

Abstract
The glycosome of the pathogenic African trypanosome Trypanosoma brucei is a specialized peroxisome that contains most of the enzymes of glycolysis and several other metabolic and catabolic pathways. The contents and transporters of this membrane-bounded organelle are of considerable interest as potential drug targets. Here we use epitope tagging, magnetic bead enrichment, and SILAC quantitative proteomics to determine a high-confidence glycosome proteome for the procyclic life cycle stage of the parasite using isotope ratios to discriminate glycosomal from mitochondrial and other contaminating proteins. The data confirm the presence of several previously demonstrated and suggested pathways in the organelle and identify previously unanticipated activities, such as protein phosphatases. The implications of the findings are discussed.
Keywords: Trypanosoma brucei, quantitative proteomics, peroxisome, glycosome
Introduction
Eukaryotic cells possess membrane-bound organelles called peroxisomes, defined by the presence of the Pex proteins responsible for their biogenesis. Peroxisomes generally contain enzymes to deal with reactive oxygen species and enzymes of fatty acid β-oxidation.1 Beyond these common functions, peroxisome content and function vary between species. The peroxisomes of the trypanosomatids are called glycosomes because, uniquely, they contain most of the enzymes of glycolysis.2,3 Ether lipid and isoprenoid/sterol biosynthesis are relatively common peroxisomal functions, and these too have been demonstrated and suggested, respectively, to be present in trypanosomatids.4,5 Other metabolic processes that are atypically compartmentalized in glycosomes include purine salvage, the latter stages of pyrimidine biosynthesis,6,7 and much of sugar nucleotide biosynthesis.8−14
An inventory of an organelle’s contents is highly desirable to help understand its functional significance, its mechanism(s) of biogenesis, and its metabolite transport systems. Given the parasitic nature of the trypanosomatids and the serious human and animal diseases inflicted by them, glycosome metabolism and organization are of considerable interest with respect to identifying therapeutic opportunities related to their unusual metabolic compartmentalization and requisite metabolite transport.15,16
Here we describe the glycosome proteome of the procyclic form of Trypanosoma brucei. This organism causes human African trypanosomiasis (African sleeping sickness) and nagana in cattle and is transmitted by the tsetse fly in much of sub-Saharan Africa. The procyclic form of the parasite normally resides in the gut of the tsetse fly vector and is the life cycle stage most readily grown to moderate densities in cell culture.
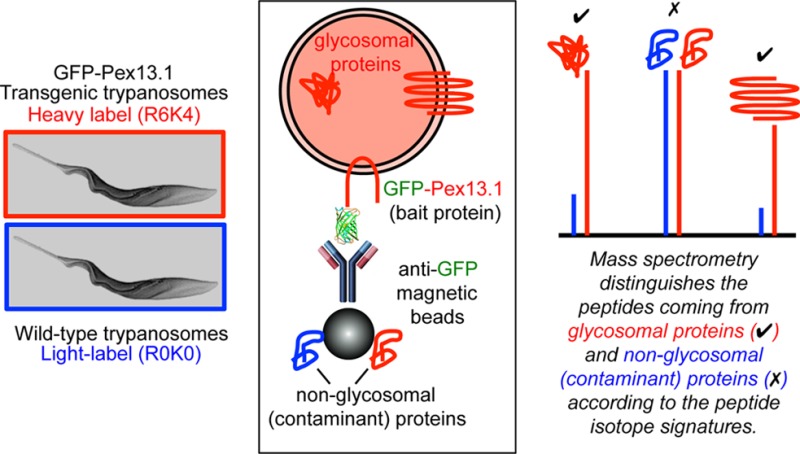
Given that subcellular fractionation can only provide an enrichment of a given organelle, the principle problem in obtaining high-confidence organelle proteomes is in deciding which of the many hundreds or thousands of protein “hits” are genuine components of the organelle rather than contaminants from other locations. We therefore decided to approach this problem for the trypanosome glycosomes by using epitope-tagged glycosomes and stable isotope labeling in cell culture (SILAC), such that in antiepitope pull-out experiments we can distinguish genuine glycosomal components from contaminants according to peptide isotopic signatures. The general approach is summarized in Figure 1, and we compare our results to previously published glycosome proteomes.17−19
Figure 1.
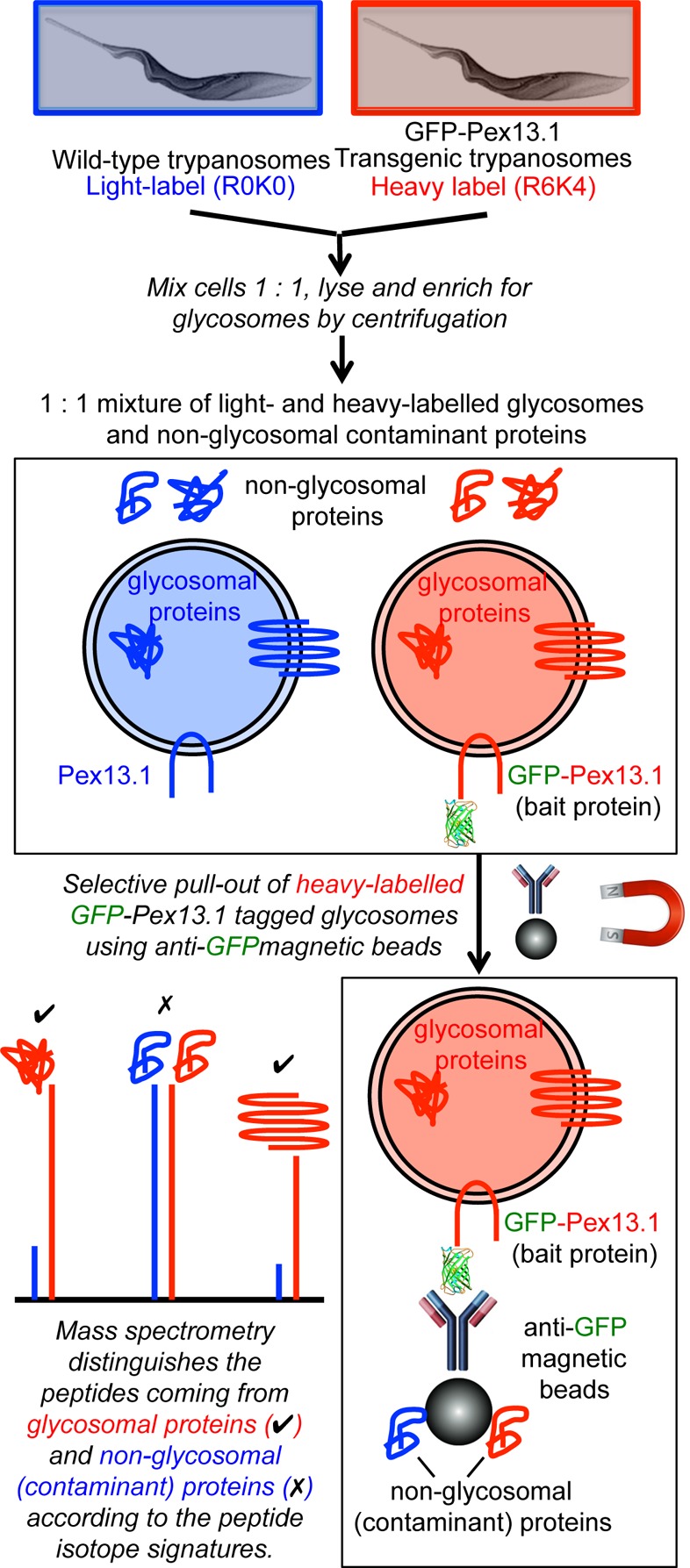
Overview of the approach taken to obtain a high-confidence glycosome proteome. Wild-type procyclic form trypanosomes were grown in normal (R0K0, light) medium, and cells transfected to express GFP-tagged Pex13.1 were labeled with heavy (R6K4) Lys and Arg. The cells were mixed 1:1, and glycosomes were enriched by centrifugation, followed by affinity-selection on anti-GFP magnetic beads. Peptides from genuine glycosome components have high heavy/light isotope ratios, whereas those from contaminants have isotope ratios close to 1:1.
Experimental Procedures
Cell Culture and Transfection of Procyclic Form T. brucei Cells
Procyclic form T. brucei 427 strain containing T7 RNA polymerase and Tet repressor protein, respectively, under control of G418 and hygromycin (clone 29.13.6 cells, kindly provided by George Cross and referred to from hereon as wild-type cells)20 were grown at 28 °C without CO2 in original SDM-79 medium,21 containing 15% (v/v) heat inactivated fetal bovine serum (FBS), 2 g/L sodium bicarbonate, 2 mM glutamax I (Invitrogen), 22.5 μg/mL hemin (added from a 10 mg/mL stock in 50 mM NaOH), and pH adjusted to 7.3. Hygromycin B (Roche) at 50 μg/mL and G418 (Invitrogen) at 15 μg/mL were also added. These cells were electroporated with 10 μg/cuvette of Not I linearized plasmid pGC1 containing an ORF encoding the full length T. brucei Pex13.1 protein sequence (Tb927.10.14720) fused to an N-terminal green fluorescent protein (eGFP) tag, a kind gift from Paul Michels.22 Electroporation conditions were as previously described,23 and selection was done using 10 μg/mL of blasticidin (Invitrogen). The resistant cells were cloned by dilution to a single cell/mL in 96-well plates using SDM-79 containing all reagents previously described but with 20% (v/v) heat-inactivated FBS and 4 g/L sodium bicarbonate and grown in a CO2 incubator at 28 °C. Thirty clones were selected and activated with tetracycline added from a freshly prepared 1 mg/mL stock in 70% ethanol. Clones were tested by Western blot and by fluorescence microscopy to confirm the appropriate GFP-tagging of Pex13.1 and its glycosomal localization.
