

Abstract
The structure of photosystem II and the catalytic intermediate states of the Mn4CaO5 cluster involved in water oxidation have been studied intensively over the past several years. An understanding of the sequential chemistry of light absorption and the mechanism of water oxidation, however, requires a new approach beyond the conventional steady-state crystallography and X-ray spectroscopy at cryogenic temperatures. In this report, we present the preliminary progress using an X-ray free-electron laser to determine simultaneously the light-induced protein dynamics via crystallography and the local chemistry that occurs at the catalytic centre using X-ray spectroscopy under functional conditions at room temperature.
Keywords: manganese, oxygen-evolving complex, photosystem II, X-ray crystallography, X-ray emission spectroscopy, X-ray free-electron laser
1. Introduction
Most of the dioxygen in the atmosphere is generated by plants, algae and cyanobacteria by the light-induced oxidation of water in photosystem II (PSII), a membrane protein complex embedded in the thylakoid membrane. This is one of the most important, life-sustaining chemical processes occurring in the biosphere. PSII is normally found in dimeric form with each monomer comprising about 20 subunits and binding around 100 cofactors. The primary light-driven charge separation takes place in the reaction centre of PSII where an array of four chlorophyll (Chl), two pheophytin and two quinone molecules are symmetrically arranged in two branches. After light-induced formation of a Chl cation, a redox-active tyrosine residue (YZ) becomes oxidized and in turn oxidizes the nearby oxygen-evolving complex (OEC) embedded in the protein framework at the lumenal side of the protein complex. Here at the OEC, formed by a Mn4CaO5 cluster and its ligand environment [1], two molecules of water are oxidized in the reaction
that couples the four-electron oxidation of water with the one-electron photochemistry occurring at the PSII reaction centre. The OEC cycles through five intermediate S-states (S0–S4) that correspond to the abstraction of four successive electrons from the OEC [2,3]. The dark stable S1 state is the first oxidized state and subsequent illumination leads to the formation of the S2 and S3 states. Finally, upon accumulation of four oxidizing equivalents (S4 state), a spontaneous reaction occurs that results in the release of O2 and the formation of the most reduced state, the S0 state. Upon one further light excitation, the initial S1 state is formed once more and the reaction can start over again.
Given the importance of PSII in maintaining life and the anticipated role of light-induced water-splitting for building a renewable energy economy, understanding the structure of the Mn4CaO5 catalyst and the mechanism of the water-oxidation reaction is considered to be one of science's grand challenges. Although details of the chemistry involved in water oxidation are slowly emerging [4–6], the mechanism of the reaction is not yet clear.
Synchrotron radiation (SR)-based X-ray diffraction (XRD) has been used over the past decade to study the structure of PSII [7–11]. The highest resolution structure of dimeric PSII at 1.9 Å [1] reveals the location and the geometry of the Mn4CaO5 cluster. However, the applied dose leads to a reduction of approximately 25% of the MnIII/IV ions to MnII [1,12], which also notably changes the atomic distances as compared to extended X-ray absorption fine structure (EXAFS) [12–14]. Thus, even at cryogenic temperatures, SR-based XRD of the Mn4CaO5 structure in PSII is fundamentally limited by the radiation damage to the redox-active metal site, making it difficult to obtain intact structures of stable or transient reaction intermediates. Not only is this radiation damage a problem for PSII, but it also presents a challenge encountered in many other redox-active metalloenzymes [15–18].
