

Abstract
Background
Patients with advanced melanoma have a poor outcome. We hypothesize that combination immunotherapy can synergistically activate host immunity to generate an effective treatment for patients with high-risk, resected stage 3, recurrent, refractory, or stage 4 melanoma.
Methods
We conducted a phase 2 clinical trial of HyperAcute Melanoma (HAM) vaccine (NLG-12036, NewLink Genetics) combined with pegylated interferon (Sylatron, Merck). Trial design consisted of a 12-week regimen with the initial 4 weekly treatments consisting of HAM alone (intradermally) followed by 8 additional treatments of HAM plus Sylatron (subcutaneously, 6 μg/kg). Trial endpoint outcomes include clinical response, overall safety, and correlative findings for observed antitumor effect.
Results
Our cohort consisted of 25 patients with a median age of 60. Twenty-one patients completed the trial and 4 stopped because of progressive disease (PD). According to the Response Evaluation Criteria in Solid Tumors, of the 16 stage 4 patients, 2 had a complete response (CR), 1 had stable disease, and 4 had no evidence of disease (NED) after resection. For stage 2/3 patients, 3 of 9 remained NED, and the 1 stage 2C patient had slow PD with a single site resected and is currently NED. The median overall survival time was 29 months, with 60% of the patients surviving for >1 year. Of the 25 patients, 12 (48%) are still alive. All evaluable patients (21/21) seroconverted, developing autoimmune antibodies. Four of 25 patients developed vitiligo, correlating with 2 CR patients and 2 NED patients.
Conclusion
Combination immunotherapy with HAM plus Sylatron shows clinical efficacy with tumor regression and concomitant immune activation. Optimization of dosing schedules and therapeutic efficacy should be further explored to enhance the benefit of this promising immunotherapeutic approach.
Keywords: Alpha-galactosyl epitope, immunotherapy, melanoma, peginterferon alfa-2b, vitiligo
INTRODUCTION
In April 2010, the US Food and Drug Administration (FDA) approved the first active immunotherapy for the treatment of cancer, Provenge (sipuleucel-T), indicated for patients with metastatic, castrate-resistant prostate cancer. Within 2 years, 2 more immunotherapeutic agents were approved for the treatment of patients deemed to be at a high risk of systemic recurrence: pegylated interferon (IFN) (Sylatron) for patients with stage 3 melanoma and ipilimumab (Yervoy) for stage 4 disease. In the present report, we evaluate a novel combination immunotherapeutic approach using the expression of α(1,3)galactosyl epitopes (αGal) to induce tumor rejection (HyperAcute Melanoma [HAM] vaccine) combined with the recently FDA-approved agent, pegylated IFN α-2b (Sylatron).
The αGal epitopes are absent in human tissues, but host immune responses against these epitopes represent a potent mechanism of xenograft rejection. Our immune system is continuously stimulated by similar epitopes expressed by intestinal flora to produce antibodies that recognize αGal epitopes.1,2 These antibodies, many of which are complement activating, initiate hyperacute rejection of xenografted tissues expressing αGal epitopes. Such a hyperacute rejection is characterized by acute tissue damage occurring within minutes to hours posttransplantation and can facilitate antibody-dependent cell-mediated cytotoxicity.3-5 Immunity to αGal epitopes expressed on α-galactosyltransferase (αGT) genetically modified melanoma cells induced antitumor immunity in αGT knockout mice.6-8 Based on this data, αGal epitope-mediated hyperacute rejection was suggested as a potential therapeutic approach to treat human malignancies, particularly melanoma.9-13
The utility of systemic adjuvant therapy with IFN α-2b in melanoma patients at high risk for a systemic recurrence has been extensively analyzed. The initial FDA approval was based on the results from the large group Eastern Cooperative Oncology Group (ECOG) trial that demonstrated statistically significant relapse-free survival (RFS) and overall survival (OS) benefits in stage 2B and stage 3 melanoma patients treated with high-dose IFN α-2b. Subsequent studies have confirmed an improvement in RFS but have produced variable results regarding the true OS benefits.14-19 The lack of an overwhelmingly proven survival benefit, in association with its high cost and numerous adverse side effects, has detracted many oncologists, both in the United States and in Europe, from treating appropriately staged patients with a standard regimen of IFN α-2b. The introduction of pegylated IFN α-2b marked a significant advance in the available standard adjuvant therapies for high-risk melanoma. The alteration in chemical structure brought significant pharmacologic benefits, including a decreased rate of drug absorption following subcutaneous injection and reduced renal and cellular clearance.20 Subsequently, improved drug exposure, efficacy, and tolerability are achieved with pegylated INF α-2b compared to IFN α-2b.21,22
Recently, the final results of the European Organization for Research and Treatment of Cancer clinical trial that examined the role of adjuvant therapy with Sylatron in resected, stage 3 melanoma patients were published.22,23 In this phase 3 controlled trial, 1,256 patients were randomized after a complete node dissection to observation vs Sylatron (at a dose of 6 μg/kg/wk for the first 8 weeks, followed by 3 μg/kg/wk) for up to 5 years of treatment. At a median follow-up point of 3.8 years, this trial showed a highly statistically significant and sustained impact on RFS, with a 4-year RFS rate of 45.6% in the Sylatron group compared to 38.9% in the observation-only arm (hazard ratio=0.82, P=0.01). The difference in distant metastasis-free survival (DMFS) and OS was not statistically significant. Additionally, several other studies have examined the utility of pegylated IFN, either as a single agent or combined with other agents.24-27 As a result, the US FDA approved Sylatron on April 11, 2011, for the adjuvant treatment of stage 3 melanoma patients with microscopic or gross nodal involvement.
In our first-in-human-subjects, unique combination immunotherapy phase 2 study, we examined the safety, efficacy, and immune responses generated by a vaccine comprised of 3 allogeneic human melanoma cell lines genetically altered to express αGT (HAM vaccine) administered with Sylatron to patients with advanced melanoma.
METHODS
Patients
All patients 18 years of age or older were enrolled in the study if they met eligibility criteria, mainly American Joint Committee on Cancer stage 3B or 3C melanoma that had been surgically resected and rendered no evidence of disease (NED). Ulceration of the primary melanoma was annotated but not exclusionary if present. The same group of pathologists confirmed the Breslow depth of invasion for all patients. Patients were considered high risk if they had a thick primary melanoma (the stage 2C patient had a Breslow depth >4.0 mm) or had resected, stage 3 disease. Patients were considered refractory if their cancer recurred after standard approaches to treatment. Patients with stage 4 melanoma had distant, metastatic disease at the time of enrollment. All patients were required to have an ECOG performance status of 0 or 1 and adequate tests of hematologic, hepatic, and renal function. The ECOG performance status is a scale of patient function that ranges from 0 to 5 (0: fully active, 1: restricted in physical strenuous activity but ambulatory, 2: ambulatory >50% of the waking hours, 3: limited self-care, confined to bed or chair >50% of waking hours, 4: completely disabled, and 5: dead).