SILAC Labeling of T. brucei Procyclic Form Cells
The SILAC labeling of T. brucei procyclic form cells was performed as described recently.24 In brief, log phase parasites were washed three times with 10 mL of SDM-79 depleted of l-lysine and l-arginine (SDM-79-RK) and used to seed cultures where the SDM-79-RK medium was supplemented with either normal isotopic abundance l-arginine and l-lysine (SDM-79 + R0K0, referred to as light label) or with [U–13C]-l-Arginine·HCl and [4,4,5,5-2H]-l-lysine·2HCl (SDM-79 + R6K4, referred to as heavy label). The stable isotope-labeled amino acids were obtained from Cambridge Isotope Laboratories, U.K. The heavy and light labels were used at the same concentration, as described in the original SDM-79 formulation.21 In general, apart from the label-swap (Table 1, experiment 4), wild-type cells were suspended at 2.5 × 106 cells/mL in SDM-79 + R0K0, and GFP-Pex13.1 expressing cells were suspended in SDM-79 + R6K4 at the same concentration. Cultures were diluted about 7.7-fold (back to ∼2.5 × 106 cells/mL) every 2 days with fresh media to allow for eight cell divisions under the labeling conditions and to enlarge cultures to about 500 mL at final concentration of ∼2.5 × 107 cells/mL. The expression of GFP-Pex13.1 was induced by adding 40 ng/mL fresh tetracycline 16 h prior to harvesting. Cells were counted, and R0K0- and R6K4-labeled cells were mixed 1:1, lysed, immunoprecipitated using anti-GFP magnetic beads, eluted, and processed for proteomics, as described later.
Table 1. Summary of Individual SILAC Experiments Performed.
| biological replicate exptsa | wild-type cell labelb | GFP-Pex13.1 transgenic cell line labelb | preclearing beads used | IP beads used | no. of fractions
prepared by FASP and SCX (no. of LC–MS/MS runs)c |
no. of fractions
prepared by SDS-PAGE (no. of LC–MS/MS runs)c |
|---|---|---|---|---|---|---|
| expt 1 | R0K0 | R6K4 | protein G dynabeads | mouse anti-GFP + protein G dynabeads | 0(0) | 9(27) |
| expt 2 | R0K0 | R6K4 | ethanolamine-inactivated Pierce NHS-magnetic beads | camelid anti-GFP coupled to NHS-magnetic beads | 0(0) | 8(24) |
| expt 3 | R0K0 | R6K4 | ethanolamine-inactivated Pierce NHS-magnetic beads | camelid anti-GFP coupled to NHS-magnetic beads | 10(30) | 0(0) |
| expt 4 | R6K4 | R0K0 | ethanolamine-inactivated Pierce NHS-magnetic beads | camelid anti-GFP coupled to NHS-magnetic beads | 10(30) | 0(0) |
Biological replicate 4 was a label-swap experiment, performed in parallel with replicate 3.
Trypanosome procyclic forms were labeled with unlabeled l-Arg and l-Lys, designated as R0K0 or with [13C6 ]-l-Arg and [2H4]-l-Lys, designated as R6K4.
Tryptic peptides were generated either by trypsin in-gel digestion of protein bands from SDS-PAGE or by filter aided sample preparation (FASP) and further fractionation using strong cation exchange chromatography (SCX).
The SILAC label-chase experiment was performed similarly, except that after steady-state labeling for eight cell divisions both cultures were split in half. One half of each culture was washed twice with, and resuspended in, R0K0 media to perform a chase for 5 h at 28 °C, while the other halves of the steady-state labeled cultures were simply harvested at the end of the labeling period and processed as described later.
Cell Lysis, Subcellular Fractionation, and Glycosome Enrichment
Light and heavy amino-acid-labeled cells were harvested separately by centrifugation at 600g for 10 min at 4 °C, washed twice, and resuspended in 250 mM sucrose, 25 mM Tris-HCl pH 7.4, and 1 mM EDTA (STE buffer), counted using a Casy Innovatis Counter (Scharfe), and mixed 1:1 to obtain a final cell suspension of 2 × 109 cells/mL in STE containing 0.1 mM 1-chloro-3-tosylamido-7-amino-2-heptone (TLCK), 1 μg/mL leupeptin, 1 μg/mL aprotinin, and complete protease inhibitor cocktail without EDTA (Roche). The combined cells were lysed with 2 shots of 10 000 psi using a One Shot (Constant Cell Disruption System; Northants, U.K.). These conditions of cell density and pressure had previously been optimized to give the best yield of glycosomes through a conventional density centrifugation procedure.25 The lysates were centrifuged twice at 2000g for 15 min at 4 °C to remove any remaining live cells, cell ghosts. and nuclei. The supernatant was then centrifuged twice at 5000g for 15 min to remove material enriched in mitochondria, known as large granular fraction. (Note: Further subcellular fractionation, by pelleting the small granular fraction, resuspending, and performing density gradient centrifugation,25 was avoided because previous experiments had shown that anti-GFP immuno-affinity enrichment of the tagged Pex13.1-GFP glycosomes was compromised when this was performed, possibly due to glycosome aggregation or membrane fusion during the high-speed pelleting step.) The supernatant from the 5000g spin, enriched in glycosomes and endoplasmic reticulum (ER) and containing cytosol, was precleared by rotating with 0.2 mL of magnetic beads without antibody for 1 h at 4 °C. The preclearing beads were removed on a magnet, and the supernatant was immunoprecipitated using magnetic beads coupled to anti-GFP, as described in Table 1, using either 16 μg of immunopure anti-GFP mouse monoclonal antibody (Roche) and 0.2 mL magnetic protein G dynabeads at 30 mg/mL (Invitrogen) for 1 h at 4 °C or camelid anti-GFP (Chromoteck, Germany) precoupled to NHS-magnetic beads according to the manufacturers instructions (Pierce, Thermo Scientific) for 1 h at 4 °C. Beads were washed three times with 1 mL of STE then once with Tris-buffered saline (TBS) and resuspended in 35 μL of TBS.
Sample Preparation for Proteomics
Samples for analysis by mass spectrometry were prepared either by SDS-PAGE separation followed by in-gel trypsin digestion or by FASP followed by strong cation exchange (SCX) chromatography. For SDS-PAGE, an equal volume of 2× SDS-sample buffer and 0.1 M fresh dithiothreitol (Cleland’s reagent, Ultrol grade, Calbiochem) was added and heated to 50 °C for 20 min. Samples were run in 10% Nupage gels with MOPS running buffer and stained with Simply Blue (Invitrogen). Gel lanes were cut in slices, as shown in Figure S1 in the Supporting Information, and washed sequentially in 50% (v/v) acetonitrile, 0.1 M NH4HCO3, 0.1 M NH4HCO3/acetonitrile (1:1 v/v), and acetonitrile. Gel pieces were dried in Speed-vac (Thermo Scientific) in-gel reduced with 0.01 M dithiothreitol, alkylated with 0.05 M iodoacetamide (Sigma) for 30 min in dark, washed in 0.1 M NH4HCO3, and digested with 12.5 μg/mL modified sequence grade trypsin (Roche) in 0.02 M NH4HCO3 for 16 h at 30 °C. Tryptic peptides were recovered in 50% acetonitrile and 2.5% formic acid and lyophilized prior to analysis by LC–MS/MS.
For filter-aided sample preparation (FASP),26 an equal volume of 4% SDS in 0.1 M Tris-HCl pH 7.2 containing 0.1 M dithiothreitol was added, and FASP protocol II was followed but using 10 000 MWCO Vivacon 500 filters (Sartorius). Tryptic peptides obtained from FASP were desalted using C18 Empore cartridges (Agilent Technologies) and freeze-dried. Prior to SCX fractionation, samples were dissolved in 200 μL of SCX solvent A (10 mM KH2PO4 pH 3.0, 30% acetonitrile) and chromatographed as previously described.24 Fractions were combined in 10 pools according to their UV absorbance at 210 nm and desalted using C18 microspin silica columns (Nest group, USA), freeze-dried, and submitted to mass spectrometry.
Mass Spectrometry Data Acquisition and Processing
Liquid chromatography tandem mass spectrometry was performed by the FingerPrints Proteomics Facility at the University of Dundee.
