

RNA-binding protein CsrA is a key regulator of a variety of cellular process in bacteria, including biofilm formation. Figueroa-Bossi et al. uncover a novel gene regulatory mechanism through which CsrA inhibits translation initiation. CsrA binding to the 5′ UTR of E. coli pgA mRNA—involved in biofilm formation—unfolds a secondary structure that sequesters an entry site for termination factor Rho, resulting in the premature stop of transcription. CsrA-induced transcription termination thus marks the switch from the sessile to the planktonic lifestyle.
Keywords: CsrA, Rho factor, transcription attenuation, bacterial gene regulation
Abstract
RNA-binding protein CsrA is a key regulator of a variety of cellular processes in bacteria, including carbon and stationary phase metabolism, biofilm formation, quorum sensing, and virulence gene expression in pathogens. CsrA binds to bipartite sequence elements at or near the ribosome loading site in messenger RNA (mRNA), most often inhibiting translation initiation. Here we describe an alternative novel mechanism through which CsrA achieves negative regulation. We show that CsrA binding to the upstream portion of the 5′ untranslated region of Escherichia coli pgaA mRNA—encoding a polysaccharide adhesin export protein—unfolds a secondary structure that sequesters an entry site for transcription termination factor Rho, resulting in the premature stop of transcription. These findings establish a new paradigm for bacterial gene regulation in which remodeling of the nascent transcript by a regulatory protein promotes Rho-dependent transcription attenuation.
Bacterial adaptation to changing environments relies on the ability of the bacterial cell to coordinately regulate groups of genes in response to chemical and/or physical signals. A variety of transcriptional and post-transcriptional mechanisms, often interconnected, underlie these global responses. One such mechanism involves a small (61-amino-acid) protein, named CsrA, acting as a post-transcriptional regulator. Originally discovered as a repressor of glycogen biosynthesis in Escherichia coli (Romeo et al. 1993), CsrA was subsequently found to regulate other growth phase-dependent processes such as biofilm formation and quorum sensing in E. coli and Vibrio cholerae (Jackson et al. 2002; Lenz et al. 2005; Wang et al. 2005; Yakhnin et al. 2011) and host invasion in Salmonella (Altier et al. 2000; Lawhon et al. 2003; Martinez et al. 2011). The CsrA structural homologs RsmA/RsmE perform similar functions in Pseudomonas and Erwinia species, where they participate in the regulation of secondary metabolism (Chatterjee et al. 1995; Cui et al. 1995; Pessi et al. 2001; Lapouge et al. 2007). In all of these systems, CrsA and its homologs act as translational repressors; they interact with messenger RNA (mRNA) sequences overlapping ribosome-binding sites and thus block translation initiation by occluding these sites (for reviews, see Babitzke and Romeo 2007; Lapouge et al. 2008; Romeo et al. 2013). Occasionally, CsrA can also act as a positive regulator, protecting mRNA against degradation by RNase E (Yakhnin et al. 2013) or remodeling translation initiation regions in ways that stimulate ribosome binding (Patterson-Fortin et al. 2013; Sterzenbach et al. 2013).
A central aspect in CsrA biology is in the mechanism that regulates CsrA activity. This occurs as a result of the production of two small RNAs, CsrB and CsrC, which contain several CsrA-binding sites and sequester the protein by molecular mimicry (Babitzke and Romeo 2007; Lapouge et al. 2008; Romeo et al. 2013). The csrB and csrC genes are under the control of the BarA/UvrY (SirA) two-component signal transduction system and are transcriptionally activated in the presence of short chain carboxylic acids and other products of carbon metabolism (Takeuchi et al. 2009; Chavez et al. 2010). In addition, CsrA indirectly activates csrB and csrC transcription via the BarA/UvrY (SirA) system by an unknown mechanism. Combined with the ability of CsrA to feedback-inhibit translation of its own mRNA (Gudapaty et al. 2001; Suzuki et al. 2002), this provides an additional homeostatic loop that likely allows CsrA activity to be finely adjusted at all times (Romeo et al. 2013). Two novel mechanisms modulating CsrA activity/availability were unveiled recently. In one system, McaS, a regulatory small RNA known to function via classical Hfq-dependent RNA pairing, was found to bind CsrA and relieve repression of some CsrA targets (Jorgensen et al. 2013). In another system, not a small RNA, but rather the 5′ leader region of fimbrial mRNA cooperates with CsrB and CsrC in titrating CsrA, preventing CsrA-mediated activation of a second hierarchically controlled fimbrial mRNA (Sterzenbach et al. 2013).
CsrA binds short RNA sequences containing a centrally positioned GGA motif that is essential for recognition (Dubey et al. 2005). The high conservation of this motif in Shine-Dalgarno sequences accounts for the predominant role of CsrA as a repressor of translation initiation. Since the functional form of CsrA is a homodimer, tandem binding sites are generally required for a productive interaction (Schubert et al. 2007; Mercante et al. 2009), with the protein bridging GGA sites anywhere between 10 and 63 nucleotides (nt) apart (Mercante et al. 2009). Some regulated genes have multiple CsrA-binding sites in their control regions. This is the case for the pgaABCD operon of E. coli, which encodes proteins needed for the synthesis and export of a polysaccharide adhesin involved in biofilm formation (Itoh et al. 2008). Work from the Romeo and Babitzke laboratories (Wang et al. 2005) identified six CsrA-binding sites in the 234-nt 5′ untranslated region (UTR) of pgaABCD mRNA and provided in vitro and in vivo evidence that two distal sites, which cover the Shine-Dalgarno sequence and the initiating AUG, are required for translational repression by CsrA. The roles of the other CsrA-binding sites or the reasons for their redundancy have remained elusive.
We became interested in the regulation of the pgaABCD locus while data-mining for links between small RNA activity and Rho-dependent transcription termination. This search, prompted by recent findings in our laboratories (Bossi et al. 2012), was made possible by the publication of a high-resolution map of Rho-dependent termination sites in the E. coli genome (Peters et al. 2012). Peters et al. (2012) used global RNA-profiling techniques to compare RNA from bacteria treated with the Rho inhibitor bicyclomycin (BCM) or left untreated and, by aligning the two sets of data, were able to identify transcripts specifically terminated by Rho. One of the “BCM significant transcripts” (BSTs) was from the pgaA leader region. We noticed that the putative termination site was near the proposed target sequence for the small regulatory RNA McaS, previously shown to activate pgaA expression (Thomason et al. 2012). This raised the intriguing possibility that McaS could act by preventing Rho from binding to the RNA. While our subsequent work invalidated this hypothesis, it led us to discover that CsrA modulates the activity of the terminator. CsrA binding to the upstream portion of the pgaA 5′ UTR prevents the formation of a secondary structure that otherwise sequesters the rut (rho utilization) sequence. Once exposed, the rut sequence is recognized by Rho factor, which terminates transcription. This is an entirely new role for CsrA protein and, to the best of our knowledge, is the first example of a regulatory protein that acts by modulating the accessibility of an RNA sequence to the binding of Rho factor.
Results
A Rho-dependent terminator in the pgaA leader region
The study by Peters et al. (2012) tentatively mapped a Rho termination site at position +55 of the pgaA 5′ UTR. Upon examining the sequence of the region, we detected the presence of a large C>G-rich “bubble,” a hallmark of Rho-dependent terminators (Alifano et al. 1991), spanning the interval between +50 and +130 (Fig. 1A). In particular, the sequence contains two sets of four regularly spaced YC dimers (Fig. 1B), either of which could allow a Rho hexamer to anchor the RNA through four contiguous subunits (Boudvillain et al. 2013). The ensemble would then constitute two superimposed (differently phased) rut sites. From their position, one would predict transcription to actually terminate around +200, ∼150 nt downstream from the site identified by Peters et al. (2012) (Supplemental Fig. S1). This discrepancy is likely ascribable to the trimming of Rho-terminated transcripts by 3′–5′ exonuclease activity in vivo (Peters et al. 2012).
