

Cardiovascular diseases (CVDs) and their treatment pose a huge economic and social burden in Western societies and in the developing world (1). Thus, there is significant interest in developing new therapeutic targets, and it is of particular interest that an old class of drugs, inhibitors of xanthine oxidoreductase (XO), may be repositioned to treat heart failure and left ventricular (LV) dysfunction. In this context, there is growing awareness that nitroso-redox balance may represent a new therapeutic target for heart failure (2). However, whether or not XO inhibitors fully address nitroso-redox imbalance in all patients remains controversial.
The hypothesis that XO inhibition could treat heart failure was tested in the OPT-CHF trial (3). In that study, the primary endpoint was not statistically different in the population of symptomatic heart failure patients enrolled; however patients with hyperuricemia appeared to be a responsive population. While hyperuricemia is classically associated with gout and nephropathy, it is clearly also a biomarker in heart failure populations (4-6). Uric acid (UA) is the end product of purine metabolism, involving the conversion of hypoxanthine to xanthine and then to UA in reactions catalyzed by XO (Figure 1) (7). In most mammals including rodents, UA is further degraded to allantoin via the enzyme urate oxidase (UO), resulting in low UA serum levels. In humans and great apes, however, this final step does not occur because of a single point mutation inactivating UO, resulting in substantially higher serum UA levels. Whether UA plays beneficial or deleterious roles remains controversial, and some arguments can be made that the impact of UA on vascular tone have played an evolutionary role (4). However, in hypertension, type 2 diabetes mellitus, coronary artery disease (CAD), heart failure, and chronic kidney disease (CKD), high UA levels correlate with an increased risk of stroke and CVDs (4,6,8-10)
Figure 1. Increased xanthine oxidase activity and nitroso-redox imbalance.
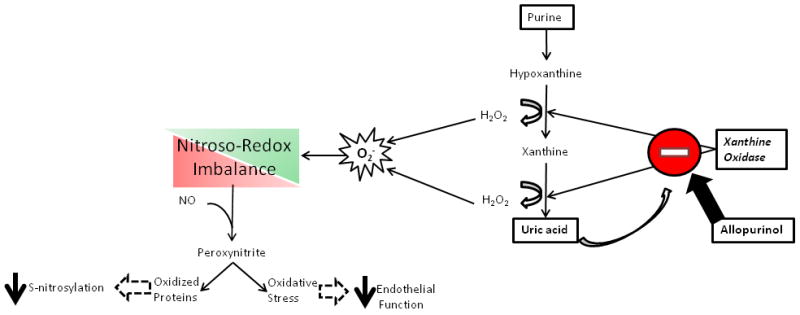
The reactive oxygen species generated as a result of purine metabolism contributes to nitroso-redox imbalance. Increased ROS production can lead to peroxynitrite formation and the oxidation of proteins blocking important post-translational nitrosylation modifications. Importantly, decreased NOS activity can activate xanthine oxidase and exacerbate nitroso-redox imbalance. As uric acid is produced in proportion to ROS by XOR, it may serve as a biomarker of increased xanthine oxidase activity and possibly nitroso-redox imbalance.
As UA levels represent a biomarker of XO activity, the OPT-CHF results suggest that patients with elevated XO activity responded preferentially (3). XO is a key oxidase participating in nitroso-redox imbalance in the heart, producing superoxide as a byproduct of purine metabolism (Figure 1). Thus, the epidemiological association of high UA levels with worse prognosis in a wide cohort of patients with CVDs likely reflects the fact that UA levels rise with increased XO activity (4-10).
XO and Nitroso-Redox Imbalance
In the heart, XO localizes to the sarcoplasmic reticulum (SR), interacting with neuronal nitric oxide (NO) synthase (nNOS, NOS1) and the ryanodine receptor (RyR2) (11,12). Depressed NOS1 activity or abundance in the SR augments XO activity (12,13), elevating oxidative stress in the SR (14-16). The functional consequences of this imbalance are evident in patients with heart failure, where inhibition of XO with allopurinol improved myocardial efficiency (reversing mechanoenergetic uncoupling) (17,18). The mechanism of this effect is attributable, at least in part, to post-translational modification of thiol moieties on proteins (19,20). In the case of the SR, nitrosylation of the RYR2 modulates the open probability of this Ca2+ pore. With nitroso-redox imbalance, formation of ROS predominates, and in turn also activates the RYR2, but in an irreversible manner, leading to Ca2+ leak (11,12,16).