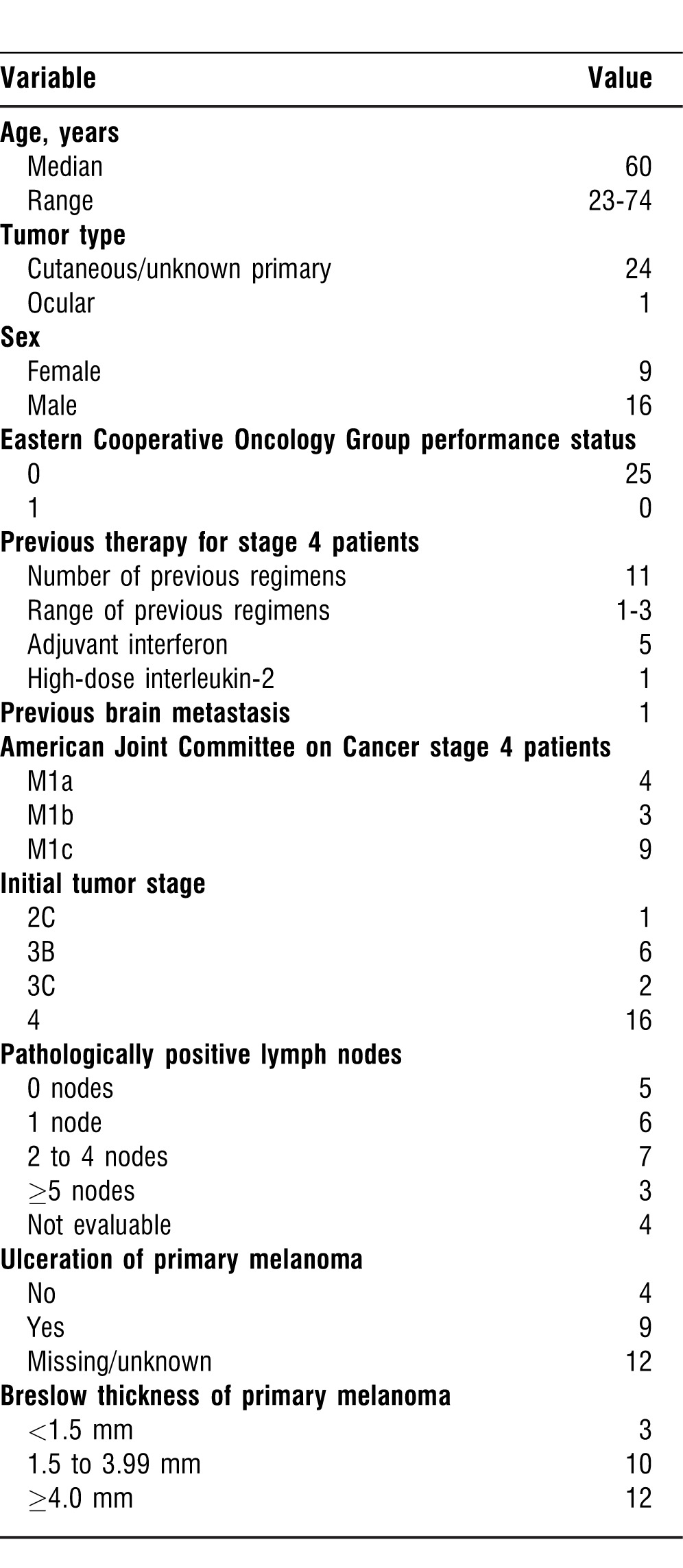
Only 1 patient with stage 2C disease was enrolled in the trial. All stage 3 patients were considered NED at the time of enrollment, with whole-body positron emission tomography (PET)/computed tomography (CT) scan, CT scan, and magnetic resonance imaging (MRI) or CT scan of the head confirming no evidence of metastatic disease. The stage 4 patients were a mixture of patients who had known metastatic disease at the time of enrollment and/or had been previously treated with surgical resection of metastatic disease. Previous adjuvant therapy or systemic therapy (chemotherapy, immunotherapy, or other clinical trials) for all patients (stages 3 and 4) was allowed for inclusion purposes. Patients with treated brain metastases without evidence of recurrence over 3 months and no new lesions identified also were allowed to participate. An institutional review board–approved written informed consent form was obtained from all patients enrolled in the trial. Table 1 outlines the patient demographics and baseline disease characteristics.
Table 1.
Patient Demographics and Characteristics (n=25)
HAM Formulation
The HAM vaccine comprises 3 components in equal doses, each containing an allogeneic melanoma tumor cell line genetically engineered to express αGT (HAM1, HAM2, and HAM3 cell lines). Each of the components was manufactured in a multistep biotechnology process beginning with transduction of a parental cancer cell line with a recombinant Moloney-based retroviral vector (LNCKG) to express the murine αGT cDNA.28,29 As a result, genetically transduced cells presented αGal residues on their cell surfaces. HAM cells were further characterized by their expression of cell surface markers, with all cell lines grown and expanded under good manufacturing practice conditions and lethally irradiated to prevent cell replication. A sterile suspension of cells mixed with 5% glycerol is cryopreserved and kept in a vapor phase of liquid nitrogen until use. Prior to administration, vaccine cells are quickly thawed and immediately drawn into syringes for administration within 30 minutes of complete thawing.
Study Design and Treatment
This study examined the safety and efficacy of the combination of the HAM vaccine and Sylatron. The study consisted of a 12-week regimen with the initial induction phase of 4 weekly treatments of HAM alone (50 million cells each of HAM1, HAM2, and HAM3), followed by 8 additional treatments of each HAM formulation (150 million cells total) of the same quantity and Sylatron (6 μg/kg). The HAM vaccine was administered as intradermal injections, while the Sylatron was administered subcutaneously. Guidelines for dose modification and reduction based upon adverse side effects were built into the protocol. Following the completion of the trial, all patients underwent regular follow-up visits and clinical examination every 3 months for the first year and every 6 months for the second year. Blood was taken for immunologic analyses from all patients at each follow-up interval.