For experiments 1 and 2, liquid chromatography was performed on a fully automated Ultimate U3000 Nano LC System (Dionex) fitted with a 300 μm id × 5 mm Acclaim PepMap 100 C18 trap column and a 75 μm × 15 cm Acclaim PepMap C18 nanocolumn (Thermo Scientific). Samples were loaded in 0.1% formic acid (buffer A) and separated using a binary gradient consisting of buffer A and buffer B (90% acetonitrile, 0.08% formic acid). Peptides were eluted with a linear gradient from 2 to 35% buffer B over 65 min. For experiments 3 and 4, liquid chromatography was performed on a fully automated Ultimate 3000 RSLCnano system (Thermo Scientific) fitted with a 100 μm × 2 cm Acclaim PepMap100 trap column (5 μm particle size) and a 75 μm × 50 cm PepMapRSLC C18 nanocolumn (2 μm particle size) (Thermo Scientific). Samples were loaded in 0.1% formic acid (buffer A) and separated using a binary gradient consisting of buffer A and buffer B (80% MeCN, 0.08% formic acid). Peptides were eluted with a linear gradient from 2 to 40% buffer B over 125 min and a column temperature of 50 °C.
The HPLC systems were coupled to an LTQ Orbitrap Velos Pro mass spectrometer (Thermo Scientific) equipped with a Proxeon Easy-Spray nanosource. The mass spectrometer was operated in data-dependent mode to perform a survey scan over a range of m/z 335–1800 in the Orbitrap analyzer (resolution = 60 000), with each MS scan triggering 15 MS2 acquisitions of the 15 most intense ions. The Orbitrap mass analyzer was internally calibrated on the fly using the lock mass of polydimethylcyclosiloxane at m/z 445.120024.
Data were processed using MaxQuant version 1.3.0.5,27 which incorporates the Andromeda search engine.28 Proteins were identified by searching a protein sequence database containing T. brucei brucei 927 annotated proteins (version 4 downloaded from TriTrypDB,28http://www.tritrypdb.org/) supplemented with the VSG221 sequence and those of frequently observed contaminants (porcine trypsin, bovine serum albumin, and human keratins). Search parameters specified an MS tolerance of 10 ppm, an MS/MS tolerance at 0.6 Da, and full trypsin specificity, allowing for up to two missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionines, N-terminal protein acetylation, and N-pyroglutamate were allowed as variable modifications. Peptides were required to be at least six amino acids in length, with a MaxQuant score >5 and false discovery rates (FDRs) of 0.01 calculated at the levels of peptides, proteins, and modification sites based on the number of hits against the reversed sequence database. SILAC ratios were calculated using only peptides that could be uniquely mapped to a given protein group and required a minimum of 2 SILAC pairs. The distribution of SILAC was normalized within MaxQuant at the peptide level so that the median of log2 ratio is zero to account for any errors in cell counting.27
Prior to statistical analysis, the outputs from MaxQuant were filtered to remove known contaminants and reverse sequences. Quantitation of the fold-enrichment of glycosomal proteins by GFP-Pex13.1 pull down versus the untagged control is derived from four SILAC-labeled biological replicates, including one label swap experiment. Replicate trimming was applied by calculating the mean and standard deviation (σ) of the distribution of the log2(H/L × L/H)30 and discarding data >2σ from the mean. Where SILAC ratios were reported in only one experiment, they were discarded if the percentage variation in the calculated SILAC ratio (calculated by MaxQuant) was >100%.30 Because the Log2(H/L) ratios were not normally distributed, with a distribution of high Log2(H/L) ratios (glycosomal proteins) superimposed onto the normal distribution (nonspecific proteins) centered about zero, an intensity-weighted local standard deviation30 of the normally distributed background (Log2H/L < |1.2|) was calculated. Data were visualized using Perseus 1.3.0.4 (www.perseus-framework.org), and further information on the identified proteins was obtained from TriTrypDB (http://www.tritrypdb.org).28 Transmembrane domain prediction for protein hits was performed using THMM analysis,34 and we searched for PTS1 and PTS2 (peroxisome targeting sequences) using the following motif searches: [STAGCN][RKH][LIVMAFY]$ and ^M.{0,20}[RK][LVI].....[HQ][ILAF], respectively, in the TriTrypDB T. brucei strain 927 predicted protein database.5
SDS-PAGE and Western Blotting
Proteins separated by SDS-PAGE (Nupage, Invitrogen) were transferred onto nitrocellulose using iBlot system (Invitrogen). After 7 min transfer in program 3, the nitrocellulose was recovered and set up on a SNAPid system (Millipore) to develop with antibodies, as recommended by manufacturer. The blocking buffer used was 50 mM Tris-HCl pH 7.4, 0.15 M NaCl, 0.25% BSA, 0.05% (w/v) Tween-20, 0.05% NaN3, and 2% (w/v) fish skin gelatin, filtered (0.2 μm) to remove particles. Mouse antiphosphomannose isomerase (PMI) serum was raised against recombinant T. brucei PMI,14 and rabbit antialdolase serum was a generous gift from Paul Michels. These sera were diluted 1:1000 and 1:4000, respectively, in blocking buffer. Purified mouse anti-GFP monoclonal antibody (Roche) was diluted to 0.4 μg/mL in blocking buffer. Secondary antibodies for the Licor Odyssey Imager were diluted in blocking buffer containing 0.01% SDS (w/v) at 1:15 000 for infrared green antimouse (IRdye 800) and 1:20 000 for infrared red (IRdye 680) antirabbit.
Light and Electron Microscopy
GFP-Pex13.1 mutant procyclic cells grown in SDM-79 were incubated with and without 40 ng/mL tetracycline for 16 h and pelleted by centrifugation at 600g for 10 min. Cells were washed in phosphate-buffered saline (PBS), fixed in 4% (w/v) paraformaldehyde in PBS at 4 °C for 30 min, and placed on a coverslip. After air-drying, coverslips were washed in PBS and mounted onto slides using Prolong Gold with DAPI (Invitrogen). GFP fluorescence images were collected in a Zeiss LSM 710 META confocal microscope, setting the red channel for cell autofluorescence.
Electron microscopy was performed on immunopurified GFP-tagged glycosomes still attached to protein G dynabeads after fixation in 4% paraformaldehyde, 0.25% glutaraldehyde in PBS overnight at 4 °C. For scanning electron microscopy (SEM), immunopurified GFP-tagged glycosome beads were retained on Track-Etch membrane filters (Nuclepore, Whatman) and processed as previously described,32 except that 8 nm gold–palladium coating was used. For immune-transmission electron microscopy (TEM), beads containing immunopurified GFP-tagged glycosomes were fixed as previously described and blocked overnight in 10% normal goat serum and 1% (w/v) fish skin gelatin (Sigma) in Tris buffered saline. The beads were washed in blocking buffer and incubated for 1 h at room temperature with 1:200 rabbit anti-Peroxin 11 (a kind gift from Christine Clayton) in blocking buffer, washed, and developed for 1 h at room temperature with 1:500 protein A conjugated to 10 nm gold (BBI Solutions). The beads were collected using a magnet and resuspended in 1% aqueous osmium tetroxide, dehydrated, and set in Durcupan epoxy resin (Sigma). Sections were cut using a Leica Ultracut UCT system and analyzed using a Philips Tecnai 12 TEM instrument. Regular TEM was processed as described, but antibody steps were omitted.
Results
Generation of Transgenic Procyclic form T. brucei Expressing GFP-Pex13.1-Tagged Glycosomes
The wild-type procyclic cell line was genetically manipulated to introduce a tetracycline-inducible ectopic copy of a gene encoding the Pex13.1 protein with a GFP tag fused to its N-terminus (GFP-Pex13.1). Because Pex13.1 is a transmembrane protein that spans the glycosome membrane twice, with both the N- and C-terminus on the surface of the glycosome (Figure 1),22 anti-GFP magnetic beads can be used to capture and enrich whole glycosomes made by transgenic parasites expressing the GFP-Pex13.1 construct. The inducible GFP-Pex13.1-transformed cells were cloned by limiting dilution, and the resulting 30 clones were induced with a range of tetracycline concentrations. Cell lysates were analyzed by Western blot for the presence of the GFP-Pex13.1 fusion protein and by fluorescence microscopy for the presence of GFP fluorescent glycosomes. We observed that full induction with 0.5 μg/mL tetracycline lead to unstable expression of GFP-Pex13.1, such that following a burst of GFP-Pex13.1 expression the cells became unresponsive to tetracycline. This may indicate that overexpression of GFP-Pex13.1 is detrimental to trypanosomes. However, we were able to select a clone that, when induced with 0.04 μg/mL tetracycline for 16 h, showed reproducible and stable expression of the GFP-Pex13.1 fusion protein (Figure 2A). This clone was analyzed by fluorescence microscopy and showed the presence of GFP tagged glycosomes in the presence but not the absence of 0.04 μg/mL tetracycline induction for 16 h (Figure 2B). Under these induction conditions, the growth of this GFP-Pex13.1 expressing clone is very similar to the parental cell line (Figure 2C), allowing us to grow the transgenic and parental cell lines in parallel for SILAC labeling.
