

Abstract
Serial femtosecond crystallography (SFX) is a new emerging method, where X-ray diffraction data are collected from a fully hydrated stream of nano- or microcrystals of biomolecules in their mother liquor using high-energy, X-ray free-electron lasers. The success of SFX experiments strongly depends on the ability to grow large amounts of well-ordered nano/microcrystals of homogeneous size distribution. While methods to grow large single crystals have been extensively explored in the past, method developments to grow nano/microcrystals in sufficient amounts for SFX experiments are still in their infancy. Here, we describe and compare three methods (batch, free interface diffusion (FID) and FID centrifugation) for growth of nano/microcrystals for time-resolved SFX experiments using the large membrane protein complex photosystem II as a model system.
Keywords: femtosecond crystallography, crystallization, nanocrystals, free-electron laser, photosystem II
1. Introduction
X-ray crystallography is the most prolific technique for solving protein structures in structural biology. Structure determination of soluble proteins has made great progress in recent years, with more than 99 000 structures solved. However, the structure determination of difficult-to-crystallize proteins, such as large multi-protein complexes and membrane proteins, is severely lagging behind, with less than 400 unique membrane protein structures determined to date [1]. One of the rate-limiting steps for the structure determination of membrane proteins with standard crystallographic methods is the growth of large well-ordered single crystals. The determination of membrane protein structures solved to date has often involved a long process taking years (or sometimes even decades) to grow large, well-ordered crystals suitable for X-ray structure determination. X-ray damage is a major problem in standard X-ray crystallography for many protein crystals [2], especially when they contain redox-active cofactors [3], therefore imposing a limitation for X-ray diffraction on microcrystals, even under cryogenic conditions [4].
The new method of serial femtosecond crystallography (SFX), overcomes many of the limitations of conventional X-ray crystallography, thereby opening an exciting new avenue for membrane protein crystallography [5–7]. It is remarkable that the first proof of principle for SFX was done not with lysozyme or any other small easy-to-crystallize protein but with photosystem I (PSI), which consists of 36 proteins to which 381 cofactors are non-covalently bound [5]. In SFX, tens of thousands of diffraction patterns can be collected in minutes on fully hydrated nano- and microcrystals in their mother liquor, at room temperature. The X-ray laser pulses are so short that they ‘outrun’ X-ray damage by the diffract-before-destroy principle [8] which also opens new avenues for time-resolved crystallography [9,10]. Unlike traditional crystallography, the SFX technique delivers thousands of small crystals of micrometre size in a liquid stream to femtosecond X-ray pulses, using an X-ray free-electron laser (XFEL). This technique is advantageous because smaller crystals have less long-range disorder compared with larger crystals and smaller crystals can be easier to grow. The decrease of long-range disorder in nanocrystals is particularly advantageous for crystals of membrane proteins and large protein complexes, which are often the most difficult to crystallize. These crystals are often plagued by long-range disorder and anisotropic resolution leading to high mosaicity. Before SFX, the growth of small crystals was undesired, making their growth and characterization relatively unexplored. The first study of crystals grown specifically for SFX described the growth of nano- and microcrystals of PSI. In this study, the crystals were grown by ultrafiltration at low ionic strength. In the ultrafiltration method, the protein solution is brought into the nucleation zone by slowly concentrating the protein at low ionic strength. This method can also be used for crystallization of proteins at high ionic strength or using other precipitants that pass through ultrafiltration membranes (C. Kupitz et al. 2014, unpublished data). It is, however, not suitable for crystallization of membrane proteins when using higher molecular weight polyethyleneglycols (PEG), as PEG is a very flexible and elongated polymer that does not easily pass through ultrafiltration membranes (even PEG1000 (with a relative molecular mass of only 1 kDa) does not pass quantitatively through ultrafiltration membranes with a 1000 kDa cuff-off). Unfortunately, PEG is one of the most commonly used precipitants in crystallography, and the majority of membrane proteins, including photosystem II (PSII), [11–13] have been crystallized in the presences of PEGs. This work uses PSII, a large membrane protein complex, to explore techniques for nano/microcrystal growth and characterization.