Toxicity and Response Assessments
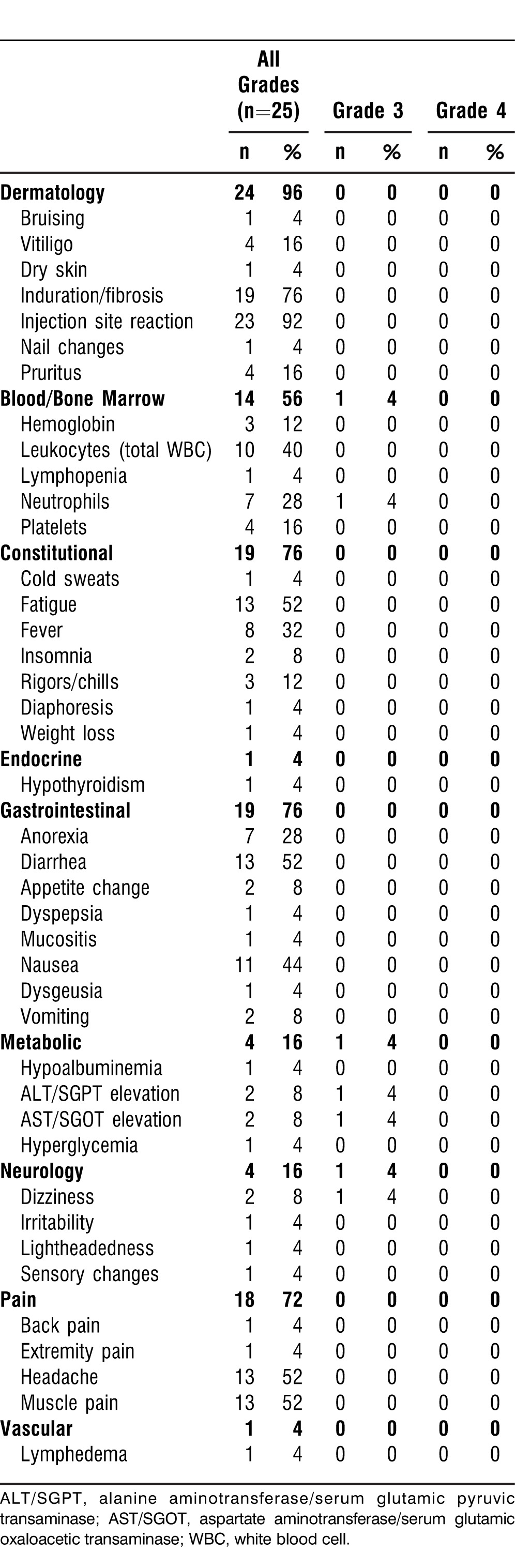
We utilized the descriptions and grading scales found in the National Cancer Institute Common Terminology Criteria for Adverse Events (version 3.0) for reporting of all adverse events (AEs). We assessed all clinical responses based upon the standard Response Evaluation Criteria in Solid Tumors (RECIST), version 1. Table 2 shows all of the reported AEs considered possibly, probably, or definitely related to the study regimen.
Table 2.
Adverse Events Attributed to the Treatment
All imaging studies were performed prior to enrollment and following the completion of the study. Clinically indicated studies were performed on an individual basis. Patients were classified as having a complete response (CR), NED, stable disease (SD), progressive disease (PD), or as dying as a direct result of their disease (DOD). Table 3 shows the individual clinical responses for the trial, and a summary of all responses is included in the text.
Table 3.
Patient Responses
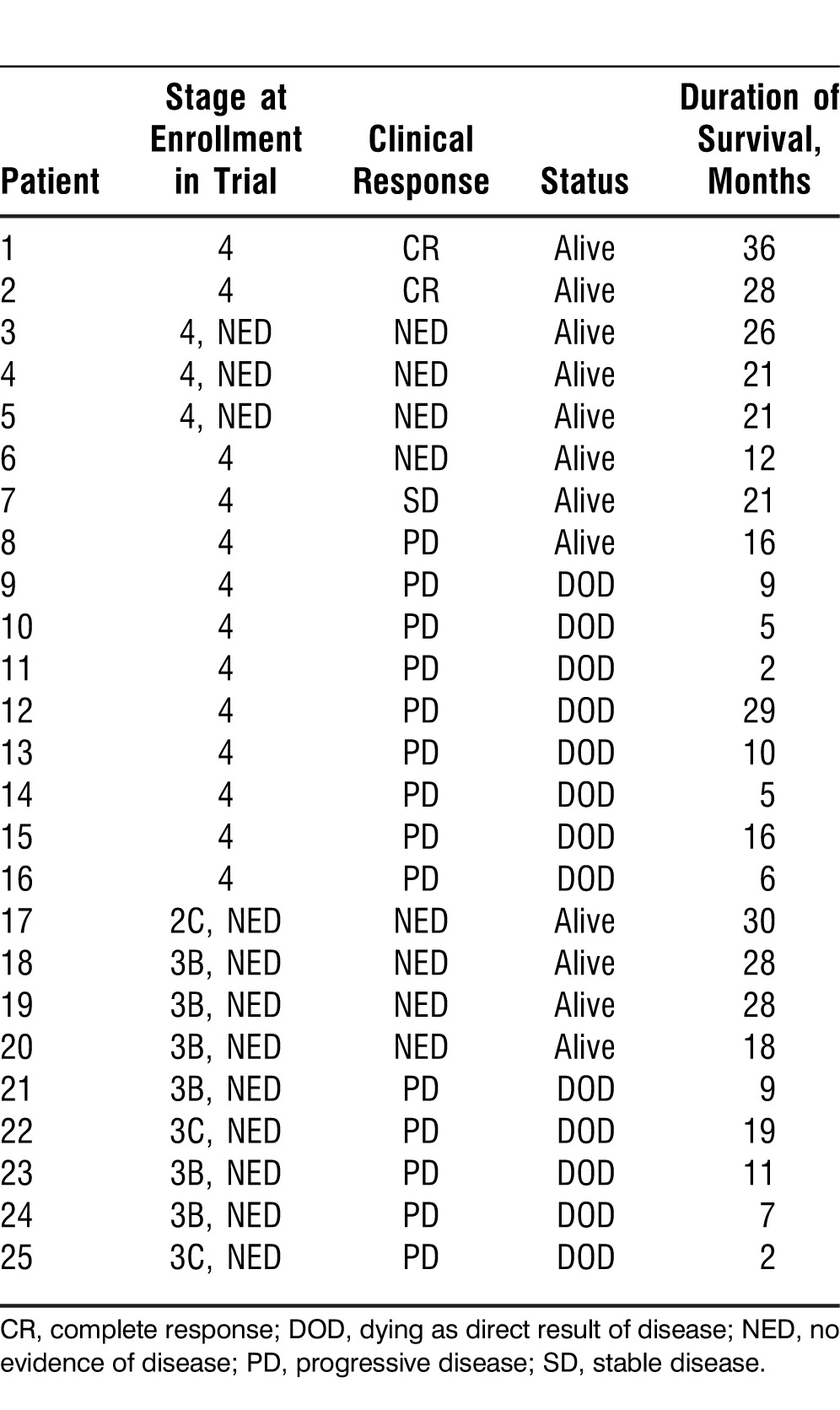
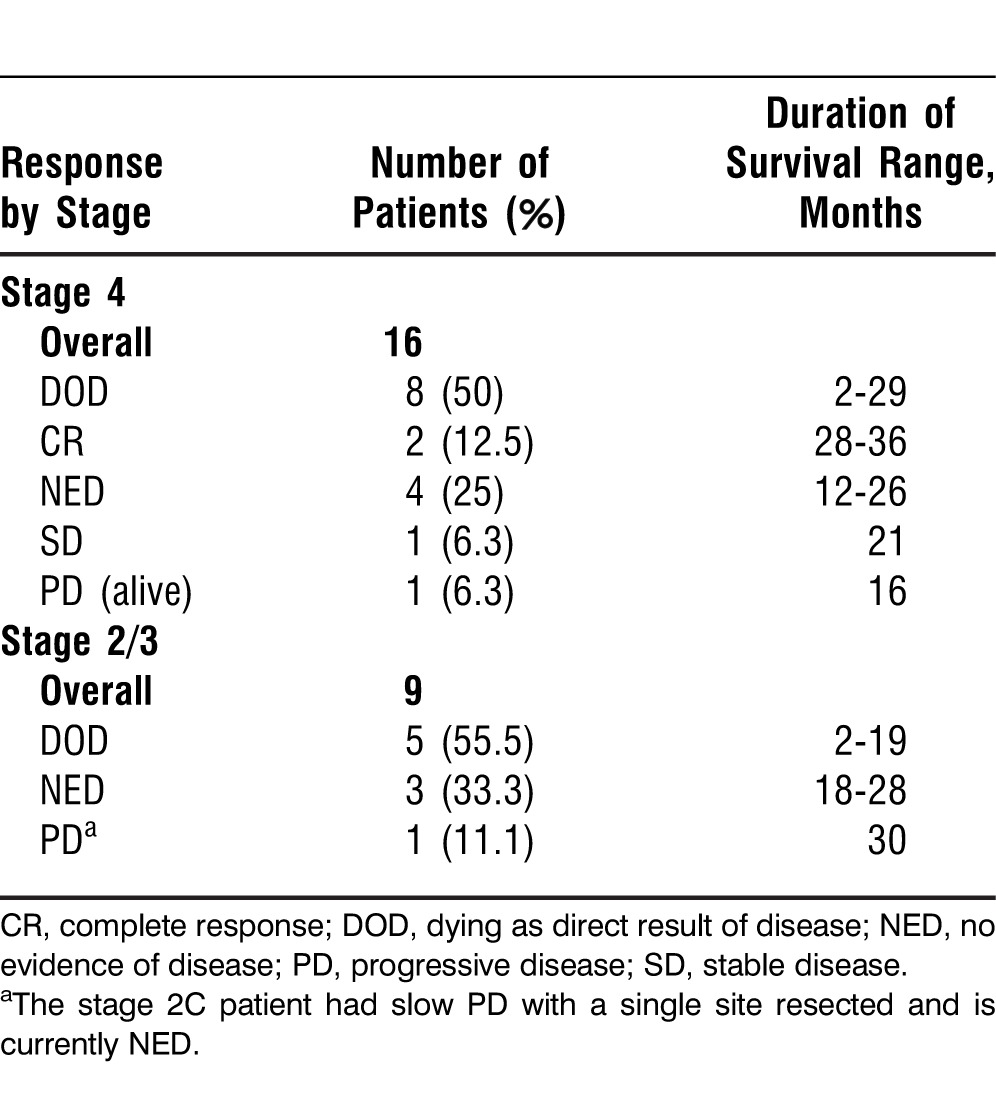
Table 4.
Summary of Clinical Responses
Statistical Methods
The study size was planned for 25 patients and designed as a single-arm phase 2 clinical trial. Based upon a previous phase 1 dose-escalation trial that used the HAM vaccine as monotherapy for 6 patients with stage 4 melanoma (Riker et al, unpublished data, 2006), we were able to ascertain a safe dose of the HAM vaccine. Similarly, safety data for the optimal maximal tolerated dose for Sylatron were readily accessible through ongoing clinical trials and published data.24-27 Performing a dose-escalation phase 1/2 trial with the combined agents was unnecessary because of the well-established safety profile of each agent. OS was defined as the time from registration to date of death, and progression-free survival (PFS) was defined as the amount of time that the patient was free of disease until the first clinical or radiographic evidence for recurrence of disease.
Kaplan-Meier methods were used to estimate median OS and PFS rates and the 12-month OS rate using SAS version 9.2 (SAS Institute). All other endpoints, including baseline characteristics, AE rates, and immunologic response parameters were summarized with descriptive statistics. The number of AEs for each patient was characterized according to the type of AE, severity (grade), and the time of onset in relation to each weekly administration.
Biomarkers
Certain biomarkers were assessed only once prior to enrollment (antigen recall skin test panel, human leukocyte antigen type, ABO/Rh blood typing, hepatitis panel, human immunodeficiency virus), while others were assessed at baseline, during each week or intermittently throughout the 12-week trial, and after the completion of the trial every year (complement activity, anti-αGal and antityrosinase antibodies, immunoglobulin [Ig] G, IgM, antinuclear antibodies, anti-DNA, antithyroglobulin, and anticardiolipin [phospholipid] antibodies). Most patients were tested throughout and after the trial for replication competent retrovirus at various time points.