Figure 2.
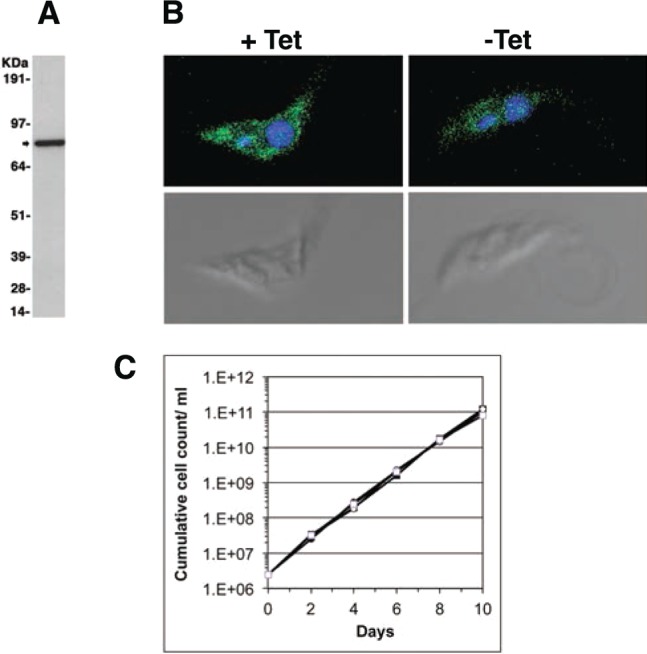
Expression of GFP-Pex13.1 in procyclic form T. brucei. (A) Cell lysate from 1.2 × 108 GFP-Pex13.1 transgenic parasites induced with 40 ng/mL tetracycline for 16 h was immunoprecipitated with GFP-trap magnetic beads, and the proteins eluted with boiling SDS-sample buffer were separated by SDS-PAGE and subjected to Western blotting with a mouse anti-GFP antibody, revealing the transgene product. The positions of Ponceau red-stained molecular weight markers are shown on the left. (B) Fluorescence imaging of the same cells for GFP (green) and DNA (blue). (C) Growth curves of wild type (open squares) and the GFP-Pex13.1 clone induced with 40 ng/mL tetracycline (open circles). The wild-type and GFP-Pex13.1 expressing cells were grown in SDM-79+R0K0 and SDM-79+R6K4 media, respectively. Cells were passaged every 2 days, and the cumulative growth is plotted against time.
To get a visual impression of our ability to enrich GFP-Pex13.1-tagged glycosomes from trypanosome lysates with anti-GFP magnetic bead preparations, we performed electron microscopy on the sample obtained using mouse anti-GFP and protein-G dynabeads. TEM showed the beads associated with round organelle structures with dimensions consistent with those of glycosomes (Figure 3A) that could be decorated with rabbit anti-Pex11 antibody to a glycosome membrane peroxin33 and detected with 10 nm gold-conjugated protein A (Figure 3B). In addition, SEM of the beads showed several such organelles associated with each magnetic bead (Figure 3C) compared with control beads that had not been incubated with trypanosome lysate (Figure 3D).
Figure 3.

Transmission and scanning electron microscopy of purified GFP-tagged glycosomes attached to magnetic beads. (A) Transmission electron micrograph showing a magnetic bead (left) attached to a spherical organelle (glycosome). (B) Transmission immunoelectron micrograph of a magnetic bead (bottom left) and attached GFP-tagged glycosome labeled with rabbit anti-Pex11 antibody and antirabbit antibody conjugated to 10 nm gold (arrow). (C) Scanning electron micrograph of magnetic beads with attached GFP-tagged glycosomes. (D) Scanning electron micrograph of a magnetic bead prior to the capture of GFP-tagged glycosomes. The scale bars correspond to 0.1 μm in panels A and B and 1 μm in panels C and D.
SILAC Labeling, GFP-Pex13.1-Tagged Glycosome Enrichment, and Sample Processing
Four separate biological replicate SILAC experiments were performed in total, and these are summarized in Table 1.
For three of these experiments, wild-type and transgenic parasites expressing GFP-Pex13.1 were grown for eight cell divisions in parallel and under identical conditions, except that the transgenic parasites expressing GFP-Pex13.1 were grown in “heavy medium” containing stable isotope-labeled Lys and Arg (R6K4), whereas the wild-type cells were grown in “light medium” containing unlabeled Lys and Arg (R0K0). In each experiment, the transgenic parasites expressing GFP-Pex13.1 and the wild-type cells were harvested, washed, counted, mixed together in a 1:1 ratio, and lysed. These lysates, containing equal amounts of labeled and unlabeled proteins, were used to capture the heavy-labeled GFP-Pex13.1 tagged glycosomes on anti-GFP magnetic beads for proteomic analysis. Using this protocol, specifically enriched glycosomal proteins can be distinguished from nonspecific contaminant (nonglycosomal) proteins by the isotope ratios of their tryptic peptides. Thus, true glycosomal protein peptides will have high heavy/light isotope ratios, whereas contaminant proteins will have heavy/light isotope ratios close to 1.
For these three experiments, the beads were recovered magnetically and washed prior to either (i) processing by SDS-PAGE into gel-slices (see Figure S1 in the Supporting Information) and performing in-gel reduction, S-alkylation, and tryptic digestion for LC–MS/MS (Table 1, experiments 1 and 2) or (ii) boiling in SDS containing buffer and performing the FASP method,26 with subsequent fractionation into subfractions by SCX HPLC prior to LC–MS/MS (Table 1, experiment 3).
The fourth experiment was a so-called “label-swap”, whereby the transgenic parasites expressing GFP-Pex13.1 were grown in light medium, and the wild-type cells were grown in the heavy medium. Otherwise, this experiment (Table 1, experiment 4) was identical to that described in Table 1, experiment 3.
For all four experiments, the peptide fractions were run in triplicate by LC–MS/MS (technical replicates). In total, data from 111 LC–MS/MS runs were used for data analysis.
Proteomic Data Analysis
The data from each individual experiment (Table 1, experiments 1–4) and the combined data for all four experiments were used to search a T. brucei predicted protein database29 using MaxQuant software.27 For the proteins identified in the combined data set, we performed a pairwise comparisons of the log2(H/L) ratios with those for the proteins identified in the four individual data sets. These showed good correlations (Pearson coefficients of 0.916, 0.872, 0.906, and 0.879; Figure S2 in the Supporting Information). Consequently, we used the combined data (after inverting the H/L ratios for experiment 4, the label swap) for all further analyses. A total of 3182 proteins were identified, at a false discovery rate of 1%, using the combined data (Table S1 in the Supporting Information).
Each protein identification was displayed on a plot (Figure 4) of the Log10 value of the combined intensities of the unique peptides of that protein (y axis) and the Log2 value of the mean heavy to light isotope ratios of the same peptides (x axis). Because of the significant number of glycosomal proteins selectively enriched by the anti-GFP bead pull-out procedure, the Log2-transformed H/L ratios were not normally distributed around zero. Essentially, there is a set of high Log2(H/L) ratio glycosomal proteins superimposed onto a normal distribution of nonspecific, contaminant, proteins with Log2(H/L) ratios centered about zero, that is, Log2(H/L) < |1.2|. We used the intensity-weighted local standard deviation of the normally distributed contaminant proteins (Log2(H/L) < |1.2|) to account for the fact that the H/L ratio of more intense ions can be measured with greater certainty.30 We have then used the calculated local standard deviation (sd) to divide the proteins into three populations (Table S2 in the Supporting Information). Group (i): A large group of 2876 proteins centered around a Log2(H/L) ratio of 0, corresponding to proteins that were not enriched by the anti-GFP pull-out, that is, contaminant proteins. Group (ii): A group of 177 proteins that are significantly enriched in Log2(H/L) ratio over the first group by >3 sd and <8 sd. Group (iii): A group of 129 proteins that are very highly enriched in Log2(H/L) ratio over the first group by >8 sd
Figure 4.
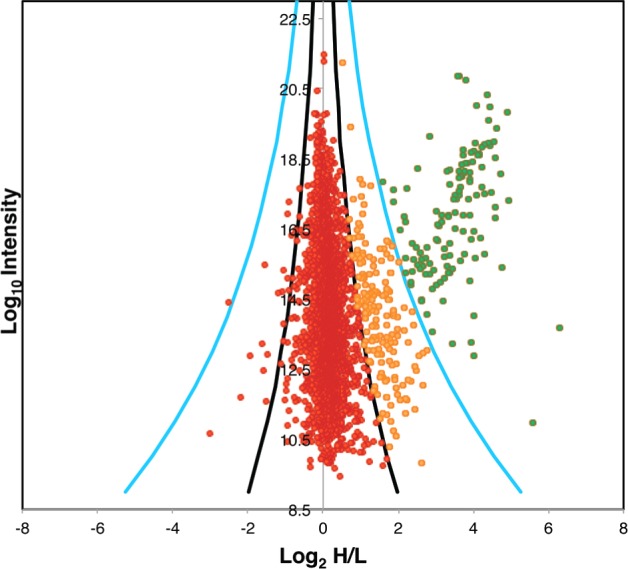
Plot of the combined glycosome proteomics data. Each protein identification is represented by a point plotted as the Log2 of their heavy-to-light isotope ratio (x axis) versus the Log10 value of the combined intensities of the peptides belonging to each protein (y axis). The black and blue curves represent three and eight standard deviations (sd) from the mean. (See the Experimental Procedures.) Proteins plotted in red within the three sd curves are designated group (i) (contaminants), those plotted in amber between the 3 and 8 sd curves are group (ii), and those beyond the 8 sd curves are designated group (iii).