PSII is one of the most important enzymes in the process of photosynthesis, converting light energy from the sun into chemical energy. It catalyses the light-driven, transmembrane electron transfer from water, which serves as the electron source, to the plastoquinone. The reaction of water oxidation involves four charge separation events, where two water molecules are oxidized, leading to the generation of four protons, four electrons and one molecule of oxygen (for a review on PSII see [14]). This reaction is unique in nature, where PSII has produced all the oxygen in the atmosphere and changed our planet from an anoxygenic to an oxygenic atmosphere 2.5 billion years ago. This reaction makes PSII an attractive candidate for renewable energy and therefore understanding its structure and function are vital [15,16]. However, PSII is an extremely difficult protein with which to work. The results of our time-resolved structural work on PSII, which were presented at the Royal Society Workshop, are published elsewhere [17]. Here, we focus on method developments for the growth of nano/microcrystals, using PSII as a model system. The techniques presented will have a broad impact on current and future projects that aim to determine the structure and dynamics of biomolecules using SFX.
The method of SFX and the success of the experiments at XFELs depends on the growth of high-quality nano- or microcrystals that are delivered to the FEL beam in a liquid stream of their mother liquor. Most SFX experiments have been performed using an injector with a virtual gas-dynamic nozzle [18,19], which is installed at the coherent X-ray imaging (CXI) beamline at the Linac Coherent Light Source (LCLS) and delivers the sample with a flow rate of 10–20 μl min−1. Alternative injector designs, with lower flow rates have also been developed; however, they require proteins to be delivered in highly viscous media. They include the injector described by Sierra et al. [20] that delivers proteins by electrospinning and the recent development of a novel injector design that allows delivery of protein crystals in lipidic cubic phase [21].
For SFX on protein crystals, large quantities of nano/microcrystals of uniform size and quality are desired. While method development of crystal growth for standard X-ray crystallography has been largely focused on new methods to increase the size and quality of the crystals, methods for growth of high-quality nanocrystals are highly desired, yet largely unexplored.
We use PSII as a test case to compare different methods of nanocrystal growth and show how diagnostic tools such as dynamic light scattering (DLS) and second-order nonlinear imaging of chiral crystals (SONICC) [22] can be used as diagnostic tools to optimize the growth of nanocrystals for SFX.
2. Material and methods
(a). Cell growth and protein purification
Currently, the method of SFX uses a liquid injector that requires large amounts of protein. Typical SFX experiments are performed at a flow rate of 10–20 µl min−1 and microcrystal suspensions with 1010–1011 crystals ml−1, depending on the desired ‘hit rate’. The concentration of protein for collection of the SFX data thereby depends on the size distribution of the crystals, and for 1–5 µm crystals is commonly in the range 10–20 mg ml−1 [5,23,24]. Thereby the amount of protein required for SFX experiments is large and one 12 h experiment can easily require 50–100 mg of protein.
PSI and PSII were some of the first proteins used as model systems for SFX [5,25], and it is often assumed that they were chosen because they are abundant in nature, can be isolated in large amounts and are easy to isolate and crystallize. Unfortunately, these assumptions are wrong. The photosystems have been used as model systems because of their extreme importance for bioenergy conversion on the Earth, despite the fact that they are probably some of the most difficult proteins to work with. PSII is a large multiprotein cofactor complex, consisting of 19 proteins and more than 50 cofactors in Thermosynechococcus elongatus [26]. It undergoes constant remodelling in the native membrane due to photodamage [27]. In the native membrane, PSII has a half-life of 30 min, with between 30 and 70% being under reconstruction depending on the environmental conditions such as light, temperature, pH and cell density. It is therefore of extreme importance to grow the cells under reproducible conditions. We grow the cells of T. elongatus at 56°C in Casteholz medium in a custom-designed 122 l photobioreactor, shown in figure 1, which controls all important parameters. It increases the light intensity in parallel with the cell density and controls the pH by feeding an air/CO2 mixture into the system. The cells are grown in a continuous mode, therefore keeping them in the logarithmic growth phase; 30 l of the cell culture is harvested each week, which corresponds to 22–28 g wet cell mass. Compared with cultures of Escherichia coli the yield of cells is low, with less than 1 g of cells harvested per litre of cell culture.
Figure 1.

Photobioreactor developed for large-scale growth of photosynthetic algae and cyanobacteria. The reactor has a capacity of 122 l. It can be sterilized in situ and allows for control of light intensity, cell density, temperature, pH and gas flow.