Anti-αGal Antibodies and Antityrosinase Antibodies
Serum samples were collected prior to immunization; on days 15, 29, 43, 57, and 71; and every 2 months at follow-up visits. Serum was separated and stored at −80°C until assayed. Anti-αGal antibodies were detected by standard enzyme-linked immunosorbent assay techniques. Antityrosinase (anti-Tyr) antibodies (Ab) were calculated as the percent change in the anti-Tyr Ab with the following formula: (optical density[OD] test − OD baseline) / OD baseline × 100.
RESULTS
Patient Characteristics
Twenty-five patients (16 men, 9 women) aged 23 to 74 years old were enrolled between July 2008 and October 2010. Sixteen patients had stage 4 melanoma, 8 patients had stage 3 disease, and 1 patient had stage 2C disease. All of the stage 2/3 patients enrolled in the trial were NED, and all stage 4 patients had received at least 1 form of previous therapy (IFN, interleukin [IL]-2, immunotherapy, chemotherapy, clinical trial). One patient had previously treated brain metastasis, and 1 patient had known ocular melanoma, metastatic to the liver (Table 1).
Treatment Details
All patients underwent the same administration of the HAM vaccine: alone for the first 4 weeks followed by combined administration of the HAM vaccine (intradermal) and Sylatron (subcutaneous). Each component of the HAM vaccine was administered separately at 3 distinct sites on the skin, rotating the extremity each week. The HAM vaccine and Sylatron were not given within an extremity that had previously undergone a complete lymph node dissection. Three injections (HAM1, HAM2, HAM3) were given weekly. Each administration site was circled with a pen and the patient subsequently reported the immediate and delayed reactions at these sites that were clinically examined each week at the time of the next administration. The Sylatron was given subcutaneously along the proximal aspect of the same extremity as the HAM vaccine.
Safety
AEs related to the study regimen are summarized in Table 2. The most common AEs related to the combination were a hyperemic, indurated, and raised area at the site of intradermal HAM vaccine injection, seen in almost all patients. Constitutional symptoms, such as fatigue, lethargy, malaise, fever, rigors, and chills were common, limited to grade 1 or 2 toxicity. Other AEs reported were diarrhea (52%), nausea (44%), anorexia (28%), headache (52%), and muscle pain (52%).
Response
Response data were available for all 25 patients, with 21 completing the 12-week trial and subsequently providing follow-up for more than 3 years. Four patients did not complete the 12-week trial because of rapid PD.
According to standard RECIST criteria, 2 patients with stage 4 disease had a CR with regression of all metastatic disease. Additionally, 2 patients had SD, and 1 of these 2 patients subsequently became NED after a resection of a single metastatic lesion. Three patients with stage 4 disease continued to be NED (Table 3).
