

Abstract
Current imaging techniques capable of tracking nanoparticles in vivo supply either a large field of view or cellular resolution, but not both. Here, we demonstrate a multimodality imaging platform of optical coherence tomography (OCT) techniques for high resolution, wide field of view in vivo imaging of nanoparticles. This platform includes the first in vivo images of nanoparticle pharmacokinetics acquired with photothermal OCT (PTOCT), along with overlaying images of microvascular and tissue morphology. Gold nanorods (51.8 ± 8.1 nm by 15.2 ± 3.3 nm) were intravenously injected into mice, and their accumulation into mammary tumors was non-invasively imaged in vivo in three dimensions over 24 hours using PTOCT. Spatial frequency analysis of PTOCT images indicated that gold nanorods reached peak distribution throughout the tumors by 16 hours, and remained well-dispersed up to 24 hours post-injection. In contrast, the overall accumulation of gold nanorods within the tumors peaked around 16 hours post-injection. The accumulation of gold nanorods within the tumors was validated post-mortem with multiphoton microscopy. This shows the utility of PTOCT as part of a powerful multimodality imaging platform for the development of nanomedicines and drug delivery technologies.
OCIS codes: (110.4500) Optical coherence tomography, (160.4236) Nanomaterials, (350.5340) Photothermal effects
1. Introduction
Clinical trials are currently underway to test gold nanoparticles as cancer therapeutics [1–3] due to their attractive physical and biological properties [4]. Gold nanoparticles have also enabled significant advances in pre-clinical cancer research as drug delivery vectors [5] and imaging contrast agents [6]. However, there is a need for improved technologies to image gold nanoparticle distributions in vivo in the context of microvessel morphology (the key delivery system for nanoparticles). Such an imaging platform could be used to optimize the size, shape, material composition, and surface chemistry of nanoparticles for improved clinical performance. High resolution, small field of view, and shallow imaging depth microscopy techniques including dark-field and multiphoton are most often used to image gold nanoparticles in vitro (and more rarely in vivo) [7–9]. For animal studies, postmortem and destructive mass spectrometry techniques [10–12] are often employed to investigate the general bio-distribution of nanoparticles. Emerging in vivo imaging modalities that can monitor gold nanoparticle delivery (e.g. computed tomography [2, 13], photoacoustic tomography [6, 14], surface enhanced Raman scattering [15], and microscopy techniques [16]) supply either a large field of view or high resolution, but not both. In addition, the majority of these existing tools cannot supply label-free contrast of the local microvasculature, an essential component for assessing drug delivery. Consequently, there is a significant gap in technologies that can provide non-destructive in vivo images of nanoparticle pharmacokinetics with high resolution at depths greater than microscopy. This spatial regime is important because heterogeneities in tumor microenvironment and microvessel morphology, for example, can drastically alter local therapeutic delivery and thus clinical efficacy [17].
We demonstrate photothermal optical coherence tomography (PTOCT), a functional extension of optical coherence tomography (OCT), to fill this spatial niche for in vivo imaging of nanoparticle delivery. OCT is a noninvasive, three-dimensional (3D) imaging tool that detects back-scattered near-infrared (NIR) photons from a sample to visualize tissue morphology [18]. OCT fills the spatial imaging niche between high resolution microscopy and whole body imaging techniques, supplying high resolution (1-30 µm) at depths greater than traditional and multiphoton microscopy (1-3 mm) [19]. In addition to tissue morphology, Doppler [20] as well as speckle [21] and phase [22] variance imaging techniques allow OCT to image microvessel morphology and quantify blood flow without the addition of contrast agents. Although OCT provides images of tissue and microvessel morphology on a spatial scale that is relevant for imaging animal models of disease, the source of OCT contrast (index of refraction mismatches) weakly differs between molecular species. Therefore, functional versions of OCT have been developed to visualize endogenous and exogenous targets [23–28]. PTOCT is an especially sensitive functional extension of OCT that identifies photon absorbing targets with high specificity over background tissue scattering [26–28]. PTOCT has been demonstrated with nanoparticle contrast agents in vitro and ex vivo including gold nanospheres [26], gold nanoshells [28], gold nanorods [27, 29], carbon nanotubes [30], and gold nanoroses [31], and PTOCT was recently demonstrated for in vivo imaging of subcutaneous injections of gold nanorods into a mouse ear [27].
This study demonstrates a novel and powerful application of PTOCT for in vivo imaging of nanoparticle delivery to tumors, and provides unique insights into heterogeneities in nanoparticle uptake and microvascular function that can be used to optimize nanoparticle delivery. This work provides the first PTOCT image volumes in vivo, as well as the first co-registered images of tissue structure, microvessel morphology, and nanoparticle delivery in the OCT spatial imaging regime. PTOCT non-destructively images nanoparticle uptake in tumors in three dimensions with microscopic resolution and a wide enough field of view to capture a large region of the tumor (multiple millimeter scan range). Therefore, PTOCT can potentially provide a robust method to study the effects of size, shape, material composition, and surface chemistry of gold nanoparticles for improved in vivo delivery. Gold nanorods (AuNRs) were studied because they exhibit tunable and narrow NIR local surface plasmon resonance peaks, with photothermal efficiencies greater than gold nanoshells [2]. Here, PTOCT is used to image the accumulation of intravenously-injected AuNRs into mouse mammary tumors via the enhanced permeability and retention (EPR) effect, a hallmark of cancer where newly formed and leaky vasculature allows extravasation of particles between 5 and 200 nm [32, 33] diameter into the tumor tissue space [17, 34].