The purification of PSII from the cells is performed in principle as described by Zouni et al. [11], which includes isolation of photosynthetic membranes by differential centrifugation, solubilization of PSII with the detergent beta-dodecylmaltoside and purification of PSII by ion-exchange chromatography. In our modification of the Zouni et al. protocol, cell disruption is performed using a microfluidizer, which breaks the cells rapidly by shear forces in a small capillary at 18 000 psi in the flow-through mode, where 80 g of cells on ice can be broken quantitatively in less than 2 min. Also, large-scale column purification is performed in a staggered mode to increase capacity. Immediately after column purification, three or four recrystallization steps (see §2b) are carried out to purify the protein further. The photobioreactor is operated in a continuous mode, where 30 l cell culture can be harvested weekly for preparation of PSII crystals, with 1.3–1.6 mg PSII being obtained from 1 l cell culture. This means that 10–20 protein preparations are required to produce sufficient amounts of PSII nanocrystals for five shifts of TR-SFX experiments.
(b). Crystallization as a last purification step
Only fully active dimeric PSII with intact subunit composition crystallizes, while PSII that is in the process of assembly/disassembly that is missing all or part of the lumenal extrinsic proteins PsbV, PsbU or PsbO does not crystallize. This occurs because PsbV is involved in crystal contacts; therefore crystallization can be used as a last purification step (P. Fromme 2003, unpublished results). In this process, PSII is precipitated three times at a Chl concentration of 0.75 mM, with decreasing concentrations of precipitant buffer Dx (100 mM Pipes pH 7.0, 5 mM CaCl2, 5 mM MgCl2, X% PEG2000), X = 7.5%, 6.5% and 5.5%, respectively. The solubility decreases as homogeneity of the protein sample increases; therefore, the PEG concentration is decreased in each of the recrystallization steps. The first and second precipitation/crystallization steps are allowed to progress for 1 h in complete darkness, on ice. The third precipitation is allowed to proceed for 8 h on ice under the same conditions.
(c). Comparison of different methods for the growth of nanocrystals
Three different techniques are compared to obtain microcrystals for SFX: the batch method, free interface diffusion (FID) and FID with centrifugation. All experiments are carried out at a chlorophyll concentration of 0.5 mM Chl at 10°C aiming for nanocrystal growth in 24–48 h.
(i). Batch method
To grow crystals in a batch experiment, the phase diagram should be known, to ensure that the solution is in the nucleation zone after mixing. The determination of the solubility curve can be quite time-consuming and is best performed by stepwise dissolution of crystals by lowering the precipitant concentration. This can be done either manually or by automated stepwise dilution of the precipitant solutions in the reservoirs. We determine the borderline between metastable and nucleation zone by small-scale batch experiments, where seeding crystals are added. In the nucleation zone, addition of seeding crystals leads to massive secondary nucleation. By contrast, the seed crystals added to the metastable zone grow without new crystals appearing. A schematic phase diagram indicating suitable starting points for batch crystallization experiments is shown in figure 2.
Figure 2.
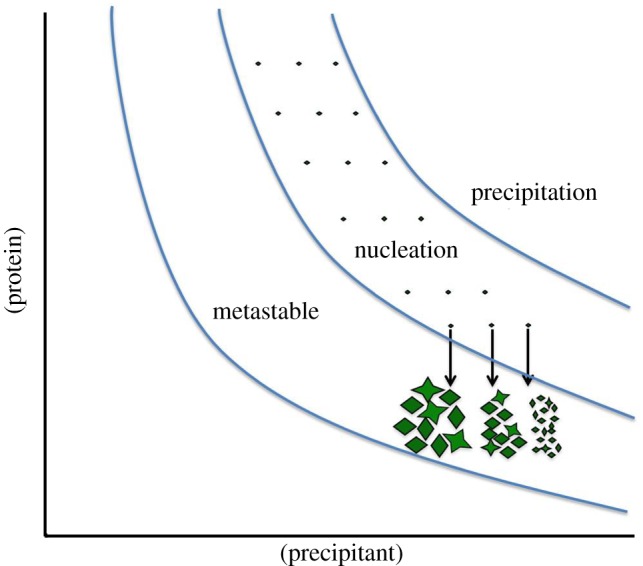
Schematic phase diagram with suitable starting points for batch experiments. All batch experiments should ideally start in the nucleation zone. The nucleation rate increases with the increase of supersaturation, leading to a larger amount of crystals.