For the 9 patients who began the trial with resected (NED) stage 2/3 disease (1 patient with stage 2C disease, 8 with stage 3 disease), 4 remained NED after the completion of the trial and during the follow-up period. One of these 4 patients developed slow PD at a single site that was subsequently resected; the patient remains alive with an ECOG performance status of 0 at approximately 30 months.
All evaluable patients (21/21) seroconverted, developing autoimmune antibodies at the completion of the trial. Vitiligo developed in 4 of 25 patients, correlating with 2 CR and 2 NED patients.
Survival
The median follow-up period was 26 months, with the total length of follow-up for those still alive ranging from 12 to 36 months. The median PFS was 8.2 months (95% confidence interval [CI], 3.0 to 14.0 months). The median OS was 29 months (95% CI, 9 months to not yet reached). Kaplan-Meier plots cannot be reasonably applied to this study because of its small sample size and marked heterogeneity of the study population with different stages of disease. We therefore did not generate such plots and instead have discussed select cases in an empiric fashion to highlight some of the more interesting findings.
Biomarkers
All patients tested had detectable levels of anti-αGal Ab before receiving HAM immunotherapy. The baseline values of anti-αGal Ab varied between patients (mean 39, range 6-156 μg/mL) with immunization increasing the anti-αGal IgG responses in all patients. Twenty-four of 25 patients had increased anti-αGal Ab after immunization (mean 23, range 3-127 μg/mL), persisting for more than 300 days in the majority of patients. Select patients' serum levels are shown in Figure 1.
Figure 1.
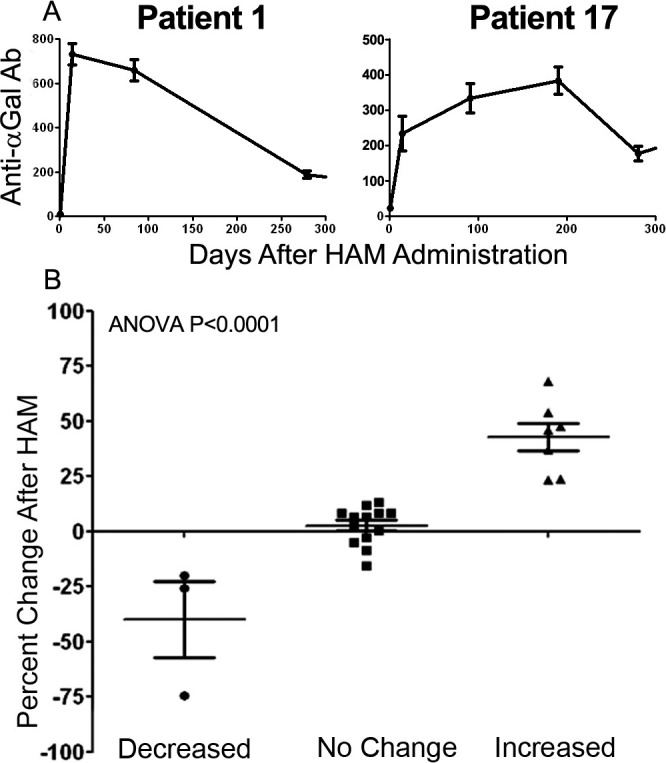
A: Level of anti-α galactosyl epitopes (Gal) antibodies induced after HyperAcute Melanoma (HAM) immunotherapy. B: Levels of anti-αGal antibodies of all 25 patients. The values of anti-αGal antibodies were determined using a standard consisting of an affinity purified human anti-αGal immunoglobulin by enzyme-linked immunosorbent assay. ANOVA, analysis of variance.
To determine if anti-Tyr Ab were induced after HAM immunotherapy, 2 serial blood samples were collected after HAM immunization. Twenty-three patients were tested for the presence of anti-Tyr Ab, with patients considered positive or reactive if they developed at least a 20% change in detectable anti-Tyr Ab. A 20% change appeared to correlate with a significant clustering of antibody response in patients (Figure 1). A total of 7 patients developed anti-Tyr Ab after immunization, and 3 patients had decreased levels during immunization.
DISCUSSION
Since the introduction of high-dose IL-2 therapy for patients with metastatic melanoma, several remarkable advances have improved our ability to treat patients with advanced melanoma. In recent years, these novel therapies have been developed as a direct result of an improved understanding of the host immune system and the mechanisms by which it is able to recognize and destroy tumor cells. Schwartzentruber et al showed that in patients with advanced metastatic melanoma, the addition of a gp100 specific peptide to high-dose IL-2 therapy significantly enhanced their PFS and overall response rates.30 This finding was soon followed by the development of 2 novel agents, vemurafenib and ipilimumab, that each use a different approach to treatment. Several landmark studies have shown these 2 agents are the first to provide a significant improvement in the OS of treated patients with metastatic melanoma.31-34
In an attempt to further the research focused upon the immunotherapy of melanoma, we designed a phase 2 study to assess the overall efficacy of a combination approach, utilizing 2 immunotherapeutic agents, specifically the HAM vaccine plus Sylatron. This small, phase 2 trial resulted in several interesting and compelling findings. Of the 16 patients with stage 4 disease, the most notable were 2 patients: 1 with dramatic tumor regression of all metastatic disease and 1 with CR of a single metastatic nodule that rendered the patient NED without evidence of recurrence at more than 28 months.
This immunotherapeutic combination appears to have a more beneficial effect than either component's individual use. However, this hypothesis is speculative because of the lack of synergism or combination effect, as the expected recurrence rate for stage 2/3 patients is clearly not 100%. In fact, a portion of these patients might have been cured by their operations that rendered them NED; therefore, the treatment strategy would have had no impact upon their ultimate long-term survival. However, select cases in this study clearly demonstrate that this treatment regimen resulted in the regression of metastatic disease and subsequently resulted in long-term survival. One patient clearly expressed this benefit, experiencing a very slow and steady CR of all disease following the completion of the trial.
For the stage 2/3 patients, several continue to be without evidence of disease, with the longest follow-up at 30 months thus far. In these high-risk patients, a longer follow-up will be necessary to determine the durability and long-term efficacy of this treatment combination. Compared to historical controls, the results of this trial suggest some possible evidence of a combined effect, compared to monotherapy, as several patients are without evidence of a systemic recurrence at >2 years following the completion of the study.
Patients Experiencing CR
The first patient with a CR began the trial in November 2008 and completed it in February 2009. This patient tolerated the 12-week regimen, with the exception of a single-dose reduction of 50% of the Sylatron secondary to dehydration. Upon completion of the trial, the patient began to develop vitiligo at the 3 original sites of the HAM vaccine along the left forearm, followed by a slow, but continuous, resolution of the in-transit disease along the left lower extremity. By 5 months posttrial, the patient had complete resolution of all disease except for a remaining large, matted, nodal mass in the left groin. Some questionable hypermetabolic lymph nodes within the celiac axis and paraaortic area were identified during whole-body PET/CT fusion scanning. As we continued to follow this patient, contemplating surgically debulking the left groin nodes, the matted nodes slowly and steadily regressed over the next 5 months. By 18 months posttrial, the left groin showed no evidence of palpable disease on physical examination. Continued follow-up for >36 months posttrial has shown complete tumor regression of all residual disease, without any evidence of recurrent disease on serial whole-body PET/CT scanning (Figure 2). This patient has returned to normal activity (ECOG performance status of 0) and continues to have several patchy areas of vitiligo.