Thus far, the geometric and electronic structural information of the intact Mn4CaO5 cluster has been primarily addressed by spectroscopic methods [19–22], especially X-ray absorption (XAS) and emission spectroscopy (XES) techniques [14,23–25]. Among these, XES is a powerful method for studying the charge and spin density and the ligand environment of the metal sites [26]. XES of the Mn Kβ transition has been used successfully to probe steady-state PSII solution samples [23]. However, following the time course of the water-oxidation reaction by these Kβ features within the threshold of radiation damage is unrealistic with SR sources. In particular, the kinetically unstable S4 state(s), in which the O–O bond formation and the evolution of molecular oxygen occurs, cannot be captured by traditional cryo-trapping methods and requires time-resolved detection at room temperature (RT) [19,24,27,28]. Within the total time-scale of approximately 1.3 ms of the S3–S4–S0 transition, which is initiated by the third flash, several sequential events occur. According to the current proposals, these may include (a) release of one proton during a lag phase (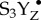 state), (b) transfer of one electron to
state), (b) transfer of one electron to 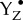 , (c) formation of O–O bond (peroxo intermediate) coupled with two-electron reduction of the Mn4CaO5 cluster, (d) formation and subsequent release of O2 under further two-electron reduction of the Mn4CaO5 cluster, (e) binding of one or two substrate water molecules to the cluster and (f) release of one proton during steps (c) to (e) (see [3,21,29] for reviews). Determining the geometric and electronic structures of intermediates of this reaction (e.g.
, (c) formation of O–O bond (peroxo intermediate) coupled with two-electron reduction of the Mn4CaO5 cluster, (d) formation and subsequent release of O2 under further two-electron reduction of the Mn4CaO5 cluster, (e) binding of one or two substrate water molecules to the cluster and (f) release of one proton during steps (c) to (e) (see [3,21,29] for reviews). Determining the geometric and electronic structures of intermediates of this reaction (e.g. 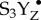 and peroxo state) is pivotal for testing such hypotheses and for deriving the water-splitting mechanism.
and peroxo state) is pivotal for testing such hypotheses and for deriving the water-splitting mechanism.
The intense and ultra-short X-ray pulses at the Linac Coherent Light Source (LCLS) provide an opportunity to overcome the above described limitations of SR sources for both crystallography and spectroscopy of biological samples with the ‘collect before destroy’ approach [30–32]. Unlike cryogenic measurements at SR, the experiments can be carried out at RT, making it possible to obtain molecular movies of the catalyst at work by recording snapshots at different time points in the catalytic cycle. In such a study, crystallography and spectroscopy can give complementary information: spectroscopy provides detailed information about changes in the Mn oxidation states, and crystallography probes the structural changes of the Mn4CaO5 cluster and the protein. As the same X-ray energy (7.1 keV) can be used for XRD and XES excitation of Mn, both methods can be applied simultaneously.
We designed an experimental set-up for the simultaneous collection of both XRD (figure 1) and XES (figure 2) data using the coherent X-ray imaging (CXI) instrument at LCLS. More details about the experimental methods can be found in the contribution by Kern et al. [35]. There, we also discuss more details about the software suite (cctbx.xfel) [36,37] written within our collaboration for processing of the X-ray free-electron laser (XFEL) data.
Figure 1.

Electron density of PSII obtained from femtosecond XRD measured at the CXI instrument of LCLS. (a) Electron density of one monomer of the dimer is shown in blue with the protein shown in yellow; view is along the membrane plane with the lumenal side on bottom and the cytoplasmic side on top. The density is contoured at 1.2σ. (b) Electron density in the vicinity of the OEC, Mn (magenta) and Ca (orange) ions are shown as spheres, the protein backbone in yellow and the electron density as grey (1.0σ) and blue (4.0σ) mesh. (c) Omit map obtained by excluding the non-haem FeII, from the phasing model. 2mFo-DFc electron density is contoured at 1.0σ (blue mesh), mFo-DFc difference density is contoured at 2.5σ (green mesh) and −2.5σ (red mesh); the protein is shown in yellow (subunit D1) and orange (subunit D2) and Fe as a red sphere. (d) Omit map obtained by excluding the Mn4Ca cluster from the phasing model. Electron density is shown as in (c), view direction and colouring of Mn and Ca is similar to panel (b). Figure is adapted from [33].