Once the phase diagram has been determined, the batch experiments are performed by simple rapid mixing of the protein with the precipitation buffer, under conditions where the solution will be in the nucleation zone after mixing. Then batch crystallization is routinely tested at five different protein concentrations combined with a fine screening of precipitant concentration. This is performed in small scale before the complete protein sample is crystallized in a large batch. The result of such a batch experiment with PSII is shown in figure 3.
Figure 3.

Batch method experiment performed using 0.5 mM Chl, and 13% PEG2000 as starting conditions. (a,b) Images of crystals created using the batch method. There are numerous large crystals, and these are obviously polycrystalline. (c) SONICC image of the entire drop of (b) confirming crystallinity. (d) DLS histogram showing that the majority of the crystals are around 10 μm radius.
Seeding is regularly used for growth of large, single crystals but is also extremely helpful for growth of nanocrystals in batch. Addition of small nanocrystals (best less than 500 nm) to the precipitant solution prior to mixing of precipitant and protein, leads to a massive increase in the nucleation rate. It also can overcome a serious problem that is observed for nanocrystal growth where proteins do not spontaneously crystallize but crystals grow out of an amorphous precipitate in the time frame of weeks. This crystallization condition is the worst-case scenario for nanocrystal growth, but can be partially overcome by adding seeds to the precipitate. The question could now be asked, how can seeds be formed when only larger crystals have been obtained? In this case, one can break the large crystals and use them as crude seeds in step 1 of the experiment. Once these large seeds induce nucleation, nice nano- and microcrystals appear in large showers which can then be used in the next step as seeds. While all these methods help to grow nano- and microcrystals for SFX in batch, the size distribution achieved is often very broad and varies between individual experiments. Furthermore, crystal growth is also very fast, so nice small crystals can grow in a few minutes into larger crystals with visible defects.
Crystal growth can progress rapidly, so crystal growth must be monitored by DLS in short time intervals to stop crystal growth at the desired size by rapid dilution in high precipitant buffer. Unfortunately, the crystals of PSII formed by the batch crystallization method tend to be polycrystalline, resembling starfish. DLS showed that the predominant size of the crystals was 5–20 µm, but optical inspection showed large crystals up to 300 µm in size, though these large crystals can be removed from the solution by prefiltering and in-line filtration. SFX data can be collected on these 5–20 μm crystals even if they are starfish shaped. Most of the diffraction patterns are single crystal diffraction patterns because in most cases the small X-ray focus of only 1 µm ‘hits’ only one of the ‘lobes’ of the starfish-shaped crystal. However, crystals of this size (even if they are perfectly shaped) are not suitable for time-resolved (TR)-SFX as data collection and evaluation face multiple problems: higher mosaicity of the crystals due to their larger size, non-uniform light excitation of PSII in the crystals due to absorption of the photons within the crystal, extremely low hit rates, high sample consumption and fluctuations in the liquid jet position. The jet is only 4 µm in diameter; if the size of the crystals exceeds the size of the jet, the jet becomes unstable and wiggles or even ‘jumps’ when a large crystal exits the nozzle. This further decreases the hit rate as the FEL X-ray beam may not ‘hit’ the jet when it moves. Furthermore, a kinked or wiggling jet path can lead to deposition of materials on the walls of the shroud. This material accumulates at the walls of the injector forming stalagmites of the precipitants (here PEG, salts and buffer) that slowly grow into the interaction region, whereupon data collection has to be stopped and the chamber has to be evacuated and cleaned.
(ii). Free interface diffusion
In this method, crystals are grown at the interface between a highly concentrated protein solution and the precipitant solution. It was designed to grow small, well-ordered crystals to overcome the problems described above for SFX data collection on 5–20 µm crystals.