Figure 1.

Rho-dependent terminator in the pgaA leader region. (A) Graph showing the distributions of cytosine and guanine residues (in 78-nt windows) (Alifano et al. 1991) in the upstream section of the pgaABCD operon. (B) Sequence of the pgaA leader region. Pyrimidine (YC) dimers (highlighted in yellow) that are “in phase” and could constitute rut subsites are linked by blue or red lines. The predicted termination site is ∼150 nt downstream from the position identified by Peters et al. (2012), a difference likely ascribable to 3′ end trimming of Rho-terminated transcripts by exonucleases in vivo (discussed by Peters et al. 2012). The positions of CsrA-binding sites (core portion highlighted in green) are from Wang et al. (2005). (C) Hairpin-like RNA motif predicted by Mfold in the upstream section of the pgaA leader rut site (see also Supplemental Fig. S2).
The rut sequence lies immediately downstream from the 5′-proximal pair of CsrA-binding sites (Fig. 1B) and includes one of the proposed target sites for the small RNA McaS (see above; Supplemental Fig. S2). Analysis of the entire pgaA leader sequence by the Mfold algorithm (Zuker 2003) predicted that the segment between +61 and +107 adopts a discontinuous, “hairpin-like” secondary structure (two double-stranded regions separated by a single-stranded bubble), with the CsrA sites on the left (L) arm of the structure, and the 5′ half of the putative rut sequence on the right (R) arm (Fig. 1C; Supplemental Fig. S2).
As a first step in this study, we sought to ascertain the presence of a Rho-dependent terminator in the 5′ UTR of pgaA by in vitro transcription. We prepared a DNA template containing the 5′ UTR and the proximal portion of the pgaA coding sequence fused to the strong promoter T7A1 (Fig. 2A). Transcription of this DNA template in the presence of purified Rho resulted in the formation of short transcripts (Fig. 2B, “term” bands) consistent with RNA release occurring in the distal portion of the 5′ UTR, downstream from the sequence encoding the putative rut motif (Fig. 2A; Supplemental Fig. S3). This Rho-mediated transcription termination event, however, was dependent on ionic strength and was nearly abolished in the presence of 150 mM KCl (Fig. 2C, lane 4). The inhibitory effect of high KCl concentrations on Rho-dependent termination is not unprecedented (Zou and Richardson 1991) and may reflect suboptimal terminator features at high ionic strength due to, for instance, stabilization of competing RNA structures. Consistent with this idea, termination in the 5′ UTR of pgaA was restored in the presence of NusG (Fig. 2C, lanes 8–10), a cofactor known to activate termination at suboptimal sites (Burns and Richardson 1995; Peters et al. 2012). CsrA completely relieved the NusG requirement, restoring termination more effectively than NusG itself (Fig. 2C, lanes 5–7). Interestingly, NusG and CsrA appear to act synergistically, inducing termination at promoter-proximal sites (up to position 138) that are not used otherwise (see below; Supplemental Fig. S3). Altogether, these data establish that the pgaA leader contains a salt-sensitive, factor-dependent termination signal that may well contribute to the regulation of the pgaABCD operon in vivo.
Figure 2.

In vitro transcription termination at the Rho-dependent pgaA terminator. (A) Schematic representation of the DNA template used for the in vitro transcriptions. Key transcript features are also depicted above the template regions. (B,C) Representative gels show the formation of truncated transcripts resulting from Rho-dependent termination at the pgaA site under “low-salt” (50 mM KCl) transcription conditions (B) or after activation by the CsrA and NusG proteins under “high-salt” (150 mM KCl) conditions (C).
CsrA targets a cis-acting anti-terminator element
We chose to carry out in vivo analyses in Salmonella—where we previously constructed a set of congenic strains designed for the study of Rho function (Bossi et al. 2012)—and optimized the techniques for “scarless” site-directed mutagenesis of chromosomal genes. Salmonella’s Rho and NusG proteins are both 99% identical to their E. coli counterparts. The Rho allele used in this study—Y80C—also exists in E. coli, where it was shown to affect ATP cofactor binding by Rho (Chalissery et al. 2007). The NusG allele—174fs, a −1 frameshift at amino acid position 174—alters the NusG C-terminal sequence (from DFSQVEKA to TSVRLRKRNRS) (Bossi et al. 2012) and presumably affects the ability of NusG to interact with Rho (Mooney et al. 2009).
Since Salmonella lacks the pgaA operon, the first step of the in vivo study involved moving a DNA fragment comprising the promoter and the 5′ UTR of the E. coli pgaA gene (from −124 to +234) into the Salmonella chromosome. The pgaA gene was placed adjacent to a resident lacZY operon, with lacZ’s initiating AUG at the exact position normally occupied by pgaA AUG. We then constructed two unmarked deletion derivatives: one lacking the sequence corresponding to the left arm of the hairpin structure (segment 61–81), hereafter referred to as the ΔL deletion, and the other, lacking the right arm (segment 82–107), named ΔR. ΔL removes both 5′-proximal CsrA-binding sites, while ΔR removes approximately the 5′ moiety of the rut region (Fig. 1B,C), including the presumptive McaS target sequence (Thomason et al. 2012), and was expected to inactivate the termination site. The effects of these alterations on pgaA-lacZY expression were analyzed in wild-type and various mutant backgrounds.
As shown in Figure 3A, the ΔR and ΔL deletions have opposite effects on the basal level of expression of the pgaA-lacZY fusion. ΔL causes the already low β-galactosidase activity to drop to nearly undetectable levels, whereas ΔR results in an ∼10-fold increase. LacZ activity is also increased in the presence of the rho and nusG mutations (12-fold and twofold, respectively), and this effect is amplified in the ΔL mutant (∼60-fold and fivefold). In contrast, the rho and nusG mutations have no significant effect on pgaA-lacZY expression in the ΔR mutant (Fig. 3A). These findings provide in vivo evidence for the involvement of Rho-dependent termination in pgaA regulation and tentatively identify the segment missing in the ΔR deletion as the site required for Rho action.
Figure 3.

Effects of cis- and trans-acting mutations on expression of pgaA-reporter gene fusions. (A) Strains carrying the wild-type pgaA-lacZ fusion or its derivative, ΔL (Δ61–81, removing the two 5′-proximal CsrA-binding sites) or ΔR (Δ82–107, removing the upstream half of the rut site), alone or in combination with relevant unlinked mutations were assayed for β-galactosidase activity. Results show that impairing Rho-dependent termination by rho or nusG mutations (Rho Y80C and NusG 174fs) leads to increased pgaA-lacZ expression. The pgaA ΔR allele is epistatic to the rho and nusG mutations. This is what was expected if ΔR inactivates the rut site. (B) Effect of deleting the csrA gene. Removal of CsrA causes a generalized increase of pgaA-lacZ expression except in the pgaA ΔL derivative, suggesting that the sequence spanning the two upstream-most CsrA-binding sites (the L region) antagonizes termination in the absence of CsrA. (C,D) A 132-base-pair (bp) fragment encompassing a tRNA gene from Staphylococcus aureus was inserted at position +235 of pgaA in the context of the natural pgaA promoter (C) or in a construct in which the pgaA promoter is replaced by the chiP promoter (D). A 17-bp spacer separates the pgaA sequence from the beginning of the mature tRNASa coding sequence. (C,D, lanes 2,3) Northern blot analysis shows that the rho Y80C mutation and the ΔcsrA deletion cause tRNASa levels to increase regardless of the promoter. ΔL prevents tRNASa expression in the ΔcsrA strains (C,D, lane 5) but not in the rho mutant (C,D, lane 6), confirming that the L sequence antagonizes termination when not bound to CsrA. (C,D, lane 7) ΔR stimulates tRNASa production. ΔcsrA further amplifies this increase in the pgaA promoter context (C, lane 8) but not in the chiP promoter context (D, lane 8). The latter finding was taken as an indication that CsrA exerts a separate inhibitory effect on the pgaA promoter (see the text). The tRNASa signals were quantified by phosphorimaging and normalized to 5S ribosomal RNA, and the results were divided by the value measured in the wild-type strain. The bars represent the means from three (C) or two (D) independent experiments, including those shown above the histograms. Error bars denote standard deviation.