Group (iii) Proteins Form the Core of a High-Confidence Glycosome Proteome
The 129 highly enriched group (iii) proteins include 9 members of the Pex proteins, involved in glycosome biogenesis, and 74 enzyme or enzyme-complex proteins, covering 65 different enzyme activities. The latter (Table S3 in the Supporting Information) include the seven glycolytic enzymes from hexokinase (HK) to phosphoglycerate kinase (PGK) as well as the NAD-dependent glycerol-3-phosphate dehydrogenase (G3PDH) and glycerol kinase (GK). The presence of the Pex and glycolytic pathway components confirms that this group of proteins includes the contents of mature glycosomes. This is further supported by the presence of all of the enzymes of purine salvage and pyrimidine biosynthesis, with the exceptions of bifunctional orotate phosphoribosyltransferase/orotidine 5′-phosphate decarboxylase (OPRT/OPDC) and phosphoribosyl pyrophosphate synthase (PRPS), although the latter appears in the group (ii) proteins. Also consistent with known glycosome biochemistry is the presence of many components of the peroxide and superoxide inactivation system, that is, trypanothione reductase (TR), tryparedoxins (TPN), tryparedoxin peroxidase (TPX), and isocitrate dehydrogenase (ICDH), which provides the NADPH necessary to provide the reducing power for the system. In addition, l-galactonolactone/d-arabinolactone oxidase (GLO), involved in the synthesis of the antioxidant ascorbate, was found, consistent with its reported glycosomal location.35 Most peroxisomes are synonymous with long-chain fatty acid beta-oxidation, and, appropriately, the trifunctional enoyl-CoA hydratase/enoyl-CoA isomerase/hydroxyacyl-CoA dehydrogenase (ECoAH/I/HACoADH) was found in the group (iii) glycosomal proteins. Glycosomes (like many peroxisomes) are also known for ether lipid synthesis,4,36 and dihydroxyacetone-3-phosphate acyltransterase (DHAPAT) and alkyl-dihydroxyacetone-3-phosphate synthase (DHPS) were both found. Finally, the presqualene segement of isoprenoid/sterol synthesis has been described in mammalian peroxisomes37 and suggested to be glycosomal based on the presence of PTS sequences on some of the components.5 Here we find two enzymes on the pathway from mevalonate to squalene, mevalonate kinase (MK) and isopentyl-diphosphate delta isomerase (IPDI), as well as some other postsqualene enzymes of sterol biosynthesis.
Group (ii) Proteins Include Glycosomal and Endoplasmic Reticulum Components
There are several proteins in group (ii) that are known to belong to the ER, for example, components of the protein N-glycosylation, ER quality control, and GPI anchor addition machineries. It is likely that Pex13.1, like many peroxisomal membrane proteins, is made in the ER,36,38 and that, therefore, using a GFP-Pex13.1 fusion protein as bait for the pull-out experiments will not only enrich glycosomal proteins but also some ER proteins that are physically close to GFP-Pex13.1 the moment of cell lysis. Discriminating bone fide glycosomal proteins from ER proteins in group (ii) is therefore complex. The prevailing model of peroxisome biogenesis is that they multiply by growing and budding. While the newly budded organelles directly take up matrix proteins and some membrane proteins (via the PEX19/PEX16 pathway) they also fuse with ER-derived vesicles that provide membrane lipids and other membrane proteins.38 On the basis of this, we performed a label and chase experiment, whereby we heavy-labeled trypanosome proteins to steady-state with R6K4, inducing the expression of GFP-Pex13.1 for the last 16 h of that labeling and then chased that label by incubating the cells in light medium containing R0Ko for 5 h. We then enriched for glycosomes using anti-GFP beads and performed quantitative proteomics, as before. As expected, most peptides, especially those from the group (iii) proteins, showed a significant reduction in their heavy/light ratios (by about 1 log2 unit; Table S4 in the Supporting Information). However, the group (ii) proteins could now be separated into subgroups: 50 with decreasing heavy/light ratios, which we suggest are ER proteins that were simply in the vicinity GFP-Pex13.1 the moment of cell rupture; 29 with increasing heavy/light ratios, which we suggest are bone fide ER-derived glycosomal proteins that, for whatever reason, take longer to reach the mature glycosomes than the group (iii) proteins; and 42 with relatively unchanged heavy/light ratios, which we would not like to call either way. Finally, there are an additional 56 group (ii) proteins in Table S2 in the Supporting Information that were not featured in this particular experiment. This brings our list of high-confidence glycosomal proteins to 158 (indicated in green in the glycosome status column; Table S2 in the Supporting Information), with a further 98 (i.e., 42 + 56) in the “possible” category (indicated by gray and white, respectively, in the glycosome status column; Table S2 in the Supporting Information) and 50 excluded (indicated in red in the glycosome status column; Table S2 in the Supporting Information). Some of the group (ii) proteins elevated to the status of high-confidence glycosomal proteins by this chase experiment include: Pex6, part of the peroxisome biosynthetic machinery; monoglyceride lipase (MGL) and acyl-CoA binding protein (ACBP), which most likely play a role in beta-oxidation by generating free fatty acid and chaperoning acyl-CoA molecules, respectively; glyoxylase II (GLXII), which presumably plays a role in the redox system, returning trypanothione thioesters to free trypanothione;39 the enzyme UDP-glucose 4′-epimerase (GALE), part of sugar nucleotide metabolism and previously shown to be glycosomal;8 and the CAAX protease, a membrane protein that is part of the protein prenylation system,40 presumably with its active site facing the cytosol. Other components, discussed further later, are associated with post-translational modification and protein folding, such as a heat-shock protein (HSP70), a protein phosphatase 2C (PP2C-2), a protein kinase A catalytic subunit (PKA) as well as a ‘nucleotide phosphate linked to X hydrolase’ (NUDIX-2),41 and a methyltransferase of unknown function (drevMT).
Implications of the High-Confidence Glycosome Proteome for Known and Suggested Aspects Glycosome Metabolism
Many of the well-annotated proteins identified in our analysis are indicated in Figure 5A,B with those belonging to group (i) “non-glycosomal” in red, those belonging to the “possibly glycosomal” category from group (ii) in amber, and those belonging to group (iii) plus the elevated group (ii) proteins belonging to “the high-confidence glycosome proteome” in green. Proteins that are expected to be glycosomal but that were either not detected or for which the Log2(H/L) ratio variance was too great are indicated in gray. All proteins that, so far as we are aware, have not been previously described to be glycosomal, are boxed. A list of the enzymes and their abbreviations shown in Figure 5A,B is provided in Table S3 in the Supporting Information.
Figure 5.