FID is used in standard crystallography to create a continuous concentration gradient leading to growth of crystals of increasing sizes along a concentration gradient. Its most common use is in Granada crystallization box experiments. Recently, microfluidic devices have been developed to use FID for mapping the phase diagram and optimization of nanocrystal growth [28]; however, they have not yet reached the capacity for large-scale growth of 50–100 mg of PSII nanocrystals for TR-SFX studies. The standard methods of FID used in traditional crystallography, where large crystals are desired, use small capillaries for the crystallization experiments to minimize the size of the interface region and create a smooth linear concentration gradient. Using this set-up, one can achieve nanocrystal growth at the interface, but the crystal size increases along the concentration gradient which leads to a very low yield of nanocrystals. Furthermore, it would be painstaking to accumulate 10 ml of nanocrystal suspension by harvesting nanocrystals from the interface of hundreds of small capillaries. To maximize the surface/volume ratio, we experimented with several different set-ups and geometries, but the best result came from the simplest method, performing the crystallization experiments in 1.5 ml reaction vessels. For the initial screening of conditions, we use volumes of 20 µl protein and 20 µl precipitant solution, which can be scaled up to 500 µl plus 500 µl. In the first sets of experiments, we tried to form a ‘perfect’ free interface by layering the protein solution (which has a lower density than the precipitate) very carefully on top of the precipitant (figure 4a). While nanocrystals formed at the interface, the yield was low and these crystals quickly grew into larger starfish-like crystals similar to what is shown for the batch experiments in figure 3.
Figure 4.

Schematic of the set-up for crystallization experiments with FID (a,b) and FID centrifugation (c). (a) Experimental set-up in which the protein solution is carefully layered on top of the precipitant solution, where only few crystals form at the interface. (b) In the inverse set-up the precipitant solution is added dropwise to the protein solution, inducing increased transient nucleation at the drop–protein interface. (c) The experiment shown in (b) is continued by centrifugation. The nuclei formed in the protein solution are accelerated by centrifugation towards the interface zone, where they grow into nano- or microcrystals. When they reach a specific size they sediment into the precipitant zone, where they stop growing. Thereby nano- or microcrystals with a very narrow size distribution can be achieved.
The nucleation rate was dramatically increased by the inversion of the set-up, shown in figure 4b. Here, the protein is pipetted into the bottom of the reaction vessel. This is followed by slowly dropping the precipitant solution through the protein layer at a rate of roughly 20 μl min−1. This procedure causes a large transient interface area where the small drops of the precipitant move through the protein to form two layers. Eventually, the two phase system forms with the precipitant solution forming a dense bottom layer with the protein on top.
The results of one of these experiments are shown in figure 5 and reveal that the crystals are significantly smaller, with an average radius of 1–2 μm. The crystals also display much less polycrystallinity, with most of the crystals being single.
Figure 5.

FID experiment using 0.5 mM Chl and 13% PEG2000 as starting conditions. (a,b) Images of crystals created using the FID method, showing that crystals are significantly smaller and more uniform in size. (c) SONICC image of the drop of (a) confirming crystallinity. (d) DLS histogram indicating that the majority of the crystals are of 2–5 μm radius.
(iii). Free interface diffusion centrifugation
This third method represents a further modification of the FID method, where the formation of the two-phase system is followed by centrifugation. A schematic drawing of the process is shown in figure 4c. Here, the reaction vessel is centrifuged after the interface has been formed, at a slow speed of 200–500 g using a swinging bucket or a fixed angle centrifuge. The centrifugation has two positive effects that support the formation of well-ordered nanocrystals: (i) the nanocrystals start sedimenting into the precipitant when they have reached a specific size. As there is no protein present in the precipitant solution, the growth of the crystals stops as soon as they enter the precipitant layer. Thereby a very uniform size distribution of small nanocrystals is achieved. (ii) Small nuclei, which form in the protein solution by diffusion of the precipitant into the protein layer, are accelerated towards the interface where the increased precipitant concentration allows them to grow into stable nanocrystals, which then enter the precipitant zone where crystal growth stops. Thereby a constant ‘stream’ of newly formed nuclei enters the interface zone and continues to grow into nanocrystals until they enter the precipitant zone. The process is fast at first and then slows down as the concentration of the protein decreases below the nucleation line. This process takes place in a time frame of 30 min to 24 h, with the majority of the crystals forming after 30 min. The crystals have a very uniform size distribution. Figure 3 shows a typical experiment where most of the crystals have a radius of 500 nm. The initial screening of conditions can be performed with the same volumes as described for the FID experiments (20 µl protein plus 20 µl precipitant). The best conditions are those scaled up to a volume ratio of 50 µl protein plus 50 µl precipitant). The results obtained with this volume ratio scale up very reproducibly to experiments at the optimal volume ratio of 1∶1. We have also twice scaled the crystallization experiments up to volumes of 6 ml protein plus 6 ml precipitant, where the experiment is performed in 15 ml Falcon tubes.