Figure 2.

Femtosecond X-ray emission spectra measured at LCLS. (a) RT Mn Kβ1,3 X-ray emission spectra of MnIICl2 (red, 500 mM Mn) and Mn2III,IVterpyridine (blue, 180 mM Mn) collected with less than 50-fs X-ray pulses; for comparison spectra of the same compounds collected at a synchrotron source are shown with symbols. The inset shows the 3d–3p exchange coupling, giving rise to the sensitivity of the Kβ1,3 spectrum to the number of unpaired electrons in the 3d orbitals, thereby providing information about the spin state of the complex. For high-spin complexes, the position of the peak is an indicator of the oxidation state on the metal. (b) X-ray emission spectra of PSII solutions in the dark state collected at the CXI instrument at RT (green) or collected using SR under cryogenic conditions with a low X-ray dose (‘8 K intact,’ light blue) or SR at RT under photoreducing conditions (‘RT damaged,’ pink). The spectrum from MnIICl2 in aqueous solution collected at RT at the CXI instrument is shown (grey) for comparison. (c) Mass-spectrometric measurements of light-induced O2 yield detected as mixed labelled 16O18O species after illumination of PSII. The data show that more than 73% of the sample occupies the S2 state after one saturating flash. (d) XFEL XES of PSII in the S2 state. The Kβ1,3 XES data collected from 362 microcrystals of PSII in the first illuminated S2 state are shown in blue asterisks. The XFEL spectrum of microcrystals of PSII in the dark stable S1 state is shown in green. For comparison, an X-ray emission spectrum of completely photoreduced (‘damaged’) PSII collected at RT at a synchrotron source is shown in pink. Figure is adapted from [34] (panel (a)) and [33] (panels (b–d)).
2. X-ray diffraction results from photosystem II at room temperature
In our initial XRD experiments at LCLS, we used PSII microcrystals about 10 μm in size. We collected more than 110 000 images containing potential diffraction signals. Diffraction was observed out to about 5.4 Å resolution, and a complete dataset at 6.5 Å was obtained by indexing and integrating the data from approximately 7200 of these diffraction images [38]. These data allowed us to prove the presence of the Mn4CaO5 OEC by phasing the structure with a molecular replacement model from which the metal ions were omitted.
The structure obtained is largely isomorphous with previously published SR structures, showing that there are no specific large-scale differences due to either radiation damage or to different temperatures (cryo-cooling for SR versus RT at LCLS) visible in the XFEL structure. The dose deposited onto the sample for each individual shot was of the order of 108 Gy (J kg−1). This dose is about one order of magnitude higher than the Henderson/Garman limit of 2–3 × 107 Gy [39,40], commonly considered as the dose limit for loss of diffractivity in cryogenic XRD, and about 100 times higher than the dose used for synchrotron XRD data collection of PSII at 100 K (approx. 1 × 106 Gy for the 1.9 Å PSII crystal structure). With our PSII microcrystals, cryogenic SR measurements are not possible due to the extent of X-ray exposure and loss of diffraction; however, the same microcrystals yield visible diffraction spots with the RT XFEL. Despite applying this high dose at RT, there appears to be no loss of diffractivity or visible differences between the RT XFEL and cryogenic SR structures; this fact demonstrates that the femtosecond XFEL pulses (approx. 45 fs) are short enough to outrun the damage processes present in conventional SR XRD.
It should be noted that, for protein structures in general, recent reports show differences between RT and cryogenic structures (e.g. [41]). PSII shows several large loop regions that are membrane extrinsic and potentially could be flexible. In addition, only a very small number of residues are engaged in crystal contacts, raising the possibility that these loop regions could adopt different conformations depending on the crystal conditions. Interestingly, no deviations in position for these loop regions are found when comparing the SR cryogenic and the XFEL RT structures, indicating that there are no large-scale effects on the structure due to the freezing for cryogenic XRD.