Figure 2.

Whole-body scans and immunohistochemistry of a patient with a complete response to therapy. A: Serial whole-body positron emission tomography/computed tomography scanning of a patient who developed a complete regression of metastatic melanoma. Scans show before and after treatment results. Mo, months. B: Immunohistochemistry of resected stable tumor mass after competing therapy. H&E, hematoxylin and eosin staining; HMB-45, melanoma marker antibody staining; CD68, macrophage lineage staining; CD56, natural killers staining; CD20, B-cell lineage staining; CD3, T-cell staining; CD4, staining for helper T cells; CD8, staining for cytotoxic T cells.
The second patient with a CR presented with a single metastatic nodule along the left forearm as the only remaining site of disease. The patient completed the 12-week trial; however, the patient also had to omit 1 week of the trial because of grade 4 toxicity with dehydration. After rehydration, the patient completed the remainder of the trial. During week 6 of the trial, the left forearm nodule began to diminish in size, and by week 10, it had completely regressed. As in the case of the previous CR, grade 4 toxicity was seen during Sylatron administration with resultant dehydration and intermittent dose reduction or stoppage. The patient completed the trial in August 2009 and remains NED at 28 months.
Tumor Destruction
An interesting patient from this trial highlights a case of direct immunologic evidence of tumor destruction from the host immune system. The patient was 23 years old, the youngest patient in the trial, and his primary melanoma was identified in the middle of the back. He underwent a wide local excision of the primary melanoma, followed by bilateral axillary lymph node dissections because of the involvement of the sentinel lymph nodes bilaterally with metastatic melanoma. Just prior to enrollment in the trial, a whole-body PET/CT identified a 1 cm hypermetabolic nodule along the right back, overlying the scapula. A fine needle aspiration biopsy of the nodule showed that it was an isolated, subcutaneous, metastatic melanoma nodule in the soft tissue overlying the right scapula.
No other lesions were identified, and he successfully completed the trial without dose reduction. Posttrial imaging with PET/CT fusion scan did not reveal any further evidence of new or progressive metastatic disease. We resected the single metastatic nodule that had remained about the same size throughout the trial and thoroughly analyzed the resected lesion with a panel of immunohistochemical studies to identify the infiltrated cells within the tumor mass. We found a massive infiltration of CD8+ T cells (99%) and few CD4+ cells (<1%) in a background of necrotic tumor cells, with little evidence of a remaining viable tumor (not shown). This patient remains without evidence of recurrent or metastatic disease at >1 year. Therefore, we report direct immunologic activation of the host immune system with a dramatic infiltration of CD8+ T cells into the tumor mass, presumably as a result of this combination immunotherapy approach.
Vitiligo
Prior to enrollment, 9 patients had an initial staging of stage 2C, 3B, or 3C disease. Of these, 5 patients died from PD, 1 had limited single-site PD and is currently NED with an OS of 30 months, and 3 (33%) remain NED with a median RFS/OS of 23 months. All 3 NED patients were staged as 3B, successfully completed the 12-week regimen, and subsequently developed autoimmune antibodies. Two of these patients had clinical manifestations of autoimmune depigmentation (vitiligo). Vitiligo did not develop within any of the nonresponders in this study.
Clinical vitiligo has been shown to develop in parallel with the clinical responses to various forms of immunotherapy regimens in melanoma patients. Vitiligo occurs via a loss of epidermal melanocytes with accompanying skin infiltration of cytotoxic T lymphocytes (CTLs).35 The exact pathogenesis of vitiligo is unknown, with hypotheses ranging from an autoimmune mechanism in response to the breaking of self-tolerance to neural, biochemical, genetic, and structural defect pathways. The association of vitiligo with other autoimmune disorders such as thyroid disease, lymphomas, and organ-specific autoantibodies has long been noted, with several immune-modulating therapies, such as IFN, implicated in the induction of vitiligo.36
Both melanocytes and melanoma cells express the shared melanocyte differentiation antigens Melan A/MART1, gp100, tyrosinase, TRP-1, and TRP-2, all of which are recognized by antigen-specific CTLs, especially CD8+ T cells.37,38 Elevated circulating levels of anti-melanocyte-specific CD8+ T cells, along with the presence of melanocyte-specific antibodies and tumor-infiltrating lymphocytes (TIL), strongly implicate a host-mediated immune response as the likely mechanism for vitiligo formation.39,40 Vitiligo affects approximately 3% of patients with melanoma, and this number increases in patients who receive various forms of immunotherapy.37
Investigation by Boasberg et al showed the development of vitiligo to be a non–time-dependent covariate for improved survival in melanoma patients, with median survival of 18.2 months for vitiligo vs 8.5 months for patients without vitiligo.35 Quaglino et al also demonstrated an increased overall 5-year survival in stage 3 patients with vitiligo (65% vs 42.5% without vitiligo) and higher DMFS over 5 years (52.4% with vitiligo vs 21.5% without), conversely citing vitiligo as a time-dependent covariate for prognosis.41 They also found a prognostic advantage in stage 4 patients who developed vitiligo both before and after the demonstration of metastatic disease.41 The results of our study lend further support for the correlation of the development of vitiligo and a clinical response after combination immunotherapy. However, this study is much too small, with the development of vitiligo seen in too few patients, to draw any conclusions about the development of vitiligo as a true predictor of clinical response.
Autoimmune Antibodies
The development of autoimmune antibodies results from similar antigenicity between self-antigens and foreign antigens. A certain level of autoimmunity is necessary for an effective immunostimulatory treatment for melanoma. Multiple studies have shown that patients commonly develop autoimmune antibodies after treatment with IFN. Bouwhuis et al suggest that the prognostic significance of the development of autoimmune antibodies after treatment with adjuvant IFN is incongruent, with several models yielding different outcomes.22
Gogas et al examined 200 patients with melanoma who were previously treated with IFN to assess the prognostic significance of developing autoimmune antibodies after treatment with IFN α-2b.42 They looked for the development of autoimmune antibodies, specifically antithyroglobulin, antinuclear, anti-DNA, and anticardiolipin (phospholipid) antibodies. Patients were also examined for the development of vitiligo after treatment and other clinical signs of autoimmunity. Of the 52 patients who developed autoimmune antibodies or developed clinical signs of autoimmunity, only 7 patients had recurrence of disease. In comparison, of 148 who did not seroconvert and develop autoimmune antibodies, 108 patients had a recurrence of disease. The patients who developed autoantibodies also had higher RFS rates and greater median OS compared to those who did not. Gogas et al concluded that the development of autoantibodies after treatment with IFN α-2b treatment had prognostic implications that translated into improved clinical outcomes.42
Based upon this trial, we chose to examine the same set of autoimmune antibodies for our study. In a striking finding, we found that all patients tested (21/21, 100%) who began the trial without evidence of autoimmune antibodies seroconverted, developing 1 or more antibodies after completing the trial. Specifically, we found that all patients developed antithyroglobulin and/or anticardiolipin antibodies. Although the overall clinical significance of this peculiar finding remains unanswered, that 100% of the patients developed autoimmune antibodies, far above the frequency of any previous study to date that has utilized IFN, is quite compelling. Interestingly, the 2 patients with CRs in our study both developed clinical signs of vitiligo, and 2 others (NED patients) also showed evidence of autoimmunity and remain NED as of spring 2014. Although several possible explanations exist for the development of autoimmune antibodies, for all our tested patients to develop autoimmune antibodies following the completion of the trial is a striking finding. The exact immunologic mechanism has yet to be determined, but this combination of agents appears to have measurable activity in terms of developing autoimmune antibodies.