2. Materials and methods
2.1 Gold nanorod (AuNR) synthesis
Methoxy-terminated poly(ethylene glycol) (PEG) AuNRs were synthesized for in vivo injection using a well-established seed-mediated growth method using the surfactant hexadecyltrimethyl ammonium bromide (CTAB) [35]. Following synthesis, CTAB surface molecules were exchanged with 5 kDa PEG molecules to increase biocompatibility and circulation time in vivo. To do so, a 20 mM solution of 5 kDa mPEG-thiol (Layson Bio) in distilled water was reduced with immobilized TCEP resin (Thermo Scientific) for one hour at room temperature. One mL of CTAB-coated AuNRs was functionalized with PEG by the sequential addition of 275 µL of 1.1 mM K2CO3 and 250 µL of reduced 5 kDa mPEG-thiol. PEG-coated AuNRs were then purified by centrifugation at 15,300 x g for 10 minutes and resuspended in 1X PBS. Efficient surface PEGylation was verified by a lack of AuNR aggregation upon resuspension in 15% FBS and 10X PBS. A stock solution of PEG-coated AuNRs with an absorption peak at 740 nm was sterile filtered using 200 nm pore syringe filters and concentrated to 9 nM for in vivo injection. A representative transmission electron microscopy (TEM) image and spectrophotometry curve for the AuNRs are shown in Fig. 1(a) . Quantitative analysis of TEM images revealed an average AuNR size of 51.8 ± 8.1 nm long by 15.2 ± 3.3 nm wide (n = 20), and an average aspect ratio of 3.4.
Fig. 1.
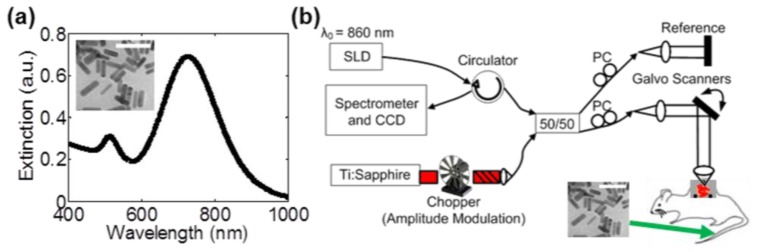
Contrast agents and PTOCT imaging instrumentation. (a) Extinction spectrum of AuNR samples with an absorption peak of approximately 740 nm, with a representative TEM (inset, scale bar = 100 nm). (b) A fiber-based PTOCT system was constructed for imaging. The titanium sapphire laser was amplitude modulated and used as a photothermal heating source. Mice with dorsal skinfold window chambers containing mammary tumors were intravenously injected with AuNRs into the tail vein and imaged with PTOCT over time. SLD: Superluminescent diode; PC: Polarization controllers; 50/50: Fiber coupler
2.2 Imaging instrumentation
A commercial fiber-coupled spectral domain OCT system (Bioptigen) was altered for PTOCT imaging (Fig. 1(b)). An 860 nm central wavelength, superluminescent diode (SLD, Fig. 1(b)) served as the OCT imaging beam (6.4 µm resolution in air), with 750 µW of power on the sample. SLD light was sent through a circulator and split between the imaging sample and a reference reflector using a 50/50 fiber coupler. Returning interference light was captured by a 2048 pixel CCD (Atmel) at a rate of 10 kHz. Incident light on the sample was scanned in the second and third spatial dimension using galvanometer mirrors, and focused to a lateral resolution of approximately 25 µm. Polarization states in the reference and sample arm were optimized using fiber-based polarization controllers (PC, Fig. 1(b)). A tunable Titanium:Sapphire NIR laser source (Coherent) served as the photothermal heating beam (note that continuous wave lasers are equally effective sources). This source was amplitude modulated with a 50% duty cycle square wave using a mechanical chopper and coupled into the sample arm fiber for co-alignment with the imaging beam. The photothermal beam was amplitude modulated at 500 Hz and tuned to 740 nm (the AuNR local surface plasmon peak wavelength), with 25 mW of average power on the sample over one modulation period.
2.3 Image acquisition and signal processing
PTOCT exploits the photothermal effect to produce changes in optical path length that can be detected with high sensitivity and specificity using phase-resolved OCT. In our experiments, the photothermal heat release from AuNRs is modulated with an amplitude-modulated laser tuned to the resonance peak of the nanoparticles (Fig. 2(a) , top). Upon heat release into the microenvironment, water in the tissue surrounding the nanoparticles undergoes thermoelastic expansion and index of refraction shifts, thus altering the optical path length over time. The phase-resolved OCT image is directly related to optical path length, allowing PTOCT to identify AuNRs within the tissue due to fluctuations in OCT phase (Fig. 2(a), bottom) [27].
Fig. 2.