3. Feasibility of X-ray emission spectroscopy
In order to monitor the electronic structure of PSII at RT and under conditions used for the XRD data collection, we developed an X-ray emission spectrometer that is compatible with the set-up at the CXI instrument at LCLS [35,42]. Although our initial XRD data did not provide any indication for radiation-induced damage to the sample during the less than 50 fs measurement, it could not be excluded that the electronic structure is modified within the pulse length. For XRD measurements using femtosecond pulses, the possibility of a self-gating mechanism was postulated [43]: the signal measured is only contributed from the initial undamaged sample; photons that hit the sample at a later part of the X-ray pulse will interact with a strongly distorted or already disrupted crystal lattice, contributing only to diffuse background scattering rather than the Bragg scattering. Hence, the signal that is collected would only reflect the mostly undamaged sample as it is seen during the initial part of the pulse [43]. By contrast, for X-ray spectroscopy, no such self-gating can be postulated, as the observed signal is the sum of all photons (within the spectral window of the spectrometer used) that are emitted from the sample during the entire length of the pulse. Therefore, the recorded spectrum would be sensitive to changes in the electronic structure of the studied element that are induced by the intense X-ray pulse.
To check for the presence of such effects of XFEL pulses to the electronic structure, we first measured XES from Mn model compounds in solution [34] using beam parameters similar to the conditions used for the femtosecond XRD experiment on PSII crystals described above. The XES data from solutions of MnIICl2 and Mn2III,IVterpyridine are shown in figure 2a. The spectra for both compounds show a good agreement with the data collected at SR sources under cryogenic conditions. In particular, the RT XFEL data from the highly oxidized MnIII,IV compound are encouraging, as collecting RT data from high-valent compounds at SR sources is challenging. We also studied the effect of pulse length, but found no differences between the spectra recorded using 50 and 100 fs pulses [34]. The absence of deviation from cryogenic SR data indicated that radiation-damage-free hard XES is possible at LCLS.
4. Simultaneous X-ray emission spectroscopy and X-ray diffraction of photosystem II
Improved microcrystallization conditions have made it possible to extend the quality of the diffraction data, although it is still at a lower resolution than the SR structure at 1.9 Å [1]. Possible reasons for this may be the quality of our PSII microcrystals or mechanical damage during the sample delivery process [38]; thus, continued efforts to optimize our PSII protein preparations, microcrystallization procedures and sample delivery methods are underway. With our current microcrystals, the best RT XFEL diffraction spots were found at 4.1 Å resolution and a complete dataset for the dark stable S1 state of PSII was obtained at a resolution of 5.7 Å by merging data from about 4400 diffraction images [33]. The obtained XFEL structure of the S1 state of PSII (figure 1a) is isomorphous to published SR XRD structures. The overall maximum of the electron density map (figure 1b) was found in the region of the Mn4CaO5 cluster, as already observed for our previous XFEL dataset. To check for the influence of model bias, we computed various omit maps, omitting either the Mn4CaO5 cluster, the non-haem iron (located at the stromal side of the PSII complex), or the four central Chl from the phasing model. The ensuing observation of an mFo-DFc difference peak in the non-haem Fe omit map (figure 1c) and the Mn4CaO5 omit map (figure 1d) at the correct position is an unbiased indication of the presence of these groups in the PSII microcrystals. Similar results were obtained when omitting the four central Chl cofactors from the phasing model. In each case, positive difference electron density for the omitted cofactors was visible in the omit map at the expected location.
In addition, improvements in the XES set-up allowed us to record XES from PSII solutions in the dark state with acceptable signal-to-noise ratio (figure 2b) [33]. The RT spectrum matches the data of undamaged PSII data collected under cryogenic conditions at a SR source. This shows that collection of undamaged RT XES is possible at LCLS even for highly radiation-sensitive, dilute biological samples.