Deleting the csrA gene causes a much larger increase in β-galactosidase activity than either the ΔR or rho mutations (Fig. 3B, note the different scale). This is not surprising, as transcripts that escape termination can be expected to remain subjected to translational repression by CsrA (Wang et al. 2005). Furthermore, CsrA down-regulates NhaR, a transcriptional activator of pgaA in E. coli, by binding to sites within its 5′ UTR and inhibiting translation (Pannuri et al. 2012). Although Salmonella lacks the pgaABCD operon, it carries a close ortholog of the nhaR gene. Thus, removal of CsrA will have the additional effect of indirectly activating pgaA transcription (see below). Interestingly, the data in Figure 3B show that most of the “activating” effects of the ΔcsrA allele are abolished in the ΔL variant. This epistasis can be interpreted to indicate that the sequence of the left arm of the putative hairpin works as a cis-acting anti-terminator element when not bound to CsrA.
In parallel with the above study, we tested our initial hypothesis that McaS might up-regulate pgaA by competing with Rho for binding to the rut site (see above). Results showed that the ΔR deletion, while removing the presumptive McaS target site (Supplemental Fig. S2), does not abolish the ability of McaS to up-regulate pgaA-lacZY (Supplemental Fig. S4). In contrast, up-regulation is lost in a ΔL ΔcsrA double mutant in spite of the fact that the McaS target sequence is still present in the mRNA (Supplemental Fig. S4). Thus, these data invalidate our hypothesis and suggest that McaS effects depend somehow on the presence of CsrA. In agreement with this conclusion, a report published while this study was under way showed CsrA protein, and not pgaA RNA, to be the primary target of McaS interaction (Jorgensen et al. 2013).
To analyze the transcriptional component of pgaA regulation separately from CsrA-mediated translational repression, the lacZ coding portion of the pgaA-lacZY fusions was replaced by a heterologous tRNA gene (tRNASa) serving as a transcriptional reporter (Bossi et al. 2012). In the new fusion, the distal CsrA-binding site (overlapping the pgaA-initiating AUG codon) (Fig. 1B) was eliminated. Northern blot detection of the tRNASa confirmed that impairing Rho function by mutation or by the ΔR deletion causes a significant increase of readthrough transcription across the pgaA leader region, thus corroborating the Rho involvement (Fig. 3C). A sharp increase in tRNASa levels is also observed in wild-type and ΔR backgrounds upon removal of the csrA gene (Fig. 3C). Two mechanisms could contribute to this increase: (1) relief of CsrA-induced transcription termination and (2) stimulation of pgaA transcription initiation by the derepressed NhaR activator (see above; Pannuri et al. 2012). The greater magnitude of the increase in the wild-type construct relative to the ΔR mutant (∼20-fold and approximately threefold, respectively) suggests that CsrA-induced transcription termination is the primary cause of the shutoff of tRNASa expression. To verify this conclusion, we sought to dissociate the promoter effects from the effects within the leader. To this end, new constructs were made with the pgaA leader sequence fused to a different promoter (the chiP promoter) (see the Materials and Methods), and the tRNASa quantification analysis was repeated with the new strains. As shown in Figure 3D, removal of CsrA still causes tRNASa levels to increase in the pgaA+ strain (cf. lanes 1 and 2). This provides compelling evidence that CsrA represses tRNASa synthesis by inducing transcription termination within the pgaA leader. The pattern from the ΔL ΔcrsA double mutant in lane 5 of Figure 3D is consistent with this interpretation. As already observed with the corresponding lacZ fusion (Fig. 3A), this construct is locked in an OFF configuration, suggesting that the L arm sequence (Fig. 1C) stimulates readthrough transcription in the absence of CsrA. CsrA binding to this sequence could then be required to counter this effect and thus promote termination.
The data in Figure 3D also show that replacing the pgaA promoter with the chiP promoter abolishes the further increase of tRNASa induced by ΔcsrA in the ΔR strain (Fig. 3, cf. C [lanes 7,8] and D [lanes 7,8] ). This confirms that CsrA also affects the activity of the pgaA promoter. To verify the NhaR involvement in these effects, an additional construct was made in which the nhaR 5′ UTR is replaced by the lacUV5 promoter and the short 5′ UTR of lacZ. In the NhaR constitutive background, ΔcsrA no longer caused tRNASa expression to further increase in the ΔR variant (Supplemental Fig. S5), confirming that this effect results from the relief of nhaR repression by CsrA (Pannuri et al. 2012).
Structure-dependent regulation of the pgaA terminator
Altogether, the above data point to a key role of RNA structure shown in Figure 1C in the regulation of termination. One alternative possibility was considered in which CsrA regulated translation of a putative leader peptide initiating at nucleotide 78 of the 5′ UTR. However, a mutation of the initiating AUG had no effect on pgaA expression (Supplemental Fig. S6, see legend for details). The hairpin-like conformation incorporates the 5′ half of the rut sequence (Fig. 1C). One might expect that RNA folding will limit the accessibility of the sequence to Rho factor. CsrA binding to the L arm of the hairpin would expose the rut site and make it fully available for termination. This model predicts that L arm mutations in the CsrA-binding sites will have opposite effects on pgaA regulation depending on whether they increase or decrease the stability of the structure. We proceeded to test the prediction by constructing double- and triple-base replacement mutants (depicted in Fig. 4A). These changes were introduced directly in the chromosome of a strain carrying the pgaA-lacZY fusion. The resulting strains were characterized by β-galactosidase assays (described below) and served as a source of template DNA for in vitro analyses. As part of the latter work, RNA transcripts from the upstream portion of the pgaA leader (interval +1 to +137) were used to measure CsrA-binding affinity by gel shift assay. This analysis offered an unforeseen yet convincing confirmation for the existence of a postulated secondary structure. We found that some of the pgaA leader mutations markedly affected RNA mobility in native gels (Fig. 4B). In particular, mutations predicted to destabilize the hairpin caused a sharp decrease in mobility. The effects could be readily rationalized considering that folding increases the compactness of the molecule, allowing it to migrate faster in the gel. Overall, the migration patterns (Fig. 4B) correlate remarkably well with the structural and free energy predictions from the Mfold program, suggesting that RNA mobility can be used to infer the presence/absence of such structure. For example, the data in Figure 4B indicate that under the conditions of the experiment, wild-type RNA adopts a folded configuration in spite of the imperfect nature of the hairpin structure.
Figure 4.

pgaA RNA hairpin mutants and their CsrA-binding properties. (A) Diagram showing the positions of the double- and triple-base changes introduced near the CsrA-binding sites in the upstream portion of the pgaA 5′ UTR and the predicted effects of these changes on the RNA secondary structure. Drawings reproduce the outputs from the Mfold program (Zuker 2003). (B) Effect of hairpin mutations on RNA mobility in native gels. Changes predicted to increase RNA folding in A cause RNA to migrate faster in the gel, while changes predicted to decrease folding cause reduced electrophoretic mobility. (C) Gel retardation assay with purified CsrA. The sequence changes in the bottom portion of the RNA hairpin lower CsrA-binding affinity regardless of their effects on secondary structure. In contrast, the behavior of mutant d1, which binds CsrA better than wild-type RNA, suggests that secondary structure determinants are more important than primary structure determinants in the upper portion of the RNA hairpin. (D,E) Effect of RNA conformational changes on pgaA-lacZ expression. Strains carrying the lacZ fusion derivatives of pgaA hairpin mutants were assayed for β-galactosidase activity in csrA+ (D) and ΔcsrA (E) backgrounds. Results show that mutations destabilizing the RNA structure inhibit lacZ expression regardless of the csrA allele. Apparently, these changes improve the accessibility of the rut site, rendering CsrA action dispensable. In contrast, stabilizing mutations promote anti-termination. The s2 and s1s2 variants show the highest levels of readthrough transcription in csrA+ but not in ΔcsrA. This can be rationalized considering that s2 and s1s2 abolish CsrA binding completely, whereas all other variants have residual CsrA-binding affinity.