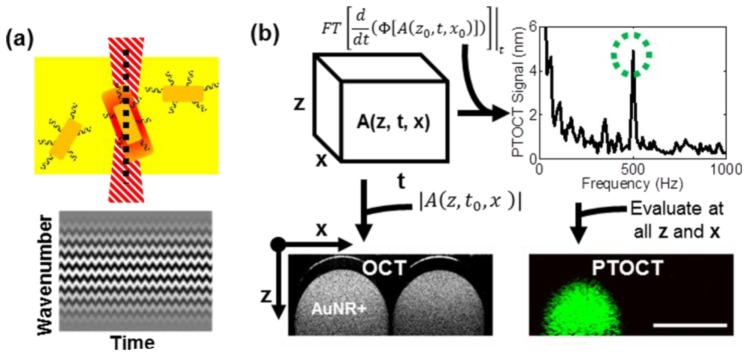
PTOCT signal analysis. (a) AuNRs (dark yellow, top) in optically scattering tissue (bright yellow, top) are identified by the OCT imaging beam (black dashed line, top) by their photothermal heat release signature after absorption of an amplitude-modulated heating beam (red dashed structure, top). Oscillations in heat release from AuNRs cause oscillations in optical path length and thus the phase of the OCT interference pattern over time (bottom). (b) For each PTOCT B-scan, a complex-valued 3D data set is constructed as a function of depth (z), space (x), and time (t) (top left). The temporal derivative of the phase (Φ) of this 3D data set is taken, followed by a temporal Fourier transform (top left). A peak in this Fourier-transformed data at 500 Hz (the amplitude-modulation frequency of the photothermal beam, green-dashed circle) reveals the presence of AuNRs (top right). This analysis is repeated at all spatial positions to reconstruct the 2D cross-sectional PTOCT image, which localizes AuNRs (green pixels, bottom right). The magnitude of the depth-resolved data set provides the traditional OCT structural image (bottom left). Bottom panels show representative OCT and PTOCT images of a solid agarose phantom with AuNRs spatially confined to the left capillary tube (scale bar = 1 mm).
Digital lock-in detection was used to isolate the photothermal oscillations within the phase-resolved OCT data. For each 1D depth-resolved PTOCT A-scan, 1000 repeated OCT A-scans were captured over time while amplitude modulating the photothermal laser at 500 Hz. Each OCT interferogram was resampled from linearity in wavelength to linearity in wavenumber, dispersion corrected [36], and background subtracted [37]. A Chirp-Z transform converted wavenumber data to depth-resolved image data, creating a three-dimensional complex-number data set as a function of depth (z), time (t), and lateral position (x) for each B-scan (Fig. 2(b)). The phase (Φ, Fig. 2(b)) at each point was used for subsequent analysis. The phase in each A-scan was referenced to the first bright reflection to increase phase stability (i.e. self-referencing) [38]. Then, the temporal derivative of the phase at each point in depth was calculated [27], and the data was Fourier transformed in the temporal dimension. The PTOCT signal for each point in depth (z) and lateral position (x) was defined as the magnitude of its temporal Fourier transform at the amplitude modulation frequency of the photothermal beam (500 Hz, green circle Fig. 2(b)), minus the background (the mean of the adjacent 200 Hz frequency components). The Fourier transform magnitude data was transformed to optical path length units according to previous methods [27]. This analysis was completed at all points in space to reconstruct PTOCT images (Fig. 2(b)). This process was expanded to a third spatial dimension to create 3D volumes. Previous work provides details of the PTOCT system, signal characterization, and signal analysis [27].
In addition to PTOCT, speckle variance OCT (SVOCT) maps of microvessel morphology (intensity-normalized variance of the magnitude image over 10 repeated A-scans) were collected over the same image volumes, and were then median filtered to reduce noise. Traditional OCT tissue morphology images were calculated as the log based ten values of the magnitude image. Three by two millimeter rectangular volume scans were acquired, with isotropic 10 µm lateral sampling for OCT and SVOCT image volumes, and 10 µm (fast scan dimension) by 100 µm (slow scan dimension) sampling for PTOCT image volumes. PTOCT cross-sectional images were filtered using a two-dimensional (2D) median filter and masked using the OCT magnitude data. The 3D image volumes where then reduced to two en face dimensions by averaging along the depth dimension (approximately 1 mm in depth). SVOCT projections were further filtered using a Gabor filter to increase contrast to vessel-like features [39]. En face projections of OCT (tissue morphology, gray), SVOCT (vessel morphology, red), and PTOCT (AuNRs, green) were used for quantitative data analysis. OCT, SVOCT, and PTOCT images were processed and quantified offline using custom Matlab software.
A spatial frequency analysis of the PTOCT en face projections was performed to characterize the diffusion of the AuNRs throughout the tissue over time. Each PTOCT en face projection was first multiplied by a raised cosine window to remove edge artifacts in the Fourier transform. PTOCT images were then mean subtracted, and a two-dimensional Fourier transform was calculated so that low spatial frequency content of the image was radially centered, with high frequency content located at the edges. Fourier transformed images were then masked to include only low or high frequency components, and the image energy (the sum of the magnitude squared) of the low and high frequency masked images were calculated. This analysis quantifies the percent of image energy that is either low (diffuse) or high (sparse) spatial frequency, and can be expanded to include directionality and multiple frequency bands. The frequency cutoff was selected to enclose spatial frequencies less than or equal to 10% of the Nyquist sampling frequency.