To advance PSII into the higher S-states, we integrated an in situ illumination set-up into our sample delivery system. The illumination set-up consists of three optical laser fibres directly coupled to the silica capillary for sample delivery and a fourth laser intersecting the jet at the X-ray interaction point [35,44]. In order to achieve a high population of the illuminated states, the illumination parameters had to be optimized. For this purpose, a replica of the set-up was built and coupled to a membrane inlet mass spectrometer. Using this device, we directly recorded the oxygen produced by illumination of the sample and found conditions where we obtained more than 73% of the sample in the S2 state (see figure 2c) [33]. Using these conditions, we then collected XES and XRD data from PSII microcrystals highly enriched in the S2 state. The XES data did not exhibit a significant difference from the S1 data (figure 2d), showing that there was no damage to the electronic structure of the Mn cluster in either state by the visible-laser pump/X-ray-probe method [33]. The small shift expected between the S1 and S2 states [23] could not be resolved within the signal/noise ratio of the spectra.
Using the same conditions, we collected XRD data for PSII enriched in the S2 state. A total of 1848 indexed diffraction patterns were merged to yield a dataset at 5.9 Å resolution. As the data are isomorphous to the S1 state data, it was possible to compute an isomorphous difference map between them. A detailed analysis of this map revealed no statistically relevant difference peaks [33]. This indicates that—within the limits of the currently available resolution of around 6 Å—there are no larger scale structural changes associated with the oxidation of the Mn4CaO5 cluster from the dark stable S1 to the first illuminated S2 state.
5. Conclusion
We established the methodology for simultaneous collection of XES and XRD data at RT from PSII microcrystals using femtosecond XFEL pulses. The diffraction data obtained show that the structure of PSII at RT is isomorphous to the structure obtained using cryogenic XRD at SR sources. Within the resolution obtained so far, no indication for the manifestation of XFEL-specific radiation damage could be found. Furthermore, the RT Mn Kβ XES data obtained from high-valent Mn model compounds and from solutions and microcrystals of PSII using the femtosecond pulses at LCLS match very well with data collected under non-damaging conditions at low temperatures using SR sources. Under the current experimental conditions, we did not find any indication for XFEL-specific changes in the electronic or geometric structure of the highly radiation-sensitive Mn4CaO5 cluster of PSII.
For resolving details of the O–O bond formation, further improvement of resolution to around 3 Å or better will be required. The combined spectroscopy and XRD approach will allow the discrimination among various proposed mechanisms for O–O bond formation. For example, the suggested formation of MnV in the S4 state should be easily detectable with XES, while O–O bond formation will lead to the development of extra electron density, allowing the identification of the two substrate-binding sites (MnA−D, Ca or free water). In addition, this method opens possibilities towards orientation-dependent polarized XES, where the XES dichroism occurs due to the relative orientation between the crystal axis and scattered X-ray vectors. The combined XRD/polarized XES will enable further detailed analysis of the spectra from the oriented single crystals, which may be beneficial towards interpreting the metal–ligand interactions occuring at the OEC during O–O bond formation. We also expect that this combined approach will be applicable to other metalloenzyme systems, enabling the collection of snapshots of the native catalytic centres of these systems during their reaction cycles.
Acknowledgements
We thank all our students, postdoctoral fellows and collaborators for their very important contributions to the PSII studies at LCLS, and the staff at LCLS for their support of the XFEL experiments.