CONCLUSION
Combination therapy using the HAM vaccine and Sylatron is feasible, safe, and shows promising efficacy data. This combination is capable of inducing complete and durable clinical responses with dramatic regression of bulky metastatic disease. This clinical effect is associated with the specific infiltration of CTL and direct evidence of TIL within metastatic melanoma lesions by the host immune system. The development of autoimmunity in all patients in this trial is significant, as our study is the first to date that shows such a high conversion rate. Future clinical trials are currently in development that will address the optimal dosing and duration of this combination, as well as other potentially synergistic combinations.
Footnotes
The following authors have financial interest in NewLink Genetics: Gabriela R. Rossi, PhD; Prerna Masih, PhD; Lucinda Tennant, RN, MS; W. Jay Ramsey, MD, PhD; Charles J. Link, MD; and Nicholas N. Vahanian, MD, MBA. The remaining authors have no financial or proprietary interest in the subject matter of this article.
This article meets the Accreditation Council for Graduate Medical Education and the American Board of Medical Specialties Maintenance of Certification competencies for Patient Care, Medical Knowledge, Systems-Based Practice, and Practice-Based Learning and Improvement.
REFERENCES
- 1.Galili U, Shohet SB, Kobrin E, Stults CL, Macher BA. Man, apes, and Old World monkeys differ from other mammals in the expression of alpha-galactosyl epitopes on nucleated cells. J Biol Chem. 1988 Nov 25;263(33):17755–17762. [PubMed] [Google Scholar]
- 2.Galili U, Mandrell RE, Hamadeh RM, Shohet SB, Griffiss JM. Interaction between human natural anti-alpha-galactosyl immunoglobulin G and bacteria of the human flora. Infect Immun. 1988 Jul;56(7):1730–1737. doi: 10.1128/iai.56.7.1730-1737.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumann BC, Forte P, Hawley RJ, Rieben R, Schneider MK, Seebach JD. Lack of galactose-alpha-1,3-galactose expression on porcine endothelial cells prevents complement-induced lysis but not direct xenogeneic NK cytotoxicity. J Immunol. 2004 May 15;172(10):6460–6467. doi: 10.4049/jimmunol.172.10.6460. [DOI] [PubMed] [Google Scholar]
- 4.Schaapherder AF, Daha MR, te Bulte MT, van der Woude FJ, Gooszen HG. Antibody-dependent cell-mediated cytotoxicity against porcine endothelium induced by a majority of human sera. Transplantation. 1994 May 15;57(9):1376–1382. doi: 10.1097/00007890-199405150-00016. [DOI] [PubMed] [Google Scholar]
- 5.Watier H, Guillaumin JM, Vallée I, et al. Human NK cell-mediated direct and IgG-dependent cytotoxicity against xenogeneic porcine endothelial cells. Transpl Immunol. 1996 Dec;4(4):293–299. doi: 10.1016/s0966-3274(96)80050-5. [DOI] [PubMed] [Google Scholar]
- 6.Rossi GR, Mautino MR, Unfer RC, Seregina TM, Vahanian N, Link CJ. Effective treatment of preexisting melanoma with whole cell vaccines expressing alpha (1,3)-galactosyl epitopes. Cancer Res. 2005 Nov 15;65(22):10555–10561. doi: 10.1158/0008-5472.CAN-05-0627. [DOI] [PubMed] [Google Scholar]
- 7.Rossi GR, Unfer RC, Seregina T, Link CJ. Complete protection against melanoma in absence of autoimmune depigmentation after rejection of melanoma cells expressing alpha(1,3)galactosyl epitopes. Cancer Immunol Immunother. 2005 Oct;54(10):999–1009. doi: 10.1007/s00262-005-0667-4. Epub 2005 May 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossi GR, Mautino MR, Awwad DZ, et al. Allogeneic melanoma vaccine expressing alphaGal epitopes induces antitumor immunity to autologous antigens in mice without signs of toxicity. J Immunother. 2008 Jul-Aug;31(6):545–554. doi: 10.1097/CJI.0b013e31817d2f45. [DOI] [PubMed] [Google Scholar]
- 9.Link CJ, Jr, Seregina T, Atchison R, Hall A, Muldoon R, Levy JP. Eliciting hyperacute xenograft response to treat human cancer: alpha(1,3) galactosyltransferase gene therapy. Anticancer Res. 1998 Jul-Aug;18((4A)):2301–2308. [PubMed] [Google Scholar]
- 10.Galili U, LaTemple DC. Natural anti-Gal antibody as a universal augmenter of autologous tumor vaccine immunogenicity. Immunol Today. 1997 Jun;18(6):281–285. doi: 10.1016/s0167-5699(97)80024-2. [DOI] [PubMed] [Google Scholar]
- 11.Posekany KJ, Pittman HK, Swanson MS, Haisch CE, Verbanac KM. Suppression of Lewis lung tumor development in alpha 1,3 galactosyltransferase knock-out mice. Anticancer Res. 2004 Mar-Apr;24((2B)):605–612. [PubMed] [Google Scholar]
- 12.Galili U. Autologous tumor vaccines processed to express alpha-gal epitopes: a practical approach to immunotherapy in cancer. Cancer Immunol Immunother. 2004 Nov;53(11):935–945. doi: 10.1007/s00262-004-0524-x. Epub 2004 Jun 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris JC, Rossi GR, Janik JE, et al. Phase I/II study of antitumor vaccination using lung cancer cells expressing murine α(1,3)galactosyltransferase (αGT) in non-small cell lung cancer (NSCLC) Cancer Res. 2010;70(8) AACR Annual Meeting abstract 2423. April 15. Suppl 1. [Google Scholar]
- 14.Hardacre JM, Mulcahy MF, Talamoni M, et al. Effect of hyperacute immunotherapy in addition to standard adjuvant therapy for resected pancreatic cancer on disease-free and overall survival: Preliminary analysis of phase II data. J Clin Oncol. 2010;28 Abstract 4059. 15 Suppl. [Google Scholar]
- 15.Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996 Jan;14(1):7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 16.Kirkwood JM, Ibrahim JG, Sondak VK, et al. High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol. 2000 Jun;18(12):2444–2458. doi: 10.1200/JCO.2000.18.12.2444. [DOI] [PubMed] [Google Scholar]
- 17.Kirkwood JM, Bender C, Agarwala S, et al. Mechanisms and management of toxicities associated with high-dose interferon alfa-2b therapy. J Clin Oncol. 2002 Sep 1;20(17):3703–3718. doi: 10.1200/JCO.2002.03.052. [DOI] [PubMed] [Google Scholar]