2.4 In vivo imaging
All animal work was approved by the Vanderbilt IACUC committee, and all surgical procedures were performed using aseptic techniques. Six nude (Foxn1nu/ Foxn1nu, The Jackson Laboratory) female mice, 12 weeks of age, underwent dorsal skinfold window chamber surgery for direct visualization of 4T1 (mouse mammary) tumors. While under inhalant isoflurane anesthesia (1.5-2%), a custom made 10 mm diameter window was placed onto the back of the mouse and secured across a dorsal skinfold with the use of three bolts and sutures at the top and bottom of the window. The skin from one side of the skinfold within the window was removed, as well as the underlying fascia layer. A 4T1 (mouse mammary) solid tumor, which was grown for 16 days in the mammary fat pad of a separate mouse, was sliced to a thin section and placed on the exposed tissue layer in the dorsal window. The window was filled with sterile saline, and overlain with a coverslip to visualize the underlying tumor. 4T1 tumors were allowed to grow in the dorsal skinfold window for 10 days prior to imaging. Following baseline (pre-injection) OCT, SVOCT, and PTOCT imaging, sterile-filtered PEG-coated AuNRs in 1X PBS (200 ml volume, ~9 nM concentration) [2] were injected into the tail vein of four mice, while sterile 1X PBS was injected as a vehicle control into two mice. After injection, three more imaging time points were performed at approximately 2, 16, and 24 hours post-injection to image the accumulation of AuNRs into the tumor tissue via the EPR effect. During imaging, mice body temperature was regulated by a circulating heating pad while the mice were under inhalant isoflurane anesthesia (1.5% in air). Dorsal windows were tightly secured to the imaging stage using a custom apparatus.
2.5 Ex vivo multiphoton microscopy
Upon completion of in vivo imaging time courses, the mice were sacrificed and tumors were harvested for further imaging. The tumor was maintained in standard cell culture media on ice and imaged ex vivo using multiphoton luminescence microscopy as an independent validation of AuNR accumulation into the tissue. A custom-built multiphoton epi-fluorescence imaging system (Prairie Technologies) was used for ex vivo tissue imaging. A Titanium:Sapphire laser was tuned to the excitation peak of the AuNRs (740 nm) with less than 1 mW of power incident on the sample. An emission filter at 607 nm with a 45 nm bandwidth was used to capture AuNR luminescence signal and reject any autofluorescence. Ex vivo images were captured with a 20X objective (NA = 0.75), and were processed offline using a custom Matlab routine. For quantitative analysis, two images for each of the six animals were analyzed (n = 12 total). All images were collected in the same imaging session, ensuring constant imaging parameters (laser power, detector settings, etc.) between samples.
3. Results
3.1 In vivo imaging of AuNR accumulation into tumors
Mice with mammary tumors in dorsal window chambers were intravenously injected with either AuNRs or 1X PBS (vehicle control), and imaged at multiple time points before and after injection. Examples of the PTOCT image volumes collected with OCT, SVOCT, and PTOCT are shown in Fig. 3 . Tissue morphology (gray channel, OCT), vessel morphology (red channel, SVOCT), and AuNR distribution (green channel, PTOCT) are inherently co-registered (Fig. 3(a)). The distribution of AuNRs at each time point is noninvasively imaged with respect to the tissue structure as well as vessel morphology. An angled slice through the depth dimension yields better visualization of all three image channels as a function of depth (Fig. 3(b)). The tissue structure (gray channel, OCT) can also be removed (Fig. 3(c)) to better visualize the structure of the AuNR distribution with respect to the vessel morphology. For quantitative comparisons between imaging data sets, image volumes were reduced to two lateral en face dimensions via averaging over the depth dimension (Fig. 3(d)).
Fig. 3.

Representative in vivo PTOCT image volumes. (a) 3D rendering of mouse 4T1 tumor 16 hours after AuNR injection via the tail vein, with OCT of tissue structure (gray channel), SVOCT of vessel morphology (red channel), and PTOCT of AuNRs (green channel). (b) An angled slice through the 3D volume reveals depth-resolved anatomy, vessel morphology, and AuNR distribution. (c) Removing the structural information reveals the underlying vessels (red) and AuNR distribution (green) in three dimensions. (d) After averaging over the depth dimension, co-registered 2D en face projections of tissue morphology, vessel morphology, and AuNR distribution are visible in the tumor tissue (scale bar = 1 mm).
Example en face projections of overlapping tissue morphology (gray channel, OCT), vessel morphology (red channel, SVOCT), and AuNR distribution (green channel, PTOCT) for control (1X PBS injected, n = 2) and experimental (AuNR injected, n = 4) animals are shown in Fig. 4 . Vessels within the tumor tissue are clearly visible in both control mice injected with 1X PBS (Fig. 4, top) as well as experimental mice with systemically injected AuNRs (Fig. 4, bottom). In addition, distinct morphological features are present from within the tumor tissue. However, the PTOCT signal is only visible in mice after systemic injection of AuNRs, indicating the ability of PTOCT to specifically image AuNRs in vivo at the tumor site, even in the presence of tissue scattering. Due to the size of the AuNRs, the particles are found mainly within close proximity of terminal microvessels over the time points imaged (Fig. 4). These findings agree with previous work using histology [40] and microscopy of superficial depths [41] that established a relationship between particle size and diffusion distance from microvessels in tumors. However, these previous studies required destructive techniques or contrast-aided visualization of vasculature.