Funding statement
The research reviewed here was supported by the NIH grant no. GM 55302 (V.K.Y.) for PSII structure and mechanism and by the Director, Office of Science, Office of Basic Energy Sciences (OBES), Division of Chemical Sciences, Geosciences, and Biosciences of the Department of Energy (DOE) under contract DE-AC02–05CH11231 (J.Y. and V.K.Y.) for X-ray instrumentation, by the NIH grant no. P41GM103393 for part of the XES instrumentation and support of U.B., NIH grant nos. GM095887 and GM102520 (N.K.S.) for data processing methods and by an LBNL Laboratory Directed Research and Development award (DOE contract DE-AC02-05CH11231) to N.K.S. The Human Frontier Research grant no. RGP0063/2013 (U.B., A.Z. and J.Y.); the DFG-Cluster of Excellence ‘UniCat’ coordinated by the Technische Universität Berlin and Sfb1078, TP A5 (A.Z., J.Hel.); the Alexander von Humboldt Foundation (J.K.); the Ruth L. Kirschstein National Research Service Award (F32GM100595, R.T.); and the Solar Fuels Strong Research Environment (Umeå University), the Artificial Leaf Project (K&A Wallenberg Foundation), VR and Energimyndigheten (J.M.) are acknowledged for supporting this project. The LCLS, and the synchrotron facilities at Stanford Synchrotron Radiation Lightsource (SSRL), the Advanced Light Source (ALS), and the Advanced Photon Source (APS), used in the course of these studies are all supported by DOE OBES.
References
- 1.Umena Y, Kawakami K, Shen JR, Kamiya N. 2011. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60. ( 10.1038/nature09913) [DOI] [PubMed] [Google Scholar]
- 2.Kok B, Forbush B, McGloin M. 1970. Cooperation of charges in photosynthetic oxygen evolution. I. A linear four step mechanism. Photochem. Photobiol. 11, 457–475. ( 10.1111/j.1751-1097.1970.tb06017.x) [DOI] [PubMed] [Google Scholar]
- 3.Renger G. 2011. Light induced oxidative water splitting in photosynthesis: energetics, kinetics and mechanism. J. Photochem. Photobiol. B 104, 35–43. ( 10.1016/j.jphotobiol.2011.01.023) [DOI] [PubMed] [Google Scholar]
- 4.Cox N, Pantazis DA, Neese F, Lubitz W. 2013. Biological water oxidation. Acc. Chem. Res. 46, 1588–1596. ( 10.1021/ar3003249) [DOI] [PubMed] [Google Scholar]
- 5.Siegbahn PEM. 2009. Structures and energetics for O2 formation in photosystem II. Acc. Chem. Res. 42, 1871–1880. ( 10.1021/ar900117k) [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi K, et al. 2013. Full geometry optimizations of the mixed-valence CaMn4O4X(H2O)4 (X=OH or O) cluster in OEC of PS II: degree of symmetry breaking of the labile Mn-X-Mn bond revealed by several hybrid DFT calculations. Int. J. Quantum Chem. 113, 525–541. ( 10.1002/qua.24117) [DOI] [Google Scholar]
- 7.Zouni A, Witt HT, Kern J, Fromme P, Krauss N, Saenger W, Orth P. 2001. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature 409, 739–743. ( 10.1038/35055589) [DOI] [PubMed] [Google Scholar]
- 8.Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. 2005. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature 438, 1040–1044. ( 10.1038/nature04224) [DOI] [PubMed] [Google Scholar]
- 9.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. 2004. Architecture of the photosynthetic oxygen-evolving center. Science 303, 1831–1838. ( 10.1126/science.1093087) [DOI] [PubMed] [Google Scholar]
- 10.Kamiya N, Shen JR. 2003. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7-Å resolution. Proc. Natl Acad. Sci. USA 100, 98–103. ( 10.1073/pnas.0135651100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guskov A, Kern J, Gabdulkhakov A, Broser M, Zouni A, Saenger W. 2009. Cyanobacterial photosystem II at 2.9-Å resolution and the role of quinones, lipids, channels and chloride. Nat. Struct. Mol. Biol. 16, 334–342. ( 10.1038/nsmb.1559) [DOI] [PubMed] [Google Scholar]