The gel shift experiments provided insight as to the primary and secondary structural elements that contribute to CsrA binding. This analysis showed that the d1 variant binds CsrA better than wild-type RNA in spite of being altered in the second CsrA-binding site (Fig. 4C). Apparently, reducing the stability of the upper portion of the hairpin structure is more important for CsrA binding than full conformity to the consensus sequence. Consistent with this conclusion, variant s3, which is unaffected in the CsrA-binding sites but is predicted to form a more stable hairpin, bound CsrA poorly. In contrast, primary sequence determinants appear critical in the bottom portion of the structure, as inferred from the finding that mutation d2 nearly completely abolishes binding (Fig. 4C). Combined with the greater effect of mutation s2 relative to mutation s1 (note also dominance in the double mutant), these findings suggest that the CsrA–RNA interaction nucleates at the first CsrA-binding site in the bottom portion of the hairpin.
The effects of these changes on the in vivo regulation of the pgaA-lacZY fusion were quantified by measuring β-galactosidase activity. This analysis showed that the destabilizing changes lower pgaA-lacZ expression in both the csrA+ (Fig. 4D) and ΔcsrA (Fig. 4E) backgrounds, as predicted if they relieve the CsrA requirement for termination. In contrast, changes that tighten the double-stranded regions of the hairpin show the opposite trend, increasing readthrough transcription. Finding that mutant s3 is partially deattenuated in the csrA+ strain (Fig. 4D) despite its sequence changes lying outside the CsrA recognition sequence is consistent with the behavior of this mutant in the gel shift assay and reiterates the importance of single strandedness for CsrA binding. Also consistent with the reduced CsrA binding to the s2 versus the s1 leader (Fig. 4C) is the higher level of readthrough transcription in mutant s2 relative to s1 (Fig. 4D), which again suggests that the CsrA interaction with the bottom part of the hairpin initiates melting of the structure. Altogether, the results from this analysis suggest that the hairpin-like secondary structure formed by the 61- to 107-nt sequence is programmed to have borderline stability to facilitate the CsrA interaction and the rapid switch to the open conformation following CsrA binding.
To complement the above study, a representative from either side of the stability spectrum (d1 and s1s2) was analyzed in the in vitro transcription system. The DNA templates that were used cover the entirety of the pgaA 5′ UTR and extend a further 100 base pairs (bp) into the lacZ gene (Fig. 5A). Experiments performed under high-salt conditions (150 mM KCl) (Fig. 5B) confirmed that CsrA is needed to activate Rho-dependent termination at the wild-type pgaA sequence (Fig. 5B, cf. lanes 2 and 3). Remarkably consistent with the in vivo data, the CsrA requirement is completely relieved in the d1 template (Fig. 5B, lanes 6,7), whereas CsrA loses the capability to activate Rho-dependent termination in the s1s2 template (Fig. 5B, lanes 10,11). Similar observations were made under “low-salt” transcription conditions (50 mM KCl), with the predicted exception that CsrA was no longer needed to activate termination with the wild-type construct (Supplemental Fig. S7). These results confirm that the degree of stability of the hairpin motif (ΔGWT = 9.9 kcal/mol) is a critical factor for the control of the Rho-dependent pgaA terminator by CsrA.
Figure 5.

Effect of representative pgaA hairpin mutations (d1 and s1s2) on the functioning of the Rho termination site in vitro. (A) Schematic representation of the DNA template used. (B,C) Representative gels show the activation of the terminator variants by CsrA (B) or NusG (C). Results show that pgaA hairpin mutation d1, expected to favor melting of the secondary structure (Fig. 4B), relieves the requirement of CsrA for in vitro termination by Rho. In contrast, the s1s2 variant abolishes termination, and CsrA cannot restore it. Thus, these data closely parallel the results from the in vivo analysis (Fig. 4D). B (lanes 4,8,12) also shows that CsrA and NusG act synergistically to stimulate Rho-dependent termination.
Intriguingly, in vitro activation of the pgaA terminator by NusG appears to be affected by hairpin stability as well (Fig. 5C, cf. lanes 3, 6, and 9). NusG only slightly activates termination at the main sites with the “strong” s1s2 hairpin construct (Fig. 5C, lane 9) while strongly activating termination at both the main sites and promoter-proximal sites with the “weak” d1 hairpin construct (Fig. 5C, lane 6, note that band signals for short transcripts are inherently weaker due to fewer internal incorporations of [α-32P]-U residues). Again, the wild-type template shows an intermediate behavior, with both CsrA and NusG needed to activate promoter-proximal termination sites (Fig. 5B, lane 4). These data suggest that NusG somehow allows Rho to bypass the requirement for a fully single-stranded rut sequence even though the combined actions of both CsrA and NusG produce maximal termination efficiency (Fig. 5, cf. B [lanes 4,8,12] and C [lanes 3,6, 9]). The NusG involvement is consistent with the in vivo data showing a moderate but reproducible increase of transcriptional readthrough at the pgaA 5′ UTR terminator in a nusG mutant (Fig. 3A,B) and with the significant overlap in the 3′ end patterns of pgaA transcripts obtained from BCM-treated wild-type E. coli and a nusG mutant, as detected on tiling arrays (Peters et al. 2012).
Discussion
The classical mechanism for regulating gene expression through the activity of transcription termination factor Rho involves the action of proteins that render the transcription elongation complex refractory to termination (anti-termination). Thus, the classical scenario (e.g., phage λ’s N protein) has termination as the default setting and anti-termination as the active step; furthermore, the regulatory target is RNA polymerase rather than Rho itself. The work described here uncovered an alternative mechanism for Rho-mediated gene regulation. In this mechanism, the active step consists of inducing termination at a rut site that is normally held invisible to Rho because it is part of a secondary structure. Termination results from the action of a protein, CsrA, that, upon binding to the 5′ arm of the structure, exposes the rut sequence, making it available for Rho binding (Fig. 6). The mechanism relies on the exquisite fine-tuning of an RNA secondary structure: stable enough to hold the rut site hidden from Rho but sufficiently unstable to allow nucleation of the CsrA–RNA complex and the opening of the structure following CsrA binding. Also important to consider is that in order to induce Rho-dependent termination, CsrA must be able to bind the RNA while the latter is being synthesized and before RNA polymerase escapes the termination “window.” Presumably, the fact that the secondary structure has not yet formed at this stage can favor the interaction. This might suggest that the 5′-proximal CsrA-binding site (the base of the hairpin stem) is the “seed” sequence. The greater effect of changes in this region on termination and CsrA binding further supports this conclusion. The extent to which other features in the region might contribute to CsrA binding and Rho-dependent termination remains to be explored. Elongation rates and interaction with RNA-binding proteins are viewed as major factors affecting cotranscriptional RNA folding (Pan and Sosnick 2006). The important role played in this study by elongation factor NusG—a protein thought to inhibit transcriptional pausing (Artsimovitch and Landick 2000) and have some RNA-binding capability (Steiner et al. 2002)—further supports the idea that cotranscriptional folding is a key component in the functioning of the pgaA attenuator structure.
Figure 6.