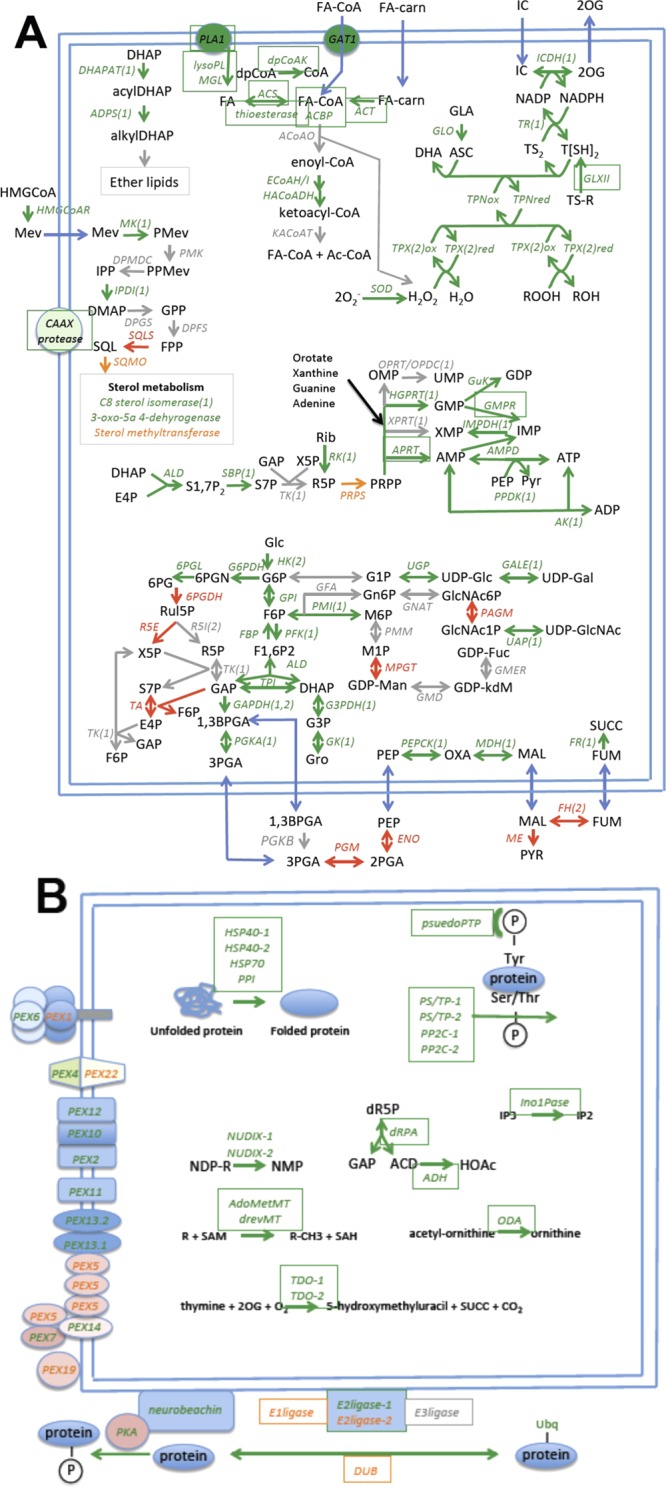
Glycosome metabolism and function according to the glycosome proteome data reported in this paper. The blue lines indicate the glycosome membrane, and the blue arrows indicate transmembrane transport of metabolites. (A) Areas of known or suggested glycosomal metabolism and (B) the glycosomal protein import machinery (Pex proteins) and areas of glycosomal metabolism that have not been previously described. Metabolites are in black. The enzyme abbreviations are in italics and appear in boxes if they have not been previously demonstrated or suggested to be glycosomal. The key to their full names appears in Table S3 in the Supporting Information, and the numbers 1 and 2 in brackets following the enzyme abbreviations indicate the presence of PTS1 and PTS2 targeting sequences. Enzyme and protein abbreviations appear green if they belong to the “high-confidence glycosome proteome”, amber if they belong to the “possibly glycosomal protein’ category, red if their SILAC ratios suggest they are without the glycosome, and gray if they were either not detected or if data variance was too great to assign location.
While the group (iii) and elevated group (ii) proteins are highly consistent with the known or suggested functions of T. brucei glycosomes, a significant number of the enzymes of the pentose phosphate pathway, generally considered to be both cytosolic and glycosomal,42 are missing. Thus, whereas glucose-6-phosphate dehydrogenase (G6PDH) and 6-phosphogluconolactonase (6PGL) are clearly present, as previously reported,43,44 along with sedheptulose 1,7-bisphosphatase (SBP),42 others (enzymes in gray) were either not detected, like ribulose 5-phosphate isomerase (R5I), or statistically excluded due to high variance between replicates, like transketolase (TK). Furthermore, some pentose phosphate pathway enzymes were identified in group (i), suggesting they are contaminants rather than bone fide glycosome components (enzymes in red). These include transaldolase (TA), ribulose 5-phosphate epimerase (R5E) and 6-phosphogluconate dehydrogenase (6PGDH). While these data do not formally exclude the presence of a complete pentose phosphate pathway in procyclic form glycosomes, they suggest that direct experimental localization studies are perhaps warranted before this is taken for granted. Indeed, the majority of transketolase has been previously reported to be in the cytosol.45
With respect to the glycolytic pathway downstream of glycerate 1,3 bisphosphate (1,3BPGA), phosphoenolpyruvate carboxykinase (PEPCK), malate dehydrogenase (MD), and NADH-dependent fumarate reductase (FR) are all clearly present in the group (iii) proteins, but fumarate hydratase (FH) is, like enolase (ENO) and malic enzyme (ME), a group (i) protein, suggesting that all are located without the glycosome. This may be significant because the presence of FH in the glycosomes as well as in the cytosol has been previously suggested.46 The absence of FH in the glycosomes would mean that the transmembrane transport of malate and fumarate would be essential for the production of glycosomal succinate, required for NAD+/NADH balance inside the procyclic glycosome.
Interestingly, in addition to the ABC transporter GAT1 that transports oleoyl-CoA into the glycosome,47 a putative carnitine O-acyltransferase (ACT) was also found, suggesting that there may be an acyl carnitine translocase/carnitine O-acyltransferase shuttle to bring acyl-carnitines into the glycosome and convert them to acyl-CoA species as substrates for beta-oxidation and other processes like ether lipid synthesis. Other possible sources of acyl-CoA substrates are the phospholipase A1 (PLA1), lyso-phospholipase (lysoPL), and monoglyceride lipase (MGL) enzymes that could provide free fatty acids to the acyl-CoA synthase (ACS). In addition, a putative thioesterase could be involved in regulating the intraglycosomal acyl-CoA to free fatty acid balance.48
Novel Glycosomal Functions Suggested by the High-Confidence Glycosome Proteome
Several of the enzymes in the 158 confidently assigned glycosomal proteins list do not obviously or directly fall into conventional glycosomal metabolic pathways, and these are mostly shown in Figure 5B. The presence of NUDIX enzymes in the glycosome (in the case of Tb927.5.4350, consistent with having a PTS2 signal sequence and with one of the Colasante proteomes17) probably makes sense with respect to the relatively high concentrations of nucleotides, sugar nucleotides, (acyl-)CoA, and NAD(H), which will be present in the organelle. Thus, NUDIX hydrolases can help to destroy nucleotides damaged by reactive oxygen species,41 and mammalian and yeast peroxisomal NUDIX proteins have been shown to act as CoA and acyl-CoA diphosphatases, preventing the buildup of CoA cofactors.48 Similarly, the presence of some protein quality control/folding machinery in such a metabolically active compartment (i.e., HSP40, HSP70, and peptidyl-prolyl isomerase (PPI)) is, in retrospect, not particularly surprising. Perhaps the most unexpected glycosomal components are members of protein phosphatase families, which include serine/threonine phosphatases and a pseudotyrosine phosphatase.49 By cross-referencing our glycosomal proteome and phosphoproteome data,31 we see that although the glycosomes contain several phosphoproteins and protein phosphatases (Table S2 in the Supporting Information), they do not contain detectable protein kinases other than PKA, which together with its putative neurobeachin membrane anchoring protein50 most likely faces the outside of the organelle. This suggests that information (e.g., relating to metabolic or cell-cycle status) might be transmitted from the cytosol to the glycosome to exert metabolic control through the import of phosphorylated cargo and that this “information” might naturally decay once in the glycosome through dephosphorylation. This is consistent with some of the hypotheses of Matthews and colleagues.51 Furthermore, one might speculate that the glycosomal catalytically inactive tyrosine phosphatase could be present to “read” the tyrosine phosphorylation status of imported phosphoproteins. Some of the phosphorylated glycosomal proteins include enzymes of carbohydrate metabolism, such as phosphofructokinase (PFK), triosephosphate isomerase (TPI), aldolase (ALD), PEPCK, FR, HK, PGK, and GK, as well as redox and other components. Many of the phosphorylation site occupancies vary considerably between bloodstream form and procyclic form trypanosomes for these enzymes and other glycosomal components (Table S2 in the Supporting Information);31 however, whether phosphorylation modulates any of these activities remains to be determined.
Discussion
Glycosome proteomes have been previously published using density gradient centrifugation enrichment prior to proteomic analyses.17−19 While the organelle enrichment in these studies may be superior to that described here by epitope-tagging and magnetic bead pull-out, only the latter is compatible with SILAC methodology, summarized in Figure 1, which greatly assists the discrimination of genuine components from contaminants. The analysis by Vertommen et al.19 preselected known and suggested glycosome proteins from the data sets and made semiquantitative comparisons, by spectral counting, between the bloodstream and procyclic forms of the parasite. This successfully highlighted metabolic changes between the two lifecycle stages but did not claim to automatically distinguish glycosomal from nonglycosomal proteins. The Colasante et al. studies17,18 also fully acknowledged the limitations of glycosome enrichment alone to unambiguously identify the glycosome proteome because contamination from other compartments, particularly the mitochondrion, is inevitable. This becomes apparent if their protein identifications are superimposed on our SILAC data set, when the extent of group (i) (likely contaminant) protein identifications in these proteomes becomes evident (Figure 6A,B).
Figure 6.