- 12.Yano J, et al. 2005. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: a case study for metalloprotein crystallography. Proc. Natl Acad. Sci. USA 102, 12 047–12 052. ( 10.1073/pnas.0505207102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grabolle M, Haumann M, Müller C, Liebisch P, Dau H. 2006. Rapid loss of structural motifs in the manganese complex of oxygenic photosynthesis by X-ray irradiation at 10–300 K. J. Biol. Chem. 281, 4580–4588. ( 10.1074/jbc.M509724200) [DOI] [PubMed] [Google Scholar]
- 14.Glöckner C, Kern J, Broser M, Zouni A, Yachandra V, Yano J. 2013. Structural changes of the oxygen evolving complex in photosystem II during the catalytic cycle. J. Biol. Chem. 288, 22 607–22 620. ( 10.1074/jbc.M113.476622) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sigfridsson KG, Chernev P, Leidel N, Popovic-Bijelic A, Graslund A, Haumann M. 2013. Rapid X-ray photoreduction of dimetal-oxygen cofactors in ribonucleotide reductase. J. Biol. Chem. 288, 9648–9661. ( 10.1074/jbc.M112.438796) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daughtry KD, Xiao Y, Stoner-Ma D, Cho E, Orville AM, Liu P, Allen KN. 2012. Quaternary ammonium oxidative demethylation: X-ray crystallographic, resonance raman, and UV-visible spectroscopic analysis of a Rieske-type demethylase. J. Am. Chem. Soc. 134, 2823–2834. ( 10.1021/ja2111898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antonyuk SV, Hough MA. 2011. Monitoring and validating active site redox states in protein crystals. Biochim. Biophys. Acta Proteins Proteomics 1814, 778–784. ( 10.1016/j.bbapap.2010.12.017) [DOI] [PubMed] [Google Scholar]
- 18.Hersleth HP, Andersson KK. 2011. How different oxidation states of crystalline myoglobin are influenced by X-rays. Biochim. Biophys. Acta Proteins Proteomics 1814, 785–796. ( 10.1016/j.bbapap.2010.07.019) [DOI] [PubMed] [Google Scholar]
- 19.Noguchi T, Suzuki H, Tsuno M, Sugiura M, Kato C. 2012. Time-resolved infrared detection of the proton and protein dynamics during photosynthetic oxygen evolution. Biochemistry 51, 3205–3214. ( 10.1021/bi300294n) [DOI] [PubMed] [Google Scholar]
- 20.Haddy A. 2007. EPR spectroscopy of the manganese cluster of photosystem II. Photosynth. Res. 92, 357–368. ( 10.1007/s11120-007-9194-9) [DOI] [PubMed] [Google Scholar]
- 21.Brudvig GW. 2008. Water oxidation chemistry of photosystem II. Phil. Trans. R. Soc. B 363, 1211–1218. ( 10.1098/rstb.2007.2217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rappaport F, Ishida N, Sugiura M, Boussac A. 2011. Ca2+ determines the entropy changes associated with the formation of transition states during water oxidation by photosystem II. Energ Environ. Sci. 4, 2520–2524. ( 10.1039/c1ee01408k) [DOI] [Google Scholar]
- 23.Messinger J, et al. 2001. Absence of Mn-centered oxidation in the S2 -> S3 transition: implications for the mechanism of photosynthetic water oxidation. J. Am. Chem. Soc. 123, 7804–7820. ( 10.1021/ja004307+) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haumann M, Liebisch P, Muller C, Barra M, Grabolle M, Dau H. 2005. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science 310, 1019–1021. ( 10.1126/science.1117551) [DOI] [PubMed] [Google Scholar]
- 25.Yano J, et al. 2006. Where water is oxidized to dioxygen: structure of the photosynthetic Mn4Ca cluster. Science 314, 821–825. ( 10.1126/science.1128186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glatzel P, Bergmann U. 2005. High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes: electronic and structural information. Coord. Chem. Rev. 249, 65–95. ( 10.1016/j.ccr.2004.04.011) [DOI] [Google Scholar]