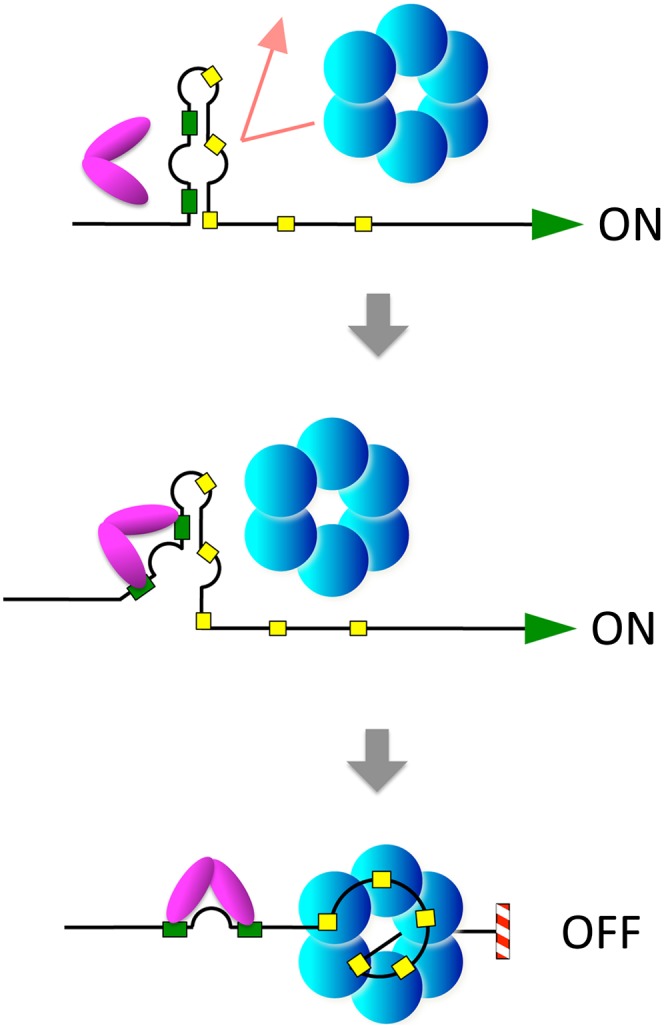
Model for CsrA-induced Rho-dependent transcription attenuation in the pgaA leader region. The initial portion of the pgaA 5′ UTR folds into an imperfect hairpin-like structure that incorporates two CsrA-binding sites in the left arm (green boxes) and part of a Rho-binding site (rut) in the right arm (row of yellow boxes). The structure renders the rut site inaccessible to Rho but is sufficiently unstable to allow nucleation of a CsrA:RNA complex (presumably starting at the base of the hairpin). Thus, when CsrA is limiting, transcription reads through the terminator and proceeds into pgaABCD structural genes, whereas, in the presence of CsrA, formation of the CsrA:RNA complex unfolds the structure, exposing the rut site and causing premature transcription termination.
Besides inducing Rho-dependent termination in the pgaA leader region, CsrA represses pgaA mRNA translation by binding to a pair of sites near the initiating AUG codon (Wang et al. 2005). The sequence layout is such that translational repression can operate only on transcripts that escape CsrA-induced termination. This raises the question of the biological role of such a redundancy, particularly since our data suggest that pgaA attenuation is sufficient to completely shut off pgaA mRNA synthesis. Perhaps the two forms of regulation target separate classes of molecules: attenuation mainly directed to nascent RNAs, and translational repression acting on transcripts that are already made at the time of the regulatory switching. This would ensure the rapidity and efficacy of pgaA silencing under conditions prompting bacteria to switch from the sessile to the planktonic lifestyle. Alternatively, the two-level regulation might be designed to do just the opposite; that is, make the transition more gradual. RNA footprint data from the initial analysis of the CsrA:pgaA mRNA interaction (Wang et al. 2005) suggest that CsrA binds the translation regulatory sites with greater affinity than the 5′-proximal sites. Thus, one might envision that pgaA translational repression would start early on during a transition and that complete shutoff will occur only after CsrA has sufficiently accumulated.
A further possibility is that the double regulatory layer could have evolved to allow independent sensing of different environmental cues. For example, one could imagine that some physical stimuli or ligands could directly influence the ability of the pgaA terminator to switch from the closed to the open configuration so as to induce termination under conditions in which CsrA is still limiting. In other words, the pgaA terminator could have the properties of a bona fide riboswitch. A precedent for this type of dual regulation was recently set by the demonstration that the molybdenum cofactor-sensing riboswitch of the moaA gene of E. coli includes binding sites for CsrA, which activates moaA expression post-transcriptionally (Patterson-Fortin et al. 2013). Other recent findings have linked riboswitch function to Rho activity. In the mgtA and ribB genes, RNA conformational changes induced by the binding of Mg2+ and flavin mononucleotide, respectively, modulate the activity of Rho-dependent transcription terminators upstream of the structural portions of the genes (Hollands et al. 2012). A “conditional” Rho-dependent terminator is also found in the chiPQ operon; here, however, the terminator is embedded within a translated region and becomes active following the inhibition of chiP mRNA translation by a regulatory small RNA (Bossi et al. 2012). The present study provides the first example of a Rho-dependent terminator activated by a protein-induced RNA conformational rearrangement.
One might predict that other genes in the CsrA network will be found to be regulated by a mechanism similar to that described here. A survey of transcriptomic data in the literature reveals some overlaps between the lists of RNAs copurified with CsrA in E. coli (Edwards et al. 2011) and transcripts found extended at their 3′ ends in BCM-treated cells (Peters et al. 2012). However, our inspection and Mfold analysis of the sequences in the 5′ regions of the shared RNAs failed to detect features reminiscent of the pgaA hairpin motif described here. The possibility remains that CsrA and Rho regulatory circuits are interconnected in a more elaborate way not immediately apparent through the above analysis. Moreover, it should be noted that pgaA is absent from the list of CsrA-bound transcripts (Edwards et al. 2011), suggesting that the list still has to be completed. Finally, the general nature and robustness of the attenuation mechanism described here suggest that structural remodeling of Rho-dependent signals may well occur in response to RNA binding by other regulatory proteins.
In terms of regulatory responses, transcription termination and anti-termination can be equated to repression and activation of initiation, respectively. A theory of gene regulation elaborated by Savageau in the late 1970s (Savageau 1977) and 1980s (Savageau 1989) posits that genes for which a high level of expression is most frequently required (high demand) will tend to be controlled in a positive manner (by activators or anti-terminators), whereas genes whose products are only required in specific instances (low demand) will be mainly controlled by negative regulatory elements (repressors or proterminators) (Savageau 1977). The theory interpreted classical patterns of gene regulation in E. coli, phage λ, and yeast. However, the lack of examples of regulatory proteins acting as transcription proterminators left the picture somewhat incomplete (Savageau 1989). The demonstration, presented here, that CsrA can perform such function allows completing the symmetry. Furthermore, since the requirement for the pgaABCD gene products may well represent a relatively infrequent (“low demand”) event in the lifestyle of most E. coli strains, our findings lend further support for Savageau’s model (Savageau 1989).
Materials and methods
Bacterial strains and culture conditions
Strains used in this study were all derived from Salmonella enterica serovar Typhimurium strain MA3409, a derivative of strain LT2 cured for the Gifsy-1 prophage (Figueroa-Bossi et al. 1997). The genotypes of strains used are listed in Supplemental Table S1. Bacteria were cultured at 37°C in liquid LB broth (Bertani 2004) or LB broth solidified by the addition of 1.5% Difco agar. Antibiotics (Sigma-Aldrich) were included at the following final concentrations: 10 µg/mL chloramphenicol, 50 µg/mL kanamycin monosulphate, 100 µg/mL sodium ampicillin, 80 µg/mL spectinomycin dihydrochloride, and 25 µg/mL tetracycline hydrochloride. LB plates containing 40 µg/mL 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside (X-gal; from Sigma) or MacConkey agar plates containing 1% lactose (Macconkey 1905) were used to monitor lacZ expression in bacterial colonies. Liquid cultures were grown in New Brunswick gyratory shakers, and growth was monitored by measuring the optical density at 600 nm with a Shimazu UV mini 1240 spectrophotometer.