Fig. 4.

PTOCT signals increase in vivo after AuNR injection. En face projection images of tissue morphology (OCT, gray), vessel morphology (SVOCT, red), and AuNR distribution (PTOCT, green) in a mouse tumor pre-injection, and 2 hours, 16 hours, and 24 hours after injection of either 1X PBS (top) or AuNRs (bottom) in 1X PBS (scale bar = 1 mm). The PTOCT signal appears only after injection of AuNRs. White arrows point to regions of positive PTOCT signal 2 hours after injection.
Specific imaging of AuNR uptake in tumors is apparent from the en face PTOCT projections from mice injected with 1X PBS control (Fig. 5(a) ) versus AuNRs (Fig. 5(b)) 16 hours post-injection. The time-course of AuNR accumulation and retention within the tumors was similar across all animals (Fig. 5(c)), and mice injected with AuNRs exhibited significantly increased PTOCT signal compared to pre-injection values (p<0.05, Wilcoxon rank sum test, Fig. 5(d)). The PTOCT signal peaked at 16 hours after AuNR injections, with a peak PTOCT signal of 145 ± 46 picometers across all experimental animals (Fig. 5(d)). Although low resolution imaging modalities can provide information on the bulk accumulation of nanoparticles in tumors, the high resolution and wide field of view of PTOCT allows for visualization of spatially distinct nanoparticle uptake over large regions of the tumor volume. The bulk accumulation of AuNRs in tumors is similar across mice (Fig. 5(c)), however there are spatial heterogeneities in uptake within individual tumors and these spatial patterns vary between tumors (Fig. 5(b)). One important attribute of PTOCT is its ability to capture these spatial heterogeneities over a wide field of view, which is difficult to accomplish with microscopy or whole body imaging techniques. Additionally, PTOCT can acquire images at depths greater than confocal and two-photon microscopy.
Fig. 5.

In vivo temporal and spatial tracking of AuNR uptake in tumors using PTOCT. En face PTOCT projection images from all mice 16 hours after injection of either (a) PBS (n = 2) or (b) AuNRs (n = 4) (scale bar = 1 mm). (c) Mean PTOCT signal at each imaging time-point for the two mice injected with PBS and the four mice injected with AuNRs (“preinj” = pre-injection). (d) Mean ± standard error PTOCT signal for all mice injected with either PBS (n = 2) or AuNRs (n = 4). *p<0.05.
The high resolution of PTOCT can be exploited for more advanced image analysis to quantify AuNR spatial distribution over time. This analysis can be applied to optimize nanoparticle delivery, and to determine the time-points at which photothermal or other therapeutic interventions would be most effective. Specifically, the sparsity of the AuNR distribution within the tumor at each time point was quantified from the spatial frequency content of each 2D PTOCT projection image. The low and high spatial frequency components correspond to AuNR distributions in the tumor that are diffuse (more dispersed throughout the tissue) and sparse (spatially confined to distinct locations), respectively.
PTOCT en face images for one mouse at the three time points after AuNR injection are shown in Fig. 6(a) , as well as the accompanying two dimensional Fourier transforms (Fig. 6(a), bottom). The red circle in each Fourier transform image (Fig. 6(a), bottom) represents the cutoff between low and high frequency content calculations. The image energy due to low frequencies is shown in Fig. 6(b) for each mouse injected with AuNRs, and the average for all four mice is plotted in Fig. 6(c). Low frequency components are at a minimum at the earliest time point, due to the sparsity of the AuNR accumulation early after injection. At later time points, the low frequency component increases (and the high frequency contribution decreases an equal amount) due to the increased accumulation and diffusion of the AuNRs throughout the tissue (p<0.05, Wilcoxon rank sum test). Between the 2 hour and 16 hour time-points, the energy of the low spatial frequencies increases by approximately 15%, then remains fairly constant, indicating that the diffusion of the AuNRs throughout the tissue peaks between those time-points. As shown previously, the mean PTOCT signal (Fig. 5(d)) due to AuNR accumulation is similar at 2 and 24 hours. However, the distribution of AuNRs is quite different between those time points (Fig. 6(c)). These distribution dynamics are helpful for understanding the pharmacokinetics of AuNRs into and out of the tumor, and for properly optimizing nanoparticles for clinical therapeutic and imaging strategies. Lower resolution imaging tools (computed tomography, photoacoustic tomography, etc.) as well as destructive techniques (e.g. ICPMS) can measure bulk accumulation of AuNRs over a large tumor section. However, the high-resolution distribution analysis provided by PTOCT is critical for understanding AuNR accumulation throughout different spatial locations in the tumor, and highlights the enabling abilities of high resolution, wide field of view, and deeply penetrating PTOCT imaging. For example, nanoparticles that are too large to diffuse away from nearby vessels can only deliver drug to perivascular tumor regions. PTOCT can resolve these intratumoral heterogeneities in nanoparticle delivery with respect to nearby vasculature, and could therefore serve as a robust tool for tracking the efficacy of nanoparticle-based drug delivery platforms that seek to homogeneously disperse throughout a tumor.