- 27.Rappaport F, Blancharddesce M, Lavergne J. 1994. Kinetics of electron-transfer and electrochromic change during the redox transitions of the photosynthetic oxygen-evolving complex. Biochim. Biophys. Acta Bioenerg. 1184, 178–192. ( 10.1016/0005-2728(94)90222-4) [DOI] [Google Scholar]
- 28.Razeghifard MR, Pace RJ. 1999. EPR kinetic studies of oxygen release in thylakoids in PSII membranes: a kinetic intermediate in the S3 to S0 transition. Biochemistry 38, 1252–1257. ( 10.1021/bi9811765) [DOI] [PubMed] [Google Scholar]
- 29.Cox N, Messinger J. 2013. Reflections on substrate water and dioxygen formation. Biochim. Biophys. Acta 1827, 1020–1030. ( 10.1016/j.bbabio.2013.01.013) [DOI] [PubMed] [Google Scholar]
- 30.Neutze R, Wouts R, van der Spoel D, Weckert E, Hajdu J. 2000. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 406, 752–757. ( 10.1038/35021099) [DOI] [PubMed] [Google Scholar]
- 31.Chapman HN, et al. 2011. Femtosecond X-ray protein nanocrystallography. Nature 470, 73–77. ( 10.1038/nature09750) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boutet S, et al. 2012. High-resolution protein structure determination by serial femtosecond crystallography. Science 337, 362–364. ( 10.1126/science.1217737) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kern J, et al. 2013. Simultaneous femtosecond X-ray spectroscopy and diffraction of photosystem II at room temperature. Science 340, 491–495. ( 10.1126/science.1234273) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alonso-Mori R, et al. 2012. Shot-by-shot energy-dispersive X-ray emission spectroscopy using an X-ray free electron laser. Proc. Natl Acad. Sci. USA 109, 19 103–19 107. ( 10.1073/pnas.1211384109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kern J, et al. 2014. Methods development for diffraction and spectroscopy studies of metalloenzymes at X-ray free-electron lasers. Phil. Trans. R. Soc. B 369, 20130590 ( 10.1098/rstb.2013.0590) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hattne J, et al. 2014. Accurate macromolecular structures using minimal measurements from X-ray free-electron lasers. Nat. Methods 11, 545–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sauter NK, Hattne J, Grosse-Kunstleve RW, Echols N. 2013. New python-based methods for data processing. Acta Crystallogr. D 69, 1274–1282. ( 10.1107/S0907444913000863) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kern J, et al. 2012. Room temperature femtosecond X-ray diffraction of photosystem II microcrystals. Proc. Natl Acad. Sci. USA 109, 9721–9726. ( 10.1073/pnas.1204598109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Owen RL, Rudino-Pinera E, Garman EF. 2006. Experimental determination of the radiation dose limit for cryocooled protein crystals. Proc. Natl Acad. Sci. USA 103, 4912–4917. ( 10.1073/pnas.0600973103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henderson R. 1990. Cryoprotection of protein crystals against radiation damage in electron and X-ray diffraction. Proc. R. Soc. Lond. B 241, 6–8. ( 10.1098/rspb.1990.0057) [DOI] [Google Scholar]
- 41.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. 2011. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc. Natl Acad. Sci. USA 108, 16 247–16 252. ( 10.1073/pnas.1111325108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alonso-Mori R, et al. 2012. A multicrystal wavelength dispersive X-ray spectrometer. Rev. Sci. Instrum. 83, 073114 ( 10.1063/1.4737630) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barty A, et al. 2012. Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nat. Photonics 6, 35–40. ( 10.1038/nphoton.2011.297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sierra RG, et al. 2012. Nanoflow electrospinning serial femtosecond crystallography. Acta Cryst. D 68, 1584–1587. ( 10.1107/S0907444912038152) [DOI] [PMC free article] [PubMed] [Google Scholar]