Correlation of the combined glycosome proteomic data in this study with other experimental and theoretical data. The plots are the same as that shown in Figure 4, except that only the three sd limit is shown (black curves) and all data points are plotted in gray unless they appear in the following data sets: (A) Red points indicate those proteins identified in the total glycosome proteome of Colasante et al. (2006).17 (B) Red points indicate those proteins identified in the total glycosome proteome of Colasante et al. (2013).18 (C) Light blue points indicate those proteins containing a PTS1 signal peptide, black points indicate those proteins containing a PTS2 signal peptide, and red points indicate those proteins containing a PTS1 and PTS2 signal peptide. (D) Red points indicate those proteins predicted to contain one transmembrane domain and blue points indicate those proteins predicted to contain more than one transmembrane domain.
A convenient way to predict potential glycosomal proteins is to search for peroxisome targeting sequences (PTS) in the predicted amino acid sequences of trypanosome ORFs.5 This approach recognizes that false positive and negative assignments are made in searching for C-terminal PTS1 and N-terminal PTS2 sequences, and the extent of the problem is evident when proteins containing one or both of these signals is superimposed on our data set (Figure 6C). Overall, 60 PTS-containing proteins were found in the 158 proteins of the high-confidence glycosome proteome, and 77 were found among the group (i) protein contaminants, suggesting that use of PTS signal sequence search in T. brucei, while useful, has a sensitivity of <40% and a specificity of <50%.
Finally, we, and others,19,52 are interested in glycosome membrane proteins that might be involved in metabolite transport into and out of these organelles. Proteins containing one or more predicted transmembrane domains are well distributed across all three protein groups, as indicated in Figure 6D. Of the 35 transmembrane proteins in the high-confidence glycosome proteome, 13 are of known glycosome location, such as eight of the Pex proteins (Pex2, 10, 11, 12, 13.1, 13.2, 14, 22), the Pex11-related proteins Gim5A and Gim5B,53 and the glycosome ABC transporters (GAT1, 2, and 3).54 The others are mostly hypothetical proteins of unknown function. It is worth noting that GAT2 was previously thought to be bloodstream form specific.47,54
In summary, we have used organelle epitope tagging, magnetic bead enrichment, and SILAC proteomics to obtain a high-confidence procyclic form T. brucei glycosome proteome of 158 proteins with a further 98 proteins in a possibly glycosomal category. These data should be a useful resource for the trypanosome biology community.
Acknowledgments
We thank Douglas Lamont and Kenny Beattie for expert assistance with LC–MS/MS, John James and Martin Kierans for expert assistance with electron microscopy, Paul Michels for antibodies, constructs, and helpful discussions, and Christine Clayton for the anti-Pex11 antibody. This work was supported by a Programme Grant (085622) and a Senior Investigator Award (101842) to M.A.J.F. from the Wellcome Trust. The necessary proteomics, microscopy, and computing infrastructure were supported by Wellcome Trust Strategic Awards (097945 and 100476).
Glossary
Abbreviations
- FASP
filter aided sample preparation
- Pex
peroxin
- PTS
peroxisome targeting sequence
- SILAC
stable isotope labeling by amino acids in cell culture
- T. brucei
Trypanosoma brucei
Supporting Information Available
Figure S1. SDS-PAGE of glycosome preparations used for proteomics. Figure S2. Correlation of biological replicate experiments. Table S1. MaxQuant output data from the combined biological and technical replicates described in Table 1. Information on the presence of PTS1 and PTS2 sequences, transmembrane domains, and occurrence in the data sets of Colasante et al.17,18 are included. Table S2. Summary of protein identifications, indicating under glycosome status (column F), the high-confidence glycosome proteome (green), the “possibly glycosomal” group (white and gray), and the contaminants (red), and phosphorylation site data (columns I and J) taken from Urbaniak et al. (2013)31 and annotated and predicted subcelluar localization information taken from the GO terms in TritrypDB29 (columns K and L). Table S3. Enzymes identified in the high-confidence glycosome proteome (green in Column C) and in the “possibly glycosomal” group (amber in Column C), together with their abbreviations (Column A). Table S4. Classification of group (ii) proteins into glycosomal (green), “possibly glycosomal” (gray) or nonglycosomal contaminants (red) using the label-chase data. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository55 with the data set identifier PXD000663. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ M.D.U.: Division of Biomedical and Life Sciences, Lancaster University, Lancaster LA1 4YG, U.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Gabaldon T. Peroxisome diversity and evolution. Philos. Trans. R. Soc., B 2010, 365, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperdoes F. R. Localization of the initial steps in alkoxyphospholipid biosynthesis in glycosomes (microbodies) of Trypanosoma brucei. FEBS Lett. 1984, 169, 35–39. [DOI] [PubMed] [Google Scholar]
- Gualdron-Lopez M.; Brennand A.; Hannaert V.; Quinones W.; Caceres A. J.; Bringaud F.; Concepcion J. L.; Michels P. A. When, how and why glycolysis became compartmentalised in the Kinetoplastea. A new look at an ancient organelle. Int. J. Parasitol. 2012, 42, 1–20. [DOI] [PubMed] [Google Scholar]
- Zomer A. W.; Opperdoes F. R.; van den Bosch H. Alkyl dihydroxyacetone phosphate synthase in glycosomes of Trypanosoma brucei. Biochim. Biophys. Acta 1995, 1257, 167–173. [DOI] [PubMed] [Google Scholar]
- Opperdoes F. R.; Szikora J. P. In silico prediction of the glycosomal enzymes of Leishmania major and trypanosomes. Mol. Biochem. Parasitol. 2006, 147, 193–206. [DOI] [PubMed] [Google Scholar]
- Parsons M. Glycosomes: parasites and the divergence of peroxisomal purpose. Mol. Microbiol. 2004, 53, 717–724. [DOI] [PubMed] [Google Scholar]
- Michels P. A.; Bringaud F.; Herman M.; Hannaert V. Metabolic functions of glycosomes in trypanosomatids. Biochim. Biophys. Acta 2006, 1763, 1463–1477. [DOI] [PubMed] [Google Scholar]
- Roper J. R.; Guther M. L.; Macrae J. I.; Prescott A. R.; Hallyburton I.; Acosta-Serrano A.; Ferguson M. A. The suppression of galactose metabolism in procylic form Trypanosoma brucei causes cessation of cell growth and alters procyclin glycoprotein structure and copy number. J. Biol. Chem. 2005, 280, 19728–19736. [DOI] [PubMed] [Google Scholar]
- Turnock D. C.; Izquierdo L.; Ferguson M. A. The de novo synthesis of GDP-fucose is essential for flagellar adhesion and cell growth in Trypanosoma brucei. J. Biol. Chem. 2007, 282, 28853–28863. [DOI] [PubMed] [Google Scholar]
- Stokes M. J.; Guther M. L.; Turnock D. C.; Prescott A. R.; Martin K. L.; Alphey M. S.; Ferguson M. A. The synthesis of UDP-N-acetylglucosamine is essential for bloodstream form trypanosoma brucei in vitro and in vivo and UDP-N-acetylglucosamine starvation reveals a hierarchy in parasite protein glycosylation. J. Biol. Chem. 2008, 283, 16147–16161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino K.; Guther M. L.; Wernimont A. K.; Amani M.; Hui R.; Ferguson M. A. Identification, subcellular localization, biochemical properties, and high-resolution crystal structure of Trypanosoma brucei UDP-glucose pyrophosphorylase. Glycobiology 2010, 20, 1619–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino K.; Guther M. L.; Wernimont A. K.; Qiu W.; Hui R.; Ferguson M. A. Characterization, localization, essentiality, and high-resolution crystal structure of glucosamine 6-phosphate N-acetyltransferase from Trypanosoma brucei. Eukaryotic Cell 2011, 10, 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandini G.; Marino K.; Guther M. L.; Wernimont A. K.; Kuettel S.; Qiu W.; Afzal S.; Kelner A.; Hui R.; Ferguson M. A. Phosphoglucomutase is absent in Trypanosoma brucei and redundantly substituted by phosphomannomutase and phospho-N-acetylglucosamine mutase. Mol. Microbiol. 2012, 85, 513–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuettel S.; Wadum M. C.; Guther M. L.; Marino K.; Riemer C.; Ferguson M. A. The de novo and salvage pathways of GDP-mannose biosynthesis are both sufficient for the growth of bloodstream-form Trypanosoma brucei. Mol. Microbiol. 2012, 84, 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyersoen J.; Choe J.; Fan E.; Hol W. G.; Michels P. A. Biogenesis of peroxisomes and glycosomes: trypanosomatid glycosome assembly is a promising new drug target. FEMS Microbiol. Rev. 2004, 28, 603–643. [DOI] [PubMed] [Google Scholar]