Fig. 6.

Spatial frequency content analysis of in vivo PTOCT images reveals an increase in low spatial frequencies over time. (a) PTOCT en face images (top) and accompanying 2D spatial Fourier transforms (bottom) from a representative mouse tumor 2 hours, 16 hours, and 24 hours after injection of AuNRs (scale bar = 1 mm). The red circle highlights the cutoff between high (outside the ring) and low (inside the ring) spatial frequencies. (b) Percent of image energy due to low spatial frequencies over time for four mice injected with AuNRs. (c) Mean ± standard error of the image energy due to low and high spatial frequencies for mice injected with AuNRs (n = 4) over time. *p < 0.05.
3.2 Ex vivo validation using multiphoton microscopy
Following the completion of the PTOCT imaging studies, the mice were sacrificed and tumors were harvested for further imaging. Gold nanoparticles possess a strong multiphoton luminescence signal that is spectrally separate from common autofluorescent proteins native to cells, so multiphoton imaging serves as a reliable validation tool for imaging AuNRs [8, 42]. Representative ex vivo multiphoton images of tumors from mice intravenously injected with PBS (Fig. 7(a) ) or AuNRs (Fig. 7(b)) after completion of the PTOCT imaging study are shown, as well as the mean signal for tissues from mice intravenously injected with AuNRs or PBS (Fig. 7(c)). All ex vivo multiphoton imaging occurred in unaltered tissue samples freshly harvested from the same mice imaged in the PTOCT study. There is a significant increase in the multiphoton signal measured in tumors from mice injected with AuNRs compared to tumors from mice injected with PBS (p<0.05, Wilcoxon rank sum test). The average value from the negative control mice indicates the noise floor of the system, since no appreciable structure or signal was visible in any PBS-injected mouse tumor. After ex vivo imaging, tumors were formalin fixed, paraffin embedded, sliced, stained with hematoxylin and eosin, and histologically verified as malignant mammary tumors (data not shown). These ex vivo analyses confirm the observed increase in PTOCT signal is due to accumulation of AuNRs in the tumor tissue.
Fig. 7.
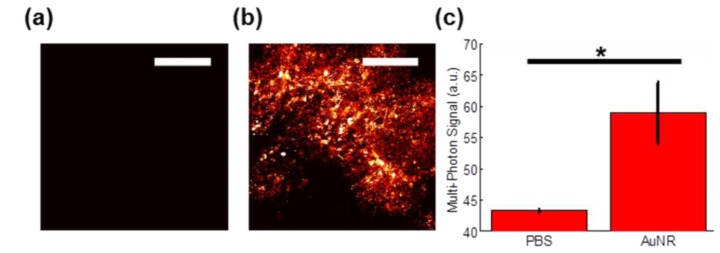
Validation of AuNR uptake into tumor tissue. Representative 20X multiphoton image of tumor tissue 24 hours after tail vein injection of (a) 1X PBS or (b) AuNRs, imaged ex vivo (scale bar = 100 µm). (b) Mean ± standard error of the multiphoton signal across all ex vivo images from mice injected with either PBS (n = 4 images) or AuNRs (n = 8 images). *p < 0.05
4. Discussion
We have shown that PTOCT can temporally track the 3D accumulation of AuNRs into tumor tissue. PTOCT identified peak AuNR accumulation in tumors around 16 hours, which agrees with temporal trends found in previous studies that employed invasive methods to quantify nanoparticle biodistribution [43]. Co-registered images of tissue structure (OCT), vessel morphology (SVOCT), and nanoparticle distribution (PTOCT) with the same imaging system could allow for comprehensive analyses of nanoparticle delivery kinetics. For example, this platform of multimodal OCT can be applied to analyze in vivo drug diffusion distances from vessels over time, or to validate in vivo drug release over a greater tumor volume than previous studies, without the need for vascular contrast agents [41, 44]. PTOCT has the potential to track not only gold nanoparticles in vivo, but any molecule with a strong NIR absorption cross section (e.g. near infrared fluorophores [45], carbon nanotubes [30]), with large fields of view, at high resolution, and at depths greater than confocal and multiphoton microscopy. This could allow for in vivo imaging of the delivery of clinically relevant agents of different size, shape, and surface chemistry in the context of the microvasculature (i.e. the drug delivery system). For example, PTOCT could track dye-tagged drugs or nanoparticles over time and over multiple doses to quantify the in vivo spatial distribution and pharmacokinetics of these agents at the target site. PTOCT could also be used to refine delivery strategies to overcome clearance by the immune system [46]. Additionally, other orthotopic tumor models including brain and mammary windows [22] can be imaged with PTOCT, further supporting widespread adoption.