We have tested several parameters that affect the growth of the crystals by the FID centrifugation method. The size of the crystals depends on the protein concentration and precipitant concentration, and is also influenced by the viscosity of the precipitant solution. The speed of centrifugation does not have a large effect on crystal size and yield within the tested range 200–500 g. The yield increases strongly with crystallization time between 5 and 30 min. The crystal yield is only slightly increased when centrifugation is continued to up to 24 h. Crystals of uniform size can be harvested after only 30 min. The size of the crystals grown with this technique can be varied by adjusting protein and precipitant concentration, but crystals grown with this method are consistently below 2 μm radius; see figure 6 for examples.
Figure 6.

Free interface diffusion centrifugation experiment using 0.5 mM Chl and 13% PEG2000 as starting conditions. (a,b) Images of crystals showing that they are smaller and very uniform in size; they grow in approximately 30 min. (c) SONICC image of the entire drop of (b) confirming crystallinity. (d) DLS histogram showing that the majority of the crystals are around 500 nm radius.
(d). Quenching of crystal growth
Ideally, crystal growth should not be quenched, as a dramatic, sudden change in conditions can lead to defects, even in nanocrystals. Data should be collected when the crystals have reached their optimal size, but while this is highly desirable, it is not practical.
For all methods mentioned earlier, crystal growth must still be quenched to prevent them becoming too large, as the diffusion of protein will eventually allow them to continue growing. Crystal growth must be quenched as soon as they reach the desired size distribution, which requires regular monitoring of the size by DLS, visualization with SONICC and optical imaging. For example, if the crystals are grown in a 15% PEG2000 precipitant, then they should be quenched in a 20% PEG2000 solution to prevent further growth.
Crystal growth is typically quenched after they reach the desired size as monitored by optical microscopy and DLS. To quench crystal growth as much supernatant is removed as possible and a high concentration precipitation buffer containing 20% PEG2000 is added on top for crystal storage. These quenched crystals are best used within 2–5 days. A word of caution must be included about transport of crystals. While the crystals maintain their size distribution for days in a laboratory setting where they are stored without vibration at 10°C, they can dramatically change their size distribution during transport in favour of larger crystals. The reason is that even under quenched conditions there is an equilibrium between protein in solution and in the crystals, where proteins on the surface dissolve while proteins in solution can bind to the surface of the crystal. The surface/volume ratio is much higher for nanocrystals than for micrometre-sized crystals, leading to a net transfer of protein from the small to the larger crystals. This process is very slow under diffusion and/or convection controlled conditions when the crystal suspension is stored with minimal vibration. However, vibration and shaking cannot be avoided during transport by air and on land, leading to changes in the size distribution; in the worst-case scenario, most of the small crystals completely dissolve and the remaining crystals are too large for data collection. Therefore, crystallization at the site of data collection is extremely important for the successful growth of well-ordered nano- and microcrystals for SFX.
3. Discussion
(a). Batch method
While the batch technique produces large quantities of small crystals, they are suboptimal for SFX as there is an indication that crystallization occurs too rapidly, producing low-quality crystals which are not ideal for X-ray diffraction. Also this technique offers no size control; crystal size can range anywhere from less than 1 µm to 300 μm. This is a definite problem for SFX because crystal size should be limited to <4 μm, corresponding to the diameter of the liquid jet [18,19] as described in §2c(i). However, there are other sample delivery techniques, including the use of the new injector developed for crystals in lipidic cubic phases [21]. This slow running jet is ideal for many SFX experiments as it requires less sample and features a much larger jet, where crystals up to 30 µm in size can be delivered. However, the slow running jet is problematic for TR-SFX experiments. Another jet type has been reported by Bogan and co-workers [20] which is based on electrospinning and also allows for the use of larger crystals; however, not all crystals may survive this procedure and the hit rates reported for use of this injector are very low.
(b). Free interface diffusion
This technique has several advantages over the batch technique, primarily the ability to control the size of the crystals grown. This could be because the nucleation and growth occur at the interface and are held by the high density of the underlaying precipitation buffer. Once the crystals achieve a certain size, depending upon the protein and the precipitation buffer, they will sink through the precipitation buffer and form a pellet. At this stage, crystal growth stops because lack of protein in the surrounding environment limits crystal size.