This colocalization of an oxidase with a NO synthase and their signaling interactions underlies the basis for nitroso-redox imbalance as a key pathophysiologic signaling mechanism (2). In this regard, we and others have demonstrated that the NOS1 deficient mouse has increased mortality, LV remodeling, and ventricular arrhythmias after myocardial infarction (MI), associated with increased XO activity and decreased S-nitrosylation of Ca2+ handling proteins (21-23). This phenotype is further substantiated in NOS1 overexpressing mice where specific myocardial NOS1 overexpression protects from remodeling and preserves Ca2+ cycling (24).
In the failing heart, NOS1 translocates from the SR to the cell membrane (25,26), an effect that is protective from Ca2+ overload. Increased NOS1 in sarcolemmal caveolae increases S-nitrosylation of the L-type calcium channel (LTCC), leading to a decreased inward Ca2+ current. In turn, the reduced Ca2+ influx within the cardiac myocyte prevents Ca2+ overload-induced injury (23,27,28). While this mechanism is largely protective, probably acutely, (29), it also results in SR nitroso-redox imbalance over the long-term, which as described above can potentiate SR Ca2+ leak, arrhythmias, and contractile dysfunction. The disruption of physiological nitrosylation of SR proteins has been shown to be deleterious (19,20,30,31).
Cardiac Function And Structure
Struthers and collaborators have made a series of contributions regarding the role of XO inhibition and cardiovascular morbidity and mortality. They have shown that allopurinol in a dose-dependent fashion (32) decreases both cardiovascular events and mortality (33), improves endothelial function in patients with CAD (34), and regresses LV mass in patients with CKD (10). In the study published in this issue of J Am Coll Cardiol (35), the authors further address a key issue, that of the regression of LV hypertrophy (LVH) in patients with CAD. Indeed, a key surrogate for clinical efficacy in disorders of LV dysfunction is the amelioration of LVH (36,37). They randomized 66 patients with CAD and LVH to receive either 600mg/day allopurinol or placebo on a background of optimal medical therapy. LV mass was reduced by 5.2 ± 5.8 g for the high dose allopurinol cohort vs. 1.3 ± 4.48g for the placebo group (p=0.007). Moreover, allopurinol significantly reduced LV end systolic volume, improved endothelial function, and reduced augmentation index, an independent predictor of LV mass regression (38). The beneficial effect of XO inhibition on LVH regression is supported by experimental animal models of heart failure (39). Furthermore, the importance of these findings are highlighted by fact that in the LIFE study (40), LVH regression alone was associated with reduced all-cause mortality (by 28%), cardiovascular mortality (by 38%), sudden cardiac death (by 19%), MI (by 15%), new heart failure (by 36%), new onset atrial fibrillation (by 12%), and stroke (by 24%).
The importance of this small single center study lies in the fact that it adds further support for the development of XO inhibitors in the treatment of heart failure. As with ACE inhibitors, beta-blockers, and the combination of isosorbide dinitrate and hydralazine, the regression of LVH augurs well for the possibility that XO inhibitors could have clinical benefits in an appropriately selected heart failure population. Thus, this work adds another piece of evidence supporting the development of therapeutic strategies targeting myocardial nitroso-redox imbalance.
Acknowledgments
Drs. Hare and Schulman are supported by NIH grants: UM1HL113460 and RO1 HL084275. Dr. Hare is also supported by NIH grants: RO1 HL094849, RO1 HL107110, and RO1 HL110737. Dr. Karantalis is supported by an American Heart Association Postdoctoral Fellowship 12POST12050346.