- 18.Brandberg Y, Aamdal S, Bastholt L, et al. Health-related quality of life in patients with high-risk melanoma randomised in the Nordic phase 3 trial with adjuvant intermediate-dose interferon alfa-2b. Eur J Cancer. 2012 Sep;48(13):2012–2019. doi: 10.1016/j.ejca.2011.11.019. Epub 2011 Dec 22. [DOI] [PubMed] [Google Scholar]
- 19.Giannelli G, Antonelli G, Fera G, et al. Biological and clinical significance of neutralizing and binding antibodies to interferon-alpha (IFN-alpha) during therapy for chronic hepatitis C. Clin Exp Immunol. 1994 Jul;97(1):4–9. doi: 10.1111/j.1365-2249.1994.tb06571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grace M, Youngster S, Gitlin G, et al. Structural and biologic characterization of pegylated recombinant IFN-alpha2b. J Interferon Cytokine Res. 2001 Dec;21(12):1103–1115. doi: 10.1089/107999001317205240. [DOI] [PubMed] [Google Scholar]
- 21.Glue P, Fang JW, Rouzier-Panis R, et al. Pegylated interferon-alpha2b: pharmacokinetics, pharmacodynamics, safety, and preliminary efficacy data. Hepatitis C Intervention Therapy Group. Clin Pharmacol Ther. 2000 Nov;68(5):556–567. doi: 10.1067/mcp.2000.110973. [DOI] [PubMed] [Google Scholar]
- 22.Bouwhuis MG, Suciu S, Testori A, et al. Phase III trial comparing adjuvant treatment with pegylated interferon Alfa-2b versus observation: prognostic significance of autoantibodies—EORTC 18991. J Clin Oncol. 2010 May 10;28(14):2460–2466. doi: 10.1200/JCO.2009.24.6264. Epub 2010 Apr 12. [DOI] [PubMed] [Google Scholar]
- 23.Eggermont AM, Suciu S, Santinami M, et al. EORTC Melanoma Group. Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet. 2008 Jul 12;372(9633):117–126. doi: 10.1016/S0140-6736(08)61033-8. [DOI] [PubMed] [Google Scholar]
- 24.Dummer R, Garbe C, Thompson JA, et al. Randomized dose-escalation study evaluating peginterferon alfa-2a in patients with metastatic malignant melanoma. J Clin Oncol. 2006 Mar 1;24(7):1188–1194. doi: 10.1200/JCO.2005.04.3216. [DOI] [PubMed] [Google Scholar]
- 25.Hwu WJ, Panageas KS, Menell JH, et al. Phase II study of temozolomide plus pegylated interferon-alpha-2b for metastatic melanoma. Cancer. 2006 Jun 1;106(11):2445–2451. doi: 10.1002/cncr.21909. [DOI] [PubMed] [Google Scholar]
- 26.Bukowski RM, Tendler C, Cutler D, Rose E, Laughlin MM, Statkevich P. Treating cancer with PEG Intron: pharmacokinetic profile and dosing guidelines for an improved interferon-alpha-2b formulation. Cancer. 2002 Jul 15;95(2):389–396. doi: 10.1002/cncr.10663. [DOI] [PubMed] [Google Scholar]
- 27.Bukowski R, Ernstoff MS, Gore ME, et al. Pegylated interferon alfa-2b treatment for patients with solid tumors: a phase I/II study. J Clin Oncol. 2002 Sep 15;20(18):3841–3849. doi: 10.1200/JCO.2002.02.051. [DOI] [PubMed] [Google Scholar]
- 28.Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989 Oct;7984-986(9):980–982. 989–990. [PMC free article] [PubMed] [Google Scholar]
- 29.Young WB, Link CJ., Jr Chimeric retroviral helper virus and picornavirus IRES sequence to eliminate DNA methylation for improved retroviral packaging cells. J Virol. 2000 Jun;74(11):5242–5249. doi: 10.1128/jvi.74.11.5242-5249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011 Jun 2;364(22):2119–2127. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010 Aug 19;363(8):711–723. doi: 10.1056/NEJMoa1003466. Epub 2010 Jun 5. Erratum in: N Engl J Med. 2010 Sep 23;363(13):1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011 Jun 30;364(26):2517–2526. doi: 10.1056/NEJMoa1104621. Epub 2011 Jun 5. [DOI] [PubMed] [Google Scholar]
- 33.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010 Aug 26;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010 Sep 30;467(7315):596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boasberg PD, Hoon DS, Piro LD, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. 2006 Dec;126(12):2658–2663. doi: 10.1038/sj.jid.5700545. Epub 2006 Aug 31. [DOI] [PubMed] [Google Scholar]
- 36.Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003 Apr;16(2):90–100. doi: 10.1034/j.1600-0749.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 37.Byrne KT, Turk MJ. New perspectives on the role of vitiligo in immune responses to melanoma. Oncotarget. 2011 Sep;2(9):684–694. doi: 10.18632/oncotarget.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byrne KT, Côté AL, Zhang P, et al. Autoimmune melanocyte destruction is required for robust CD8+ memory T cell responses to mouse melanoma. J Clin Invest. 2011 May;121(5):1797–1809. doi: 10.1172/JCI44849. Epub 2011 Apr 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palermo B, Garbelli S, Mantovani S, Giachino C. Transfer of efficient anti-melanocyte T cells from vitiligo donors to melanoma patients as a novel immunotherapeutical strategy. J Autoimmune Dis. 2005 Aug 31;2:7. doi: 10.1186/1740-2557-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garbelli S, Mantovani S, Palermo B, Giachino C. Melanocyte-specific, cytotoxic T cell responses in vitiligo: the effective variant of melanoma immunity? Pigment Cell Res. 2005 Aug;18(4):234–242. doi: 10.1111/j.1600-0749.2005.00244.x. [DOI] [PubMed] [Google Scholar]
- 41.Quaglino P, Marenco F, Osella-Abate S, et al. Vitiligo is an independent favourable prognostic factor in stage III and IV metastatic melanoma patients: results from a single-institution hospital-based observational cohort study. Ann Oncol. 2010 Feb;21(2):409–414. doi: 10.1093/annonc/mdp325. Epub 2009 Jul 21. [DOI] [PubMed] [Google Scholar]
- 42.Gogas H, Ioannovich J, Dafni U, et al. Prognostic significance of autoimmunity during treatment of melanoma with interferon. N Engl J Med. 2006 Feb 16;354(7):709–718. doi: 10.1056/NEJMoa053007. [DOI] [PubMed] [Google Scholar]