Remaining optimizations including phase accumulation [47] and imaging speed [48] can also be incorporated for more streamlined acquisition and analysis. In its current implementation, PTOCT contains axial image artifacts due to phase accumulation. Published models address this issue in layered homogeneous phantoms [47], and models for heterogeneous in vivo images are currently under development. In addition, the current PTOCT implementation uses digital sampling and lock-in, which required a 14-minute scan time per volume. However, with the development of optical lock-in techniques for low coherence interferometry [48], PTOCT can potentially be performed with significantly reduced acquisition times. Long in vivo scan times can also cause image artifacts due to respiratory and cardiac motion. However, a few key data acquisition and signal processing considerations were used in this study to minimize the impact of motion artifact on PTOCT images. First, the photothermal heating laser was modulated at 500 Hz, which lies outside of the frequency range of motion artifact (<500 Hz). Second, the phase at all points in depth was digitally referenced to the most shallow reflection in the image (i.e. the dorsal window coverslip), thus improving phase stability [38]. Last, any remaining motion artifact was background subtracted using the average of the frequency components adjacent to the photothermal frequency.
In conclusion, PTOCT is a promising imaging platform for the study of gold nanoparticles and other photon-absorbing molecules in vivo. PTOCT provides non-invasive images of 3D drug delivery dynamics, along with corresponding tissue and vessel morphology images. As the technology continues to develop, PTOCT could have broad applications to assess in vivo delivery of nanomedicines and imaging contrast agents, greatly impacting drug discovery and molecular imaging.
Acknowledgments
This work was supported by grants from the National Institutes of Health under award numbers R00CA142888 and R25CA136440-05. In addition, the authors wish to acknowledge Victoria Youngblood and Dr. Jin Chen for assistance with in vivo tumor systems, Spencer Crowder for supplying 4T1 tumor cells, and Tianwei Shen and Travis Meyer for assistance in gold nanorod synthesis.
References and links
- 1.Schwartz J. A., Shetty A. M., Price R. E., Stafford R. J., Wang J. C., Uthamanthil R. K., Pham K., McNichols R. J., Coleman C. L., Payne J. D., “Feasibility Study of Particle-Assisted Laser Ablation of Brain Tumors in Orthotopic Canine Model,” Cancer Res. 69(4), 1659–1667 (2009). 10.1158/0008-5472.CAN-08-2535 [DOI] [PubMed] [Google Scholar]
- 2.von Maltzahn G., Park J. H., Agrawal A., Bandaru N. K., Das S. K., Sailor M. J., Bhatia S. N., “Computationally Guided Photothermal Tumor Therapy Using Long-Circulating Gold Nanorod Antennas,” Cancer Res. 69(9), 3892–3900 (2009). 10.1158/0008-5472.CAN-08-4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gobin A. M., Lee M. H., Halas N. J., James W. D., Drezek R. A., West J. L., “Near-infrared resonant nanoshells for combined optical imaging and photothermal cancer therapy,” Nano Lett. 7(7), 1929–1934 (2007). 10.1021/nl070610y [DOI] [PubMed] [Google Scholar]
- 4.Thakor A. S., Jokerst J., Zavaleta C., Massoud T. F., Gambhir S. S., “Gold Nanoparticles: A Revival in Precious Metal Administration to Patients,” Nano Lett. 11(10), 4029–4036 (2011). 10.1021/nl202559p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Libutti S. K., Paciotti G. F., Byrnes A. A., Alexander H. R., Jr, Gannon W. E., Walker M., Seidel G. D., Yuldasheva N., Tamarkin L., “Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine,” Clin. Cancer Res. 16(24), 6139–6149 (2010). 10.1158/1078-0432.CCR-10-0978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agarwal A., Huang S. W., O’Donnell M., Day K. C., Day M., Kotov N., Ashkenazi S., “Targeted gold nanorod contrast agent for prostate cancer detection by photoacoustic imaging,” J. Appl. Phys. 102(6), 064701 (2007). 10.1063/1.2777127 [DOI] [Google Scholar]
- 7.Oyelere A. K., Chen P. C., Huang X. H., El-Sayed I. H., El-Sayed M. A., “Peptide-conjugated gold nanorods for nuclear targeting,” Bioconjug. Chem. 18(5), 1490–1497 (2007). 10.1021/bc070132i [DOI] [PubMed] [Google Scholar]
- 8.Durr N. J., Larson T., Smith D. K., Korgel B. A., Sokolov K., Ben-Yakar A., “Two-photon luminescence imaging of cancer cells using molecularly targeted gold nanorods,” Nano Lett. 7(4), 941–945 (2007). 10.1021/nl062962v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park J., Estrada A., Schwartz J. A., Diagaradjane P., Krishnan S., Dunn A. K., Tunnell J. W., “Intra-Organ Biodistribution of Gold Nanoparticles Using Intrinsic Two-photon Induced Photoluminescence,” Lasers Surg. Med. 42(7), 630–639 (2010). 10.1002/lsm.20935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qian X. M., Peng X. H., Ansari D. O., Yin-Goen Q., Chen G. Z., Shin D. M., Yang L., Young A. N., Wang M. D., Nie S. M., “In vivo tumor targeting and spectroscopic detection with surface-enhanced Raman nanoparticle tags,” Nat. Biotechnol. 26(1), 83–90 (2008). 10.1038/nbt1377 [DOI] [PubMed] [Google Scholar]