Enzymes and chemicals
T4 polynucleotide kinase, T4 DNA ligase, Taq DNA polymerase, and restriction enzymes were purchased from New England Biolabs. Pfu-Turbo DNA polymerase was from Stratagene. Sigma-saturated RNA polymerase from E. coli was obtained from Epicentre. DNA oligonucleotides were custom-synthesized by Sigma-Aldrich, Eurogentec, or Eurofins MWG/Operon. Most chemicals were purchased from Sigma-Aldrich. Acrylamide–bisacrylamide and other electrophoresis reagents were from Bio-Rad and Amresco. Agarose was from Invitrogen. Hybond-N+ membranes and hybridization buffer used for Northern blot analysis were from GE Healthcare and Applied Biosystems-Ambion, respectively. The rNTPs were from Promega, and the 32P-NTPs were from PerkinElmer or Hartmann Analytic. 32P-labeled nucleic acids were detected by phosphorimaging using ImageQuant software.
Genetic techniques
Generalized transduction was performed using the high-frequency transducing mutant of phage P22, HT 105/1 int-201 (Schmieger 1972), as described (Lemire et al. 2011). Chromosomal engineering (recombineering) was carried out by the λ red recombination method (Datsenko and Wanner 2000; Murphy et al. 2000; Yu et al. 2000), implemented as in Datsenko and Wanner (2000). “Scarless” recombineering was performed as described (Bossi et al. 2012). Donor DNA fragments were generated by PCR using plasmid DNA, chromosomal DNA, or DNA oligonucleotides as templates. Amplified fragments were electroporated into appropriate strains harboring the conditionally replicating plasmid pKD46, which carries the λ red operon under the control of the PBAD promoter (Datsenko and Wanner 2000). Bacteria carrying pKD46 were grown at 30°C in the presence of ampicillin and exposed to 10 mM arabinose for 3 h prior to preparation of electrocompetent cells. Electroporation was carried out using a Bio-Rad MicroPulser under the conditions specified by the manufacturer. Recombinant colonies were selected on LB plates containing the appropriate antibiotic. Constructs were verified by PCR and DNA sequence analysis (performed by GATC Company). The complete list of primers used in this study are in Supplemental Table S2, and details on the construction of several alleles are in Supplemental Table S3.
Construction of relevant alleles
pgaA-lacZY
Two constructs were made that convert a chromosomal chiP-lacZY gene fusion (Figueroa-Bossi et al. 2009) into a pgaA-lacYZ fusion. In the first construct, a DNA fragment spanning the portion of the pgaABCD operon from −124 to + 234 (amplified from E. coli strain MG1655 with primers ppL56 and ppL59) (Supplemental Table S2) was used to replace the region between −253 and +89 of the chiP gene (spanning the transcription regulatory region and the 5′ UTR) (Plumbridge et al. 2014). This yielded strain MA11469. In the second construct, the exchanged segment covers only the 5′ UTR of chiP (from +1 to +89), replaced by the pgaA segment from +1 to + 234 (amplified with primers ppL57 and ppL59); thus, the fusion is transcribed from the chiP promoter (strain MA11470). In both constructs, the lacZ-initiating AUG falls at the exact same position normally occupied by pgaA AUG. Derivatives of MA11469 and/or MA11470 used in this study include strains carrying (1) tRNASa gene fusions in place of lacZ, (2) deletions in relevant portions of the pgaA 5′ UTR (ΔL and ΔR), and (3) point mutations affecting the hairpin structure in the 5′ UTR (see below).
pgaA hairpin mutants
Short DNA fragments (typically ∼100 bp) containing the desired mutations were obtained by annealing suitably designed, partially complementary DNA oligonucleotides and filling in the single-stranded regions by PCR under standard amplification conditions (“reciprocal priming”). DNA fragments were introduced into a strain MA11469 derivative, a tetRA insertion in the pgaA leader (Supplemental Tables S1, S3), and tetracycline-sensitive recombinants were selected for resistance to fusaric acid (Bochner et al. 1980).
ΔcsrA∷cat
A deletion removing nearly the entirety of the Salmonella csrA coding sequence (167 out of 183 bp) was obtained by replacing this region with the FRT-cat-FRT module of plasmid pKD3 (Datsenko and Wanner 2000), amplified with primers ppM17 and ppM18 (Supplemental Table S2). As previously reported (Altier et al. 2000), we found the csrA deletion to severely affect growth. A deletion of glgCAP operon, reported to suppress this growth defect (Timmermans and Van Melderen 2009), failed to restore normal growth. However, suppressors of the growth defect arose spontaneously at high frequency (Altier et al. 2000). Introduction of the pgaA-lacZ fusion in four independent fast-growing isolates showed all four of them to express high levels of β-galactosidase activity. However, when the ΔcsrA∷cat allele was replaced by wild-type csrA, pgaA-lacZ expression returned to normally repressed levels in all four strains, indicating that the defect in pgaA regulation is entirely ascribable to the loss of CsrA activity. One of the ΔcsrA∷cat sup strains (MA11529) was used throughout this study.
Δ[nhaA-nhaR′]∷aph Plac-nhaR
A DNA fragment carrying the aph (KanR) gene and a lacUV5 promoter was amplified from plasmid pNFB20, with primers ppP23 and ppP24 (Supplemental Table S2) used to replace the region encompassing the nhaA gene and the 5′ UTR of the adjacent nhaR gene in the Salmonella chromosome by λ red recombination (Supplemental Table S3). In the strain obtained (MA11914), the nhaR gene is expressed from the lacUV5 promoter and is no longer sensitive to translational repression by CsrA (Pannuri et al. 2012). Plasmid pNFB20 is an oriR6K plasmid carrying the aph gene and lacUV5 side by side in parallel orientation.
Construction of plasmid pBR-mcaS
Recombinant plasmid pBR-mcaS, which carries the mcaS gene under the control of the lac promoter, was constructed by cloning a DNA fragment amplified from E. coli strain MG1655 (with primers ppL67 and ppL68) into plasmid pBR-plac (Guillier and Gottesman 2006) as described by Thomason et al. (2012).
Measurement of β-galactosidase activity
β-Galactosidase activity was assayed in toluene-permeabilized cells as described (Miller 1992) and is expressed in Miller units throughout this study. Typically, measurements were performed on duplicate or triplicate cultures grown in late exponential phase (OD600 ≈ 0.7). All experiments included parental or reference strains as normalization controls. Standard deviations were generally <5% of the mean.
RNA extraction and Northern analysis
RNA was prepared by the acid hot phenol method from exponentially growing cells (OD600 of 0.35) as previously described (Bossi and Figueroa-Bossi 2007). RNA was fractionated on an 8% polyacrylamide/8 M urea gel and transferred to Hybond-N+ membranes. Blots were hybridized to DNA oligonucleotides ppJ62 and ppB10, complementary to Staphylococcus aureus tRNAArg and Salmonella 5S ribosomal RNA, respectively (Supplemental Table S2), labeled at the 5′ ends with T4 polynucleotide kinase. Hybridization patterns were analyzed by phosphorimaging, and RNA bands were quantified using the ImageQuant program.
Preparation of proteins
Plasmid pEt28b-CsrA, encoding CsrA fused to a C-terminal His6 tag, was obtained by inserting phosphorylated oligonucleotides Csra1 to Csra3 (annealed to complementary oligonucleotides Csra4 to Csra6) (Supplemental Table S2) between the NcoI and XhoI sites of plasmid pET28b (EMD Millipore) in a “one-pot” ligation reaction. CsrA protein was overexpressed in BL21(DE3):pEt28b-CsrA and purified by affinity chromatography on HisPur cobalt resin (Thermo Scientific) following a protocol similar to that previously described (Liu and Romeo 1997). Recombinant Rho and NusG proteins were overexpressed and purified as described previously (Rabhi et al. 2011). Concentrations of active oligomeric forms (Rho hexamers, CsrA dimers, and NusG monomers) are indicated throughout the article.