Abbreviations
- CVDs
Cardiovascular diseases
- CKD
chronic kidney disease
- CAD
coronary artery disease
- LV
left ventricular
- LVH
left ventricular hypertrophy
- LTCC
L-type calcium channel
- nNOS, NOS1
neuronal nitric oxide synthase
- NO
nitric oxide
- RyR2
ryanodine receptor
- SR
sarcoplasmic reticulum
- UO
urate oxidase
- UA
Uric acid
- XO
xanthine oxidoreductase
Footnotes
Disclosures
Dr. Hare is an inventor of a patent related to the content in this Editorial. Drs. Karantalis and Schulman have nothing to disclose.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hare JM. Nitroso-redox balance in the cardiovascular system. N Engl J Med. 2004;351:2112–4. doi: 10.1056/NEJMe048269. [DOI] [PubMed] [Google Scholar]
- 3.Hare JM, Mangal B, Brown J, et al. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol. 2008;51:2301–9. doi: 10.1016/j.jacc.2008.01.068. [DOI] [PubMed] [Google Scholar]
- 4.Hare JM, Johnson RJ. Uric acid predicts clinical outcomes in heart failure: insights regarding the role of xanthine oxidase and uric acid in disease pathophysiology. Circulation. 2003;107:1951–3. doi: 10.1161/01.CIR.0000066420.36123.35. [DOI] [PubMed] [Google Scholar]
- 5.George J, Carr E, Davies J, Belch JJ, Struthers A. High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation. 2006;114:2508–16. doi: 10.1161/CIRCULATIONAHA.106.651117. [DOI] [PubMed] [Google Scholar]
- 6.Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359:1811–21. doi: 10.1056/NEJMra0800885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004;555:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franse LV, Pahor M, Di Bari M, et al. Serum uric acid, diuretic treatment and risk of cardiovascular events in the Systolic Hypertension in the Elderly Program (SHEP) J Hypertens. 2000;18:1149–54. doi: 10.1097/00004872-200018080-00021. [DOI] [PubMed] [Google Scholar]
- 9.Lehto S, Niskanen L, Ronnemaa T, Laakso M. Serum uric acid is a strong predictor of stroke in patients with non-insulin-dependent diabetes mellitus. Stroke. 1998;29:635–9. doi: 10.1161/01.str.29.3.635. [DOI] [PubMed] [Google Scholar]
- 10.Kao MP, Ang DS, Gandy SJ, et al. Allopurinol benefits left ventricular mass and endothelial dysfunction in chronic kidney disease. JASN. 2011;22:1382–9. doi: 10.1681/ASN.2010111185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hare JM. Nitric oxide and excitation-contraction coupling. J Mol Cell Cardiol. 2003;35:719–29. doi: 10.1016/s0022-2828(03)00143-3. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez DR, Treuer AV, Castellanos J, Dulce RA, Hare JM. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J Biol Chem. 2010;285:28938–45. doi: 10.1074/jbc.M110.154948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A. 2007;104:20612–7. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan SA, Lee K, Minhas KM, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–8. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kinugawa S, Huang H, Wang Z, Kaminski PM, Wolin MS, Hintze TH. A defect of neuronal nitric oxide synthase increases xanthine oxidase-derived superoxide anion and attenuates the control of myocardial oxygen consumption by nitric oxide derived from endothelial nitric oxide synthase. Circ Res. 2005;96:355–62. doi: 10.1161/01.RES.0000155331.09458.A7. [DOI] [PubMed] [Google Scholar]
- 16.Cutler MJ, Plummer BN, Wan X, et al. Aberrant S-nitrosylation mediates calcium-triggered ventricular arrhythmia in the intact heart. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:18186–91. doi: 10.1073/pnas.1210565109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cappola TP, Kass DA, Nelson GS, et al. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation. 2001;104:2407–11. doi: 10.1161/hc4501.098928. [DOI] [PubMed] [Google Scholar]
- 18.Saavedra WF, Paolocci N, St John ME, et al. Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res. 2002;90:297–304. doi: 10.1161/hh0302.104531. [DOI] [PubMed] [Google Scholar]
- 19.Schulman IH, Hare JM. Regulation of cardiovascular cellular processes by S-nitrosylation. Biochimica et biophysica acta. 2012;1820:752–62. doi: 10.1016/j.bbagen.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lima B, Forrester MT, Hess DT, Stamler JS. S-Nitrosylation in Cardiovascular Signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saraiva RM, Minhas KM, Raju SV, et al. Deficiency of neuronal nitric oxide synthase increases mortality and cardiac remodeling after myocardial infarction: role of nitroso-redox equilibrium. Circulation. 2005;112:3415–22. doi: 10.1161/CIRCULATIONAHA.105.557892. [DOI] [PubMed] [Google Scholar]