(c). Free interface diffusion centrifugation
The major difference between this technique and FID becomes apparent when comparing results obtained using a swinging bucket or fixed angle centrifuge rotor. When using a swinging bucket rotor, the crystals form a pellet at the bottom of the centrifuge tube where there is no protein but only high precipitant which immediately quenches crystal growth. When using a fixed angle rotor, the crystals coat the sides of the walls away from the centripetal force, causing a gradient in crystal sizes. Larger crystals are found towards the top of the microcentrifuge tube and smaller crystals are found towards the bottom, probably because the crystals at the top of the pellet are exposed to the mixing zone for longer and therefore acquire more protein, resulting in larger crystals.
Advantages of this technique are twofold: the first is the increased speed with which crystallization occurs. Rather than taking 24 h, a comparable yield can be achieved in approximately 30 min. The second advantage is that the crystals are much smaller, typically 0.5–2 μm.
4. Conclusion
We have presented several methods for nanocrystal growth. These have been tested primarily using PSII as a model protein for large unit cell membrane proteins. These techniques require a higher density precipitant to be used in the crystallization process, and many proteins are crystallized in various PEGs and other high-density media. For cases in which the protein is denser than its precipitant, this process can be reversed, with the precipitant being added to the protein. The crystallization techniques presented here have been verified on other proteins to demonstrate the generality of the method; therefore we are confident that these techniques can be used for the growth of nano- or microcrystals for SFX experiments.
Funding statement
We acknowledge support by the Center for Bio-Inspired Solar Fuel Production, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001016, which supported the work of Raimund Fromme and Shatabdi Roy-Chowdhury as well as part of work of Chelsie Conrad and paid for part of the summer salary devoted to this project for Petra Fromme. This work was supported by the National Institutes of Health Common Fund in Structural Biology grant R01 GM095583, the National Institute of General Medical Sciences PSI:Biology grants U54 GM094599 as well as the National Science Foundation grant MCB-1021557 which supported the work of Christopher Kupitz. Recent support for participation of the work of Chelsie Conrad as well as support for publication costs from the NSF BioXFEL Science and Technology Center award no. 1231306 is acknowledged.
References
- 1.Moraes I, Evans G, Sanchez-Weatherby J, Newstead S, Stewart PD. 2014. Membrane protein structure determination—the next generation. Biochim. Biophys. Acta 1838, 78–87. ( 10.1016/j.bbamem.2013.07.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt M, Srajer V, Purwar N, Tripathi S. 2012. The kinetic dose limit in room-temperature time-resolved macromolecular crystallography. J. Synchrotron Radiat. 19, 264–273. ( 10.1107/S090904951105549X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yano J, et al. 2005. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: a case study for metalloprotein crystallography. Proc. Natl Acad. Sci. USA 102, 12 047–12 052. ( 10.1073/pnas.0505207102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burmeister WP. 2000. Structural changes in a cryo-cooled protein crystal owing to radiation damage. Acta Crystallogr. D Biol. Crystallogr. 56, 328–341. ( 10.1107/S0907444999016261) [DOI] [PubMed] [Google Scholar]
- 5.Chapman HN, et al. 2011. Femtosecond X-ray protein nanocrystallography. Nature 470, 73–77. ( 10.1038/nature09750) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fromme P, Spence JC. 2011. Femtosecond nanocrystallography using X-ray lasers for membrane protein structure determination. Curr. Opin. Struct. Biol. 21, 509–516. ( 10.1016/j.sbi.2011.06.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spence JCH, Weierstall U, Chapman HN. 2012. X-ray lasers for structural and dynamic biology. Rep. Progr. Phys. 75, 102601 ( 10.1088/0034-4885/75/10/102601) [DOI] [PubMed] [Google Scholar]