In vitro transcription termination experiments
DNA templates were prepared by standard PCR amplification of genomic DNA using forward primer pgaA1, which contains the sequence of the T7A1 promoter fused to the 31-nt upstream segment of the pgaA leader sequence, and reverse primer pgaA2 (Fig. 2A) or pgaA6 (Fig. 5A). Standard transcription termination experiments were performed as described previously (Rabhi et al. 2011), with minor modifications. Briefly, 18-µL mixtures containing the DNA templates (5 nM final concentration), 20 nM RNA polymerase, 0 or 70 nM Rho, 0.2 U/µL SUPERase-In (Ambion), 0 or 70 nM CsrA (unless indicated otherwise), and 0 or 140 nM NusG (unless indicated otherwise) in transcription buffer (40 mM Tris-HCl at pH 8.0, 50 or 150 mM KCl, 5 mM MgCl2, 1.5 mM DTT) were preincubated for 10 min at 37°C. Next, 2 µL of transcription initiation mix (2 mM ATP, GTP, and CTP; 0.2 mM UTP; 2.5 µCi/µL [a-32P]-UTP; 250 µg/mL rifampicin in transcription buffer) was added before incubation for 20 min at 37°C. Transcription reactions were stopped with 4 µL of 0.5 M EDTA and 2 µL of 0.25 mg/mL tRNA before extraction with a 5:24:1 phenol:chloroform:isoamyl alcohol mix and precipitation with ethanol. Reaction pellets were dissolved in denaturing loading buffer (95% formamide, 15 mM EDTA) and analyzed by denaturing 6% polyacrylamide gel electrophoresis. RNA release at the pgaA terminator was also monitored in single-round “chase” transcription experiments with bead-affixed transcription elongation complexes, as detailed in the Supplemental Material.
Electrophoretic mobility assays
RNA transcripts corresponding to the 1–137 region of the pgaA leader (including wild-type or mutant hairpin variants) were prepared by standard in vitro transcription with T7 RNA polymerase and PCR-generated DNA templates. Transcripts were purified by denaturing PAGE and stored at −20°C in ME buffer (10 mM MOPS at pH 6.5, 1 mM EDTA). Transcripts were dephosphorylated in small batches (∼10 pmol) with alkaline phosphatase (Roche Applied Science) before being labeled with [32P-g]-ATP and T4 polynucleotide kinase and subsequently purified by 7% denaturing PAGE. Concentrations of transcripts were determined from their absorptions at 260 nm using a Nanodrop spectrophotometer. For electrophoretic mobility assays, the 32P-labeled transcripts (0.1 nM final concentration) were incubated for 20 min at 30°C in EMSA buffer (40 mM Tris-HCl at pH 8.0, 150 mM KCl, 5 mM MgCl2, 0.2 mM DTT, 50 µg/mL BSA, 30 µg/mL tRNA, 2% glycerol) in the presence or absence of CsrA protein (final concentrations are indicated in the figures). The mixtures were then mixed with Ficoll-400 (4%, final concentration) and analyzed by 6% native PAGE. Gels were run at 10 V/cm for 5 h at 19°C.
Acknowledgments
We thank Damien Le Minoux for help in strain construction during the late stages of this work, and Emilie Soares for the preparation of materials used in in vitro experiments. We are grateful to Maude Guillier (IBPC, CNRS, Paris) for the gift of plasmid pBR-plac. This work was supported by CNRS core funding and grants from the French Agence Nationale de la Recherche (ANR-13-BSV3-0005-01 to N.F.-B., and ANR-13-BSV3-0005-02 to M.B.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.240192.114.
References
- Alifano P, Rivellini F, Limauro D, Bruni CB, Carlomagno MS 1991. A consensus motif common to all Rho-dependent prokaryotic transcription terminators. Cell 64: 553–563 [DOI] [PubMed] [Google Scholar]
- Altier C, Suyemoto M, Lawhon SD 2000. Regulation of Salmonella enterica serovar Typhimurium invasion genes by csrA. Infect Immun 68: 6790–6797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artsimovitch I, Landick R 2000. Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc Natl Acad Sci 97: 7090–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babitzke P, Romeo T 2007. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol 10: 156–163 [DOI] [PubMed] [Google Scholar]
- Bertani G 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J Bacteriol 186: 595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochner BR, Huang HC, Schieven GL, Ames BN 1980. Positive selection for loss of tetracycline resistance. J Bacteriol 143: 926–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi L, Figueroa-Bossi N 2007. A small RNA downregulates LamB maltoporin in Salmonella. Mol Microbiol 65: 799–810 [DOI] [PubMed] [Google Scholar]
- Bossi L, Schwartz A, Guillemardet B, Boudvillain M, Figueroa-Bossi N 2012. A role for Rho-dependent polarity in gene regulation by a noncoding small RNA. Genes Dev 26: 1864–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudvillain M, Figueroa-Bossi N, Bossi L 2013. Terminator still moving forward: expanding roles for Rho factor. Curr Opin Microbiol 16: 118–124 [DOI] [PubMed] [Google Scholar]
- Burns CM, Richardson JP 1995. NusG is required to overcome a kinetic limitation to Rho function at an intragenic terminator. Proc Natl Acad Sci 92: 4738–4742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalissery J, Banerjee S, Bandey I, Sen R 2007. Transcription termination defective mutants of Rho: role of different functions of Rho in releasing RNA from the elongation complex. J Mol Biol 371: 855–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Cui Y, Liu Y, Dumenyo CK, Chatterjee AK 1995. Inactivation of rsmA leads to overproduction of extracellular pectinases, cellulases, and proteases in Erwinia carotovora subsp. carotovora in the absence of the starvation/cell density-sensing signal, N-(3-oxohexanoyl)-L-homoserine lactone. Appl Environ Microbiol 61: 1959–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez RG, Alvarez AF, Romeo T, Georgellis D 2010. The physiological stimulus for the BarA sensor kinase. J Bacteriol 192: 2009–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Chatterjee A, Liu Y, Dumenyo CK, Chatterjee AK 1995. Identification of a global repressor gene, rsmA, of Erwinia carotovora subsp. carotovora that controls extracellular enzymes, N-(3-oxohexanoyl)-L-homoserine lactone, and pathogenicity in soft-rotting Erwinia spp. J Bacteriol 177: 5108–5115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey AK, Baker CS, Romeo T, Babitzke P 2005. RNA sequence and secondary structure participate in high-affinity CsrA–RNA interaction. RNA 11: 1579–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AN, Patterson-Fortin LM, Vakulskas CA, Mercante JW, Potrykus K, Vinella D, Camacho MI, Fields JA, Thompson SA, Georgellis D, et al. 2011. Circuitry linking the Csr and stringent response global regulatory systems. Mol Microbiol 80: 1561–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Bossi N, Coissac E, Netter P, Bossi L 1997. Unsuspected prophage-like elements in Salmonella typhimurium. Mol Microbiol 25: 161–173 [DOI] [PubMed] [Google Scholar]
- Figueroa-Bossi N, Valentini M, Malleret L, Fiorini F, Bossi L 2009. Caught at its own game: regulatory small RNA inactivated by an inducible transcript mimicking its target. Genes Dev 23: 2004–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudapaty S, Suzuki K, Wang X, Babitzke P, Romeo T 2001. Regulatory interactions of Csr components: the RNA binding protein CsrA activates csrB transcription in Escherichia coli. J Bacteriol 183: 6017–6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillier M, Gottesman S 2006. Remodelling of the Escherichia coli outer membrane by two small regulatory RNAs. Mol Microbiol 59: 231–247 [DOI] [PubMed] [Google Scholar]