- 22.Dawson D, Lygate CA, Zhang MH, Hulbert K, Neubauer S, Casadei B. nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation. 2005;112:3729–37. doi: 10.1161/CIRCULATIONAHA.105.539437. [DOI] [PubMed] [Google Scholar]
- 23.Burger DE, Lu X, Lei M, et al. Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation. 2009;120:1345–54. doi: 10.1161/CIRCULATIONAHA.108.846402. [DOI] [PubMed] [Google Scholar]
- 24.Loyer X, Gomez AM, Milliez P, et al. Cardiomyocyte overexpression of neuronal nitric oxide synthase delays transition toward heart failure in response to pressure overload by preserving calcium cycling. Circulation. 2008;117:3187–98. doi: 10.1161/CIRCULATIONAHA.107.741702. [DOI] [PubMed] [Google Scholar]
- 25.Beigi F, Oskouei BN, Zheng M, Cooke CA, Lamirault G, Hare JM. Cardiac nitric oxide synthase-1 localization within the cardiomyocyte is accompanied by the adaptor protein, CAPON. Nitric Oxide. 2009;21:226–33. doi: 10.1016/j.niox.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Damy T, Ratajczak P, Shah AM, et al. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet. 2004;363:1365–7. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 27.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–63. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 28.Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res. 2006;98:403–11. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Chen X, Gao E, et al. Increasing cardiac contractility after myocardial infarction exacerbates cardiac injury and pump dysfunction. Circ Res. 2010;107:800–9. doi: 10.1161/CIRCRESAHA.110.219220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beigi F, Gonzalez DR, Minhas KM, et al. Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:4314–9. doi: 10.1073/pnas.1113319109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez DR, Treuer A, Sun QA, Stamler JS, Hare JM. S-Nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:188–95. doi: 10.1097/FJC.0b013e3181b72c9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei L, Fahey T, Struthers AD, MacDonald TM. Association between allopurinol and mortality in heart failure patients: a long-term follow-up study. Int J Clin Pract. 2009;63:1327–33. doi: 10.1111/j.1742-1241.2009.02118.x. [DOI] [PubMed] [Google Scholar]
- 33.Wei L, Mackenzie IS, Chen Y, Struthers AD, MacDonald TM. Impact of allopurinol use on urate concentration and cardiovascular outcome. British journal of clinical pharmacology. 2011;71:600–7. doi: 10.1111/j.1365-2125.2010.03887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noman A, Ang DS, Ogston S, Lang CC, Struthers AD. Effect of high-dose allopurinol on exercise in patients with chronic stable angina: a randomised, placebo controlled crossover trial. Lancet. 2010;375:2161–7. doi: 10.1016/S0140-6736(10)60391-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rekhraj S, Gandy S, Szwejkowski B, et al. High dose allopurinol reduces left ventricular mass in patients with ischemic heart disease. J Am Coll Cardiol. 2012 doi: 10.1016/j.jacc.2012.09.066. [DOI] [PubMed] [Google Scholar]
- 36.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. The New England journal of medicine. 1990;322:1561–6. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 37.Mancini GB, Dahlof B, Diez J. Surrogate markers for cardiovascular disease: structural markers. Circulation. 2004;109:IV22–30. doi: 10.1161/01.CIR.0000133443.77237.2f. [DOI] [PubMed] [Google Scholar]
- 38.Hashimoto J, Imai Y, O’Rourke MF. Indices of pulse wave analysis are better predictors of left ventricular mass reduction than cuff pressure. Am J Hypertens. 2007;20:378–84. doi: 10.1016/j.amjhyper.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 39.Minhas KM, Saraiva RM, Schuleri KH, et al. Xanthine Oxidoreductase Inhibition Causes Reverse Remodeling in Rats With Dilated Cardiomyopathy. Circ Res. 2006;98:271–279. doi: 10.1161/01.RES.0000200181.59551.71. [DOI] [PubMed] [Google Scholar]
- 40.Devereux RB, Dahlof B, Gerdts E, et al. Regression of hypertensive left ventricular hypertrophy by losartan compared with atenolol: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) trial. Circulation. 2004;110:1456–62. doi: 10.1161/01.CIR.0000141573.44737.5A. [DOI] [PubMed] [Google Scholar]