- 8.Barty A, et al. 2012. Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nat. Photonics 6, 35–40. ( 10.1038/nphoton.2011.297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aquila A, et al. 2012. Time-resolved protein nanocrystallography using an X-ray free-electron laser. Opt. Express 20, 2706–2716. ( 10.1364/OE.20.002706) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neutze R, Moffat K. 2012. Time-resolved structural studies at synchrotrons and X-ray free electron lasers: opportunities and challenges. Curr. Opin. Struct. Biol. 22, 651–659. ( 10.1016/j.sbi.2012.08.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zouni A, Jordan R, Schlodder E, Fromme P, Witt HT. 2000. First photosystem II crystals capable of water oxidation. Biochim. Biophys. Acta 1457, 103–105. ( 10.1016/S0005-2728(00)00100-6) [DOI] [PubMed] [Google Scholar]
- 12.Shen JR, Kawakami K, Koike H. 2011. Purification and crystallization of oxygen-evolving photosystem II core complex from thermophilic cyanobacteria. Methods Mol. Biol. 684, 41–51. ( 10.1007/978-1-60761-925-3_5) [DOI] [PubMed] [Google Scholar]
- 13.Kern J, Loll B, Luneberg C, DiFiore D, Biesiadka J, Irrgang KD, Zouni A. 2005. Purification, characterisation and crystallisation of photosystem II from Thermosynechococcus elongatus cultivated in a new type of photobioreactor. Biochim. Biophys. Acta 1706, 147–157. ( 10.1016/j.bbabio.2004.10.007) [DOI] [PubMed] [Google Scholar]
- 14.Renger G. 2012. Mechanism of light induced water splitting in photosystem II of oxygen evolving photosynthetic organisms. Biochim. Biophys. Acta 1817, 1164–1176. ( 10.1016/j.bbabio.2012.02.005) [DOI] [PubMed] [Google Scholar]
- 15.Gust D, Moore TA, Moore AL. 2009. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 42, 1890–1898. ( 10.1021/ar900209b) [DOI] [PubMed] [Google Scholar]
- 16.Barber J. 2009. Photosynthetic energy conversion: natural and artificial. Chem. Soc. Rev. 38, 185–196. ( 10.1039/b802262n) [DOI] [PubMed] [Google Scholar]
- 17.Kupitz C, et al. In press. Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature. ( 10.1038/nature13453) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DePonte DP, Weierstall U, Schmidt K, Warner J, Starodub D, Spence JCH, Doak RB. 2008. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J. Phys. D Appl. Phys. 41, 195505 ( 10.1088/0022-3727/41/19/195505) [DOI] [Google Scholar]
- 19.Weierstall U, Spence JCH, Doak RB. 2012. Injector for scattering measurements on fully solvated biospecies. Rev. Sci. Instrum. 83, 035108 ( 10.1063/1.3693040) [DOI] [PubMed] [Google Scholar]
- 20.Sierra RG, et al. 2012. Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallogr. D Biol. Crystallogr. 68, 1584–1587. ( 10.1107/S0907444912038152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weierstall U, et al. 2014. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309 ( 10.1038/ncomms4309) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wampler RD, Kissick DJ, Dehen CJ, Gualtieri EJ, Grey JL, Wang HF, Thompson DH, Cheng JX, Simpson GJ. 2008. Selective detection of protein crystals by second harmonic microscopy. J. Am. Chem. Soc. 130, 14 076–14 077. ( 10.1021/ja805983b) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boutet S, et al. 2012. High-resolution protein structure determination by serial femtosecond crystallography. Science 337, 362–364. ( 10.1126/science.1217737) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Redecke L, et al. 2013. Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X-ray laser. Science 339, 227–230. ( 10.1126/science.1229663) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kern J, et al. 2012. Room temperature femtosecond X-ray diffraction of photosystem II microcrystals. Proc. Natl Acad. Sci. USA 109, 9721–9726. ( 10.1073/pnas.1204598109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Umena Y, Kawakami K, Shen JR, Kamiya N. 2011. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60. ( 10.1038/nature09913) [DOI] [PubMed] [Google Scholar]
- 27.Takahashi S, Badger MR. 2011. Photoprotection in plants: a new light on photosystem II damage. Trends Plant Sci. 16, 53–60. ( 10.1016/j.tplants.2010.10.001) [DOI] [PubMed] [Google Scholar]
- 28.Abdallah BG, Kupitz C, Fromme P, Ros A. 2013. Crystallization of the large membrane protein complex photosystem I in a microfluidic channel. ACS Nano 7, 10 534–10 543. ( 10.1021/nn402515q) [DOI] [PMC free article] [PubMed] [Google Scholar]