipid peroxidation and as peroxy radical scavengers. Arch. Biochem. Biophy. 1994;315:161–169. doi: 10.1006/abbi.1994.1485. [DOI] [PubMed] [Google Scholar]
- Ellman G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- El-Saadani M., Esterbauer H., El-Sayed M., Goher M., Nassar A.Y., Juergens G.A. Spectrophotometric assay for lipid peroxides in serum lipoproteins using a commercially available reagent. J. Lipid Res. 1989;30:627–630. [PubMed] [Google Scholar]
- Fogliano V., Verde V., Randazzo G., Ritieni A. Method for measuring antioxidant activity and its application to monitoring antioxidant capacity of wines. J. Agric. Food Chem. 1999;47:1035–1040. doi: 10.1021/jf980496s. [DOI] [PubMed] [Google Scholar]
- Frei B., Stocker R., England L., Ames B.N. Ascorbate the most effective antioxidant in human blood plasma. Adv. Med. Exp. Biol. 1990;264:155–163. doi: 10.1007/978-1-4684-5730-8_24. [DOI] [PubMed] [Google Scholar]
- Ghafourifar P., Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005;26:190–195. doi: 10.1016/j.tips.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Ghiselli A., Serafini M., Maiani G., Azzini E., Ferro-Luzzi A. A fluorescence-based method for measuring total plasma antioxidant capability. Free Radic. Biol. Med. 1995;18:29–36. doi: 10.1016/0891-5849(94)00102-p. [DOI] [PubMed] [Google Scholar]
- Guidi I., Galimberti D., Lonati S., Novembrino C., Bamonti F., Tiriticco M., Fenoglio C., Venturelli E., Baron P., Bresolin N. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging. 2006;27:262–269. doi: 10.1016/j.neurobiolaging.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Hyun D.H., Hernandez J.O., Mattson M.P., de Cabo R. The plasma membrane redox system in aging. Aging Res. Rev. 2006;5:209–220. doi: 10.1016/j.arr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Jayaprakash G.K., Singh R.P., Sakariah K.K. Antioxidant activity of grape seed extracts on peroxidation models in-vitro. J. Agric. Food Chem. 2001;55:1018–1022. [Google Scholar]
- Jocelyn P.C. Academic Press; London: 1972. Biochemistry of the SH Group. pp. 10. [Google Scholar]
- Kakkar P., Das B., Viswanathan P.N. A modified spectrophotometric assay of superoxide dismutase. Ind. J. Biochem. Biophys. 1984;21:131–132. [PubMed] [Google Scholar]
- Kabouche A., Kabouche Z., Ôzturk M., Kolal U., Topçu G. Antioxidant abietane diterpenoids from Salvia barrelieri. Food. Chem. 2007;102:1281–1287. [Google Scholar]
- Kikuzaki H., Usuguchi J., Nakatani N. Constituents of Zingiberaceae I. Diarylheptanoid from the rhizomes of ginger (Zingiber officinale Roscoe) Chem. Pharm. Bull. 1991;39:120. [Google Scholar]
- Kinnula V.L., Crapo J.D. Superoxide dismutases in malignant cells and human tumors. Free Radic. Biol. Med. 2004;36:718–744. doi: 10.1016/j.freeradbiomed.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Kooy N.W., Royall J.A., Ischiropoulos H., Beckman J.S. Peroxynitrite-mediated oxidation of dihydrorhodamine 123. Free Radic. Biol. Med. 1994;16:149–156. doi: 10.1016/0891-5849(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Kunchandy E., Rao M.N.A. Oxygen radical scavenging activity of curcumin. Int. J. Pharm. 1990;58:237–240. [Google Scholar]
- Manzocco L., Anese M., Nicoli M.C. Antioxidant properties of tea extracts as affected by processing. Lebens-mittel-Wissenschaft Und-Technologie. 1998;31(7–8):694–698. [Google Scholar]
- Marcocci I., Marguire J.J., Droy-lefaiz M.T., Packer L. The nitric oxide scavenging properties of Ginkgo biloba extract. Biochem. Biophys. Res. Commun. 1994;201:748–755. doi: 10.1006/bbrc.1994.1764. [DOI] [PubMed] [Google Scholar]
- Marletta M.A. Nitric oxide: biosynthesis and biological significance. Trends Biochem. Sci. 1989;14(12):488–492. doi: 10.1016/0968-0004(89)90181-3. [DOI] [PubMed] [Google Scholar]
- McCord J., Fridovich I. Superoxide dismutase, an enzymic function for erythrocuprin. J. Biol. Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- Moncada S., Palmer R.M., Jr, Higgs E.A. Biosynthesis of nitric oxide from l-arginine: a pathway for the regulation of cell function and communication. Biochem. Pharmacol. 1989;38:1709–1715. doi: 10.1016/0006-2952(89)90403-6. [DOI] [PubMed] [Google Scholar]
- Meyer A.S., Isaksen A. Application of enzymes as food antioxidants. Trends Food Sci. Technol. 1995;6:300–304. [Google Scholar]
- Noro T., Oda Y., Miyase T., Ueno A., Fukushima S. Inhibitors of xanthine oxidase from the flowers and buds of Daphne genkwa. Chem. Pharm. Bull. 1983;31:3984–3987. doi: 10.1248/cpb.31.3984. [DOI] [PubMed] [Google Scholar]