- Hollands K, Proshkin S, Sklyarova S, Epshtein V, Mironov A, Nudler E, Groisman EA 2012. Riboswitch control of Rho-dependent transcription termination. Proc Natl Acad Sci 109: 5376–5381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Rice JD, Goller C, Pannuri A, Taylor J, Meisner J, Beveridge TJ, Preston JF 3rd, Romeo T 2008. Roles of pgaABCD genes in synthesis, modification, and export of the Escherichia coli biofilm adhesin poly-β-1,6-N-acetyl-D-glucosamine. J Bacteriol 190: 3670–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DW, Suzuki K, Oakford L, Simecka JW, Hart ME, Romeo T 2002. Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J Bacteriol 184: 290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen MG, Thomason MK, Havelund J, Valentin-Hansen P, Storz G 2013. Dual function of the McaS small RNA in controlling biofilm formation. Genes Dev 27: 1132–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge K, Sineva E, Lindell M, Starke K, Baker CS, Babitzke P, Haas D 2007. Mechanism of hcnA mRNA recognition in the Gac/Rsm signal transduction pathway of Pseudomonas fluorescens. Mol Microbiol 66: 341–356 [DOI] [PubMed] [Google Scholar]
- Lapouge K, Schubert M, Allain FH, Haas D 2008. Gac/Rsm signal transduction pathway of γ-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol 67: 241–253 [DOI] [PubMed] [Google Scholar]
- Lawhon SD, Frye JG, Suyemoto M, Porwollik S, McClelland M, Altier C 2003. Global regulation by CsrA in Salmonella typhimurium. Mol Microbiol 48: 1633–1645 [DOI] [PubMed] [Google Scholar]
- Lemire S, Figueroa-Bossi N, Bossi L 2011. Bacteriophage crosstalk: coordination of prophage induction by trans-acting antirepressors. PLoS Genet 7: e1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz DH, Miller MB, Zhu J, Kulkarni RV, Bassler BL 2005. CsrA and three redundant small RNAs regulate quorum sensing in Vibrio cholerae. Mol Microbiol 58: 1186–1202 [DOI] [PubMed] [Google Scholar]
- Liu MY, Romeo T 1997. The global regulator CsrA of Escherichia coli is a specific mRNA-binding protein. J Bacteriol 179: 4639–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macconkey A 1905. Lactose-fermenting bacteria in faeces. J Hyg (Lond) 5: 333–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez LC, Yakhnin H, Camacho MI, Georgellis D, Babitzke P, Puente JL, Bustamante VH 2011. Integration of a complex regulatory cascade involving the SirA/BarA and Csr global regulatory systems that controls expression of the Salmonella SPI-1 and SPI-2 virulence regulons through HilD. Mol Microbiol 80: 1637–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercante J, Edwards AN, Dubey AK, Babitzke P, Romeo T 2009. Molecular geometry of CsrA (RsmA) binding to RNA and its implications for regulated expression. J Mol Biol 392: 511–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. 1992. A short course in bacterial genetics. a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Mooney RA, Schweimer K, Rosch P, Gottesman M, Landick R 2009. Two structurally independent domains of E. coli NusG create regulatory plasticity via distinct interactions with RNA polymerase and regulators. J Mol Biol 391: 341–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KC, Campellone KG, Poteete AR 2000. PCR-mediated gene replacement in Escherichia coli. Gene 246: 321–330 [DOI] [PubMed] [Google Scholar]
- Pan T, Sosnick T 2006. RNA folding during transcription. Annu Rev Biophys Biomol Struct 35: 161–175 [DOI] [PubMed] [Google Scholar]
- Pannuri A, Yakhnin H, Vakulskas CA, Edwards AN, Babitzke P, Romeo T 2012. Translational repression of NhaR, a novel pathway for multi-tier regulation of biofilm circuitry by CsrA. J Bacteriol 194: 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson-Fortin LM, Vakulskas CA, Yakhnin H, Babitzke P, Romeo T 2013. Dual posttranscriptional regulation via a cofactor-responsive mRNA leader. J Mol Biol 425: 3662–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessi G, Williams F, Hindle Z, Heurlier K, Holden MT, Camara M, Haas D, Williams P 2001. The global posttranscriptional regulator RsmA modulates production of virulence determinants and N-acylhomoserine lactones in Pseudomonas aeruginosa. J Bacteriol 183: 6676–6683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Mooney RA, Grass JA, Jessen ED, Tran F, Landick R 2012. Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev 26: 2621–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumbridge J, Bossi L, Oberto J, Wade JT, Figueroa-Bossi N 2014. Interplay of transcriptional and small RNA-dependent control mechanisms regulates chitosugar uptake in Escherichia coli and Salmonella. Mol Microbiol doi: 10.1111/mmi.12573 [DOI] [PubMed] [Google Scholar]
- Rabhi M, Espeli O, Schwartz A, Cayrol B, Rahmouni AR, Arluison V, Boudvillain M 2011. The Sm-like RNA chaperone Hfq mediates transcription antitermination at Rho-dependent terminators. EMBO J 30: 2805–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo T, Gong M, Liu MY, Brun-Zinkernagel AM 1993. Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J Bacteriol 175: 4744–4755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo T, Vakulskas CA, Babitzke P 2013. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ Microbiol 15: 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savageau MA 1977. Design of molecular control mechanisms and the demand for gene expression. Proc Natl Acad Sci 74: 5647–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savageau MA. 1989. Are there rules governing patterns of gene regulation? In Theoretical biology—epigenetic and evolutionary order from complex systems (ed. Goodwin BC, Saunders PT), pp. 42–66. Edinburgh University Press, Edinburgh. [Google Scholar]
- Schmieger H 1972. Phage P22-mutants with increased or decreased transduction abilities. Mol Gen Genet 119: 75–88 [DOI] [PubMed] [Google Scholar]
- Schubert M, Lapouge K, Duss O, Oberstrass FC, Jelesarov I, Haas D, Allain FH 2007. Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat Struct Mol Biol 14: 807–813 [DOI] [PubMed] [Google Scholar]
- Steiner T, Kaiser JT, Marinkovic S, Huber R, Wahl MC 2002. Crystal structures of transcription factor NusG in light of its nucleic acid- and protein-binding activities. EMBO J 21: 4641–4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterzenbach T, Nguyen KT, Nuccio SP, Winter MG, Vakulskas CA, Clegg S, Romeo T, Baumler AJ 2013. A novel CsrA titration mechanism regulates fimbrial gene expression in Salmonella typhimurium. EMBO J 32: 2872–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Wang X, Weilbacher T, Pernestig AK, Melefors O, Georgellis D, Babitzke P, Romeo T 2002. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J Bacteriol 184: 5130–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Kiefer P, Reimmann C, Keel C, Dubuis C, Rolli J, Vorholt JA, Haas D 2009. Small RNA-dependent expression of secondary metabolism is controlled by Krebs cycle function in Pseudomonas fluorescens. J Biol Chem 284: 34976–34985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason MK, Fontaine F, De Lay N, Storz G 2012. A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli. Mol Microbiol 84: 17–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans J, Van Melderen L 2009. Conditional essentiality of the csrA gene in Escherichia coli. J Bacteriol 191: 1722–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Dubey AK, Suzuki K, Baker CS, Babitzke P, Romeo T 2005. CsrA post-transcriptionally represses pgaABCD, responsible for synthesis of a biofilm polysaccharide adhesin of Escherichia coli. Mol Microbiol 56: 1648–1663 [DOI] [PubMed] [Google Scholar]
- Yakhnin H, Baker CS, Berezin I, Evangelista MA, Rassin A, Romeo T, Babitzke P 2011. CsrA represses translation of sdiA, which encodes the N-acylhomoserine-L-lactone receptor of Escherichia coli, by binding exclusively within the coding region of sdiA mRNA. J Bacteriol 193: 6162–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakhnin AV, Baker CS, Vakulskas CA, Yakhnin H, Berezin I, Romeo T, Babitzke P 2013. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol Microbiol 87: 851–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci 97: 5978–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou LL, Richardson JP 1991. Enhancement of transcription termination factor rho activity with potassium glutamate. J Biol Chem 266: 10201–10209 [PubMed] [Google Scholar]
- Zuker M 2003. Mfold Web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]