- Gualdron-Lopez M.; Brennand A.; Avilan L.; Michels P. A. Translocation of solutes and proteins across the glycosomal membrane of trypanosomes; possibilities and limitations for targeting with trypanocidal drugs. Parasitology 2013, 140, 1–20. [DOI] [PubMed] [Google Scholar]
- Colasante C.; Ellis M.; Ruppert T.; Voncken F. Comparative proteomics of glycosomes from bloodstream form and procyclic culture form Trypanosoma brucei brucei. Proteomics 2006, 6, 3275–3293. [DOI] [PubMed] [Google Scholar]
- Colasante C.; Voncken F.; Manful T.; Ruppert T.; Tielens A. G. M.; van Hellemond J. J.; Clayton C. Proteins and lipids of glycosmal membranes from Leishmania tarentole and Trypanosoma brucei. F1000Research 2013, 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertommen D.; Van Roy J.; Szikora J. P.; Rider M. H.; Michels P. A.; Opperdoes F. R. Differential expression of glycosomal and mitochondrial proteins in the two major life-cycle stages of Trypanosoma brucei. Mol. Biochem. Parasitol. 2008, 158, 189–201. [DOI] [PubMed] [Google Scholar]
- Wirtz E.; Leal S.; Ochatt C.; Cross G. A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 99, 89–101. [DOI] [PubMed] [Google Scholar]
- Brun R.; Schonenberger Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Short communication. Acta Trop. 1979, 36, 289–292. [PubMed] [Google Scholar]
- Brennand A.; Rigden D. J.; Michels P. A. Trypanosomes contain two highly different isoforms of peroxin PEX13 involved in glycosome biogenesis. FEBS Lett. 2012, 586, 1765–1771. [DOI] [PubMed] [Google Scholar]
- Guther M. L.; Lee S.; Tetley L.; Acosta-Serrano A.; Ferguson M. A. GPI-anchored proteins and free GPI glycolipids of procyclic form Trypanosoma brucei are nonessential for growth, are required for colonization of the tsetse fly, and are not the only components of the surface coat. Mol. Biol. Cell 2006, 17, 5265–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbaniak M. D.; Guther M. L.; Ferguson M. A. Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS One 2012, 7, e36619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperdoes F. R.; Baudhuin P.; Coppens I.; De Roe C.; Edwards S. W.; Weijers P. J.; Misset O. Purification, morphometric analysis, and characterization of the glycosomes (microbodies) of the protozoan hemoflagellate Trypanosoma brucei. J. Cell Biol. 1984, 98, 1178–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualised p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J.; Neuhausert N.; Michalskit A.; Scheltemat R. A.; Olsen J. V.; Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- Aslett; et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acid Res. 2010, 38, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. Microarray data normalization and transformation. Nat. Genet. 2002, 32Suppl496–501. [DOI] [PubMed] [Google Scholar]
- Urbaniak M. D.; Martin D. M.; Ferguson M. A. Global quantitative SILAC phosphoproteomics reveals differential phosphorylation is widespread between the procyclic and bloodstream form lifecycle stages of Trypanosoma brucei. J. Proteome Res. 2013, 12, 2233–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbaniak M. D.; Turnock D. C.; Ferguson M. A. Galactose starvation in a bloodstream form Trypanosoma brucei UDP-glucose 4′-epimerase conditional null mutant. Eukaryotic Cell 2006, 5, 1906–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voncken F.; van Hellemond J. J.; Pfisterer I.; Maier A.; Hillmer S.; Clayton C. Depletion of GIM5 causes cellular fragility, a decreased glycosome number, and reduced levels of ether-linked phospholipids in trypanosomes. J. Biol. Chem. 2003, 278, 35299–35310. [DOI] [PubMed] [Google Scholar]
- Krogh A.; Larsson B.; von Heijne G.; Sonnhammer E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [DOI] [PubMed] [Google Scholar]
- Wilkinson S. R.; Prathalingam S. R.; Taylor M. C.; Horn D.; Kelly J. M. Vitamin C biosynthesis in trypanosomes: a role for the glycosome. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11645–11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels P. A.; Moyersoen J.; Krazy H.; Galland N.; Herman M.; Hannaert V. Peroxisomes, glyoxysomes and glycosomes (review). Mol. Membr. Biol. 2005, 22, 133–145. [DOI] [PubMed] [Google Scholar]
- Kovacs W. J.; Tape K. N.; Shackelford J. E.; Duan X.; Kasumov T.; Kelleher J. K.; Brunengraber H.; Krisans S. K. Localization of the pre-squalene segment of the isoprenoid biosynthetic pathway in mammalian peroxisomes. Histochem. Cell Biol. 2007, 127, 273–290. [DOI] [PubMed] [Google Scholar]
- Nuttall J. M.; Motley A.; Hettema E. H. Peroxisome biogenesis: recent advances. Curr. Opin Cell Biol. 2011, 23, 421–426. [DOI] [PubMed] [Google Scholar]
- Wendler A.; Irsch T.; Rabbani N.; Thornalley P. J.; Krauth-Siegel R. L. Glyoxalase II does not support methylglyoxal detoxification but serves as a general trypanothione thioesterase in African trypanosomes. Mol. Biochem. Parasitol. 2009, 163, 19–27. [DOI] [PubMed] [Google Scholar]
- Gillespie J. R.; Yokoyama K.; Lu K.; Eastman R. T.; Bollinger J. G.; Van Voorhis W. C.; Gelb M. H.; Buckner F. S. C-terminal proteolysis of prenylated proteins in trypanosomatids and RNA interference of enzymes required for the post-translational processing pathway of farnesylated proteins. Mol. Biochem. Parasitol. 2007, 153, 115–124. [DOI] [PubMed] [Google Scholar]
- McLennan A. G. The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 2006, 63, 123–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannaert V.; Bringaud F.; Opperdoes F. R.; Michels P. A. Evolution of energy metabolism and its compartmentation in Kinetoplastida. Kinetoplastid. Biol. Dis. 2003, 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise N.; Opperdoes F. R. Purification, localisation and characterisation of glucose-6-phosphate dehydrogenase of Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 99, 21–32. [DOI] [PubMed] [Google Scholar]
- Duffieux F.; Van Roy J.; Michels P. A.; Opperdoes F. R. Molecular characterization of the first two enzymes of the pentose-phosphate pathway of Trypanosoma brucei. Glucose-6-phosphate dehydrogenase and 6-phosphogluconolactonase. J. Biol. Chem. 2000, 275, 27559–27565. [DOI] [PubMed] [Google Scholar]
- Stoffel S. A.; Alibu V. P.; Hubert J.; Ebikeme C.; Portais J. C.; Bringaud F.; Schweingruber M. E.; Barrett M. P. Transketolase in Trypanosoma brucei. Mol. Biochem. Parasitol. 2011, 179, 1–7. [DOI] [PubMed] [Google Scholar]
- Bringaud F.; Riviere L.; Coustou V. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol. Biochem. Parasitol. 2006, 149, 1–9. [DOI] [PubMed] [Google Scholar]
- Igoillo-Esteve M.; Mazet M.; Deumer G.; Wallemacq P.; Michels P. A. Glycosomal ABC transporters of Trypanosoma brucei: characterisation of their expression, topology and substrate specificity. Int. J. Parasitol. 2011, 41, 429–438. [DOI] [PubMed] [Google Scholar]
- Antonenkov V. D.; Hiltunen J. K. Transfer of metabolites across the peroxisomal membrane. Biochim. Biophys. Acta 2012, 1822, 1374–1386. [DOI] [PubMed] [Google Scholar]
- Brenchley R.; Tariq H.; McElhinney H.; Szoor B.; Huxley-Jones J.; Stevens R.; Matthews K.; Tabernero L. The TriTryp phosphatome: analysis of the protein phosphatase catalytic domains. BMC Genomics 2007, 8, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Herberg F. W.; Laue M. M.; Wullner C.; Hu B.; Petrasch-Parwez E.; Kilimann M. W. Neurobeachin: A protein kinase A-anchoring, beige/Chediak-higashi protein homolog implicated in neuronal membrane traffic. J. Neurosci. 2000, 20, 8551–8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szoor B.; Ruberto I.; Burchmore R.; Matthews K. R. A novel phosphatase cascade regulates differentiation in Trypanosoma brucei via a glycosomal signaling pathway. Genes Dev. 2010, 24, 1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualdron-Lopez M.; Vapola M. H.; Miinalainen I. J.; Hiltunen J. K.; Michels P. A.; Antonenkov V. D. Channel-forming activities in the glycosomal fraction from the bloodstream form of Trypanosoma brucei. PLoS One 2012b, 7, e34530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voncken F.; van Hellemond J. J.; Pfisterer I.; Maier A.; Hillmer S.; Clayton C. Depletion of GIM5 causes cellular fragility, a decreased glycosome number, and reduced levels of ether-linked phospholipids in trypanosomes. J. Biol. Chem. 2003, 278, 35299–35310. [DOI] [PubMed] [Google Scholar]
- Yernaux C.; Fransen M.; Brees C.; Lorenzen S.; Michels P. A. Trypanosoma brucei glycosomal ABC transporters: identification and membrane targeting. Mol. Membr. Biol. 2006, 23, 157–172. [DOI] [PubMed] [Google Scholar]
- Vizcaino J. A.; Cote R.; Csordas A.; Dianes J. A.; Fabregat A.; Foster J. M.; Griss J.; Alpi E.; Biri M.; Contell J.; O’Kelly G.; Schoenegger A.; Ovelleiro D.; Perez-Riverol Y.; Reisinger F.; Rios D.; Wang R.; Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013, 41, D1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.