- Ohkawa H., Onishi N., Yagi K. Assay for lipid peroxidation in animal tissue by thiobarbituric acid reaction. Anal. Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- Ottolenghi A. Interaction of ascorbic acid and mitochondria lipids. Arch. Biochem. Biophys. 1959;79:355. [Google Scholar]
- Ou B.X., Huang D.J., Hampsch-Woodill M., Flanagan J.A., Deemer E.K. Analysis of antioxidant activities of common vegetables employing oxygen radical absorbance capacity (ORAC) and ferric reducing antioxidant power (FRAP) assays: a comparative study. J. Agric. Food Chem. 2002;50(11):3122–3128. doi: 10.1021/jf0116606. [DOI] [PubMed] [Google Scholar]
- Oyaizu M. Studies on products of browning reactions: antioxidant activities of products of browning reaction prepared from glucosamine. J. Nutrit. 1986;44:307–315. [Google Scholar]
- Paglia, D.E., Valentin, W.N., l967. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 70, 158–169. [PubMed]
- Prieto P., Pineda M., Aguilar M. Spectrophotometric quantitation of antioxidant capacity through the formation of a phosphomolybdenum complex: specific application to the determination of vitamin E. Anal. Biochem. 1999;269:337–341. doi: 10.1006/abio.1999.4019. [DOI] [PubMed] [Google Scholar]
- Prior R.L., Hoang H., Gu L. Assays for hydrophilic and lipophilic antioxidant capacity (oxygen radical absorbance capacity (ORAC (FL)) of plasma and other biological and food samples. J. Agric. Food Chem. 2003;51:3273–3279. doi: 10.1021/jf0262256. [DOI] [PubMed] [Google Scholar]
- Ramakrishna B.S., Varghese R., Jayakumar S., Mathan M., Balasubramanian K.A. Circulating antioxidants in ulcerative colitis and their relationship to disease severity and activity. J. Gastroenterol. Hepatol. 1997;12:490–494. doi: 10.1111/j.1440-1746.1997.tb00471.x. [DOI] [PubMed] [Google Scholar]
- Robak J., Gryglewski R.J. Flavonoids are scavengers of superoxide anions. Biochem. Pharmacol. 1988;37:837–841. doi: 10.1016/0006-2952(88)90169-4. [DOI] [PubMed] [Google Scholar]
- Ruch R.J., Cheng S.J., Klaunig J.E. Prevention of cytotoxicity and inhibition of intercellular communication by antioxidant catechins isolated from Chinese green tea. Carcinogen. 1989;10:1003–1008. doi: 10.1093/carcin/10.6.1003. [DOI] [PubMed] [Google Scholar]
- Sapakal V.D., Shikalgar T.S., Ghadge R.V., Adnaik R.S., Naikwade N.S., Magdum C.S. In vivo screening of antioxidant profile: a review. J. Herbal Med. Toxicol. 2008;2(2):1–8. [Google Scholar]
- Sas K., Robotka H., Toldi J., Vecsei L. Mitochondrial, metabolic disturbances, oxidative stress and kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007;257:221–239. doi: 10.1016/j.jns.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Seeram N.P., Henning S.M., Lee R., Niu Y., Scheuller H.S., Heber D. Catechin and caffeine contents of green tea dietary supplements and correlation with antioxidant activity. J. Agric. Food Chem. 2006;54:1599–1603. doi: 10.1021/jf052857r. [DOI] [PubMed] [Google Scholar]
- Singh U., Jialal I. Oxidative stress and atherosclerosis. Pathophysiology. 2006;13:129–142. doi: 10.1016/j.pathophys.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Singhal P.K., Kapur N., Dhillon K.S., Beamish R.E., Dhalla N.S. Role of free radicals in catecholamine cardiomyopathy. Canadian J. Physiol. Pharmacol. 1982;60:1390. doi: 10.1139/y82-207. [DOI] [PubMed] [Google Scholar]
- Smith M.A., Rottkamp C.A., Nunomura A., Raina A.K., Perry G. Oxidative stress in Alzheimer’s disease. Biochim. Biophys. Acta. 2000;1502:139–144. doi: 10.1016/s0925-4439(00)00040-5. [DOI] [PubMed] [Google Scholar]
- Soler-Rivas C., Espin J.C., Wichers H.J. An easy and fast test to compare total free radical scavenger capacity of foodstuffs. Phytochem. Anal. 2000;11:330–338. [Google Scholar]
- Umesh B., Jagtap S.N., Panaskar, Bapat V.A. Evaluation of antioxidant capacity and phenol content in jackfruit (Artocarpus heterophyllus Lam.) fruit pulp. Plant Foods Hum. Nutr. 2010;65:99–104. doi: 10.1007/s11130-010-0155-7. [DOI] [PubMed] [Google Scholar]
- Upston J.M., Kritharides L., Stocker R. The role of vitamin E in atherosclerosis. Prog. Lipid Res. 2003;42:405–422. doi: 10.1016/s0163-7827(03)00024-9. [DOI] [PubMed] [Google Scholar]
- Virginia H., Sarah L.E., Rachel J.S., Nathaniel T., Joseph S., Adam E., Cecilia G. Mitochondrial nitric-oxide synthase: role in pathophysiology. IUBMB Life. 2003;55(10 &11):599–603. doi: 10.1080/15216540310001628681. [DOI] [PubMed] [Google Scholar]
- Wood J.L. In: Fishman W.H., editor. vol. II. Academic Press; New York: 1970. pp. 61–299. (Metabolic Conjugation and Metabolic Hydrolysis). [Google Scholar]
- Yang G., Chen J., Wen Z., Ge K., Zhu L., Chen X. The role of selenium in Keshan disease. Adv. Nutr. Res. 1984;6:203–220. doi: 10.1007/978-1-4613-2801-8_8. [DOI] [PubMed] [Google Scholar]