- 11.De Jong W. H., Hagens W. I., Krystek P., Burger M. C., Sips A. J. A. M., Geertsma R. E., “Particle size-dependent organ distribution of gold nanoparticles after intravenous administration,” Biomaterials 29(12), 1912–1919 (2008). 10.1016/j.biomaterials.2007.12.037 [DOI] [PubMed] [Google Scholar]
- 12.Akiyama Y., Mori T., Katayama Y., Niidome T., “The effects of PEG grafting level and injection dose on gold nanorod biodistribution in the tumor-bearing mice,” J. Control. Release 139(1), 81–84 (2009). 10.1016/j.jconrel.2009.06.006 [DOI] [PubMed] [Google Scholar]
- 13.Hainfeld J. F., Slatkin D. N., Focella T. M., Smilowitz H. M., “Gold nanoparticles: a new X-ray contrast agent,” Br. J. Radiol. 79(939), 248–253 (2006). 10.1259/bjr/13169882 [DOI] [PubMed] [Google Scholar]
- 14.Chen Y. S., Frey W., Kim S., Kruizinga P., Homan K., Emelianov S., “Silica-Coated Gold Nanorods as Photoacoustic Signal Nanoamplifiers,” Nano Lett. 11(2), 348–354 (2011). 10.1021/nl1042006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zavaleta C. L., Smith B. R., Walton I., Doering W., Davis G., Shojaei B., Natan M. J., Gambhir S. S., “Multiplexed imaging of surface enhanced Raman scattering nanotags in living mice using noninvasive Raman spectroscopy,” Proc. Natl. Acad. Sci. U.S.A. 106(32), 13511–13516 (2009). 10.1073/pnas.0813327106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong L., Wei Q. S., Wei A., Cheng J. X., “Gold Nanorods as Contrast Agents for Biological Imaging: Optical Properties, Surface Conjugation and Photothermal Effects,” Photochem. Photobiol. 85(1), 21–32 (2009). 10.1111/j.1751-1097.2008.00507.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang J., Nakamura H., Maeda H., “The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect,” Adv. Drug Deliv. Rev. 63(3), 136–151 (2011). 10.1016/j.addr.2010.04.009 [DOI] [PubMed] [Google Scholar]
- 18.Huang D., Swanson E. A., Lin C. P., Schuman J. S., Stinson W. G., Chang W., Hee M. R., Flotte T., Gregory K., Puliafito C. A., Fujimoto J. G., “Optical Coherence Tomography,” Science 254(5035), 1178–1181 (1991). 10.1126/science.1957169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujimoto J. G., “Optical coherence tomography for ultrahigh resolution in vivo imaging,” Nat. Biotechnol. 21(11), 1361–1367 (2003). 10.1038/nbt892 [DOI] [PubMed] [Google Scholar]
- 20.Liu G. J., Lin A. J., Tromberg B. J., Chen Z. P., “A comparison of Doppler optical coherence tomography methods,” Biomed. Opt. Express 3(10), 2669–2680 (2012). 10.1364/BOE.3.002669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mariampillai A., Standish B. A., Moriyama E. H., Khurana M., Munce N. R., Leung M. K. K., Jiang J., Cable A., Wilson B. C., Vitkin I. A., Yang V. X. D., “Speckle variance detection of microvasculature using swept-source optical coherence tomography,” Opt. Lett. 33(13), 1530–1532 (2008). 10.1364/OL.33.001530 [DOI] [PubMed] [Google Scholar]
- 22.Vakoc B. J., Lanning R. M., Tyrrell J. A., Padera T. P., Bartlett L. A., Stylianopoulos T., Munn L. L., Tearney G. J., Fukumura D., Jain R. K., Bouma B. E., “Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging,” Nat. Med. 15(10), 1219–1223 (2009). 10.1038/nm.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John R., Rezaeipoor R., Adie S. G., Chaney E. J., Oldenburg A. L., Marjanovic M., Haldar J. P., Sutton B. P., Boppart S. A., “In vivo magnetomotive optical molecular imaging using targeted magnetic nanoprobes,” Proc. Natl. Acad. Sci. U.S.A. 107(18), 8085–8090 (2010). 10.1073/pnas.0913679107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacob D., Shelton R. L., Applegate B. E., “Fourier domain pump-probe optical coherence tomography imaging of Melanin,” Opt. Express 18(12), 12399–12410 (2010). 10.1364/OE.18.012399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oldenburg A. L., Hansen M. N., Ralston T. S., Wei A., Boppart S. A., “Imaging gold nanorods in excised human breast carcinoma by spectroscopic optical coherence tomography,” J. Mater. Chem. 19(35), 6407–6411 (2009). 10.1039/b823389f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skala M. C., Crow M. J., Wax A., Izatt J. A., “Photothermal Optical Coherence Tomography of Epidermal Growth Factor Receptor in Live Cells Using Immunotargeted Gold Nanospheres,” Nano Lett. 8(10), 3461–3467 (2008). 10.1021/nl802351p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tucker-Schwartz J. M., Meyer T. A., Patil C. A., Duvall C. L., Skala M. C., “In vivo photothermal optical coherence tomography of gold nanorod contrast agents,” Biomed. Opt. Express 3(11), 2881–2895 (2012). 10.1364/BOE.3.002881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adler D. C., Huang S. W., Huber R., Fujimoto J. G., “Photothermal detection of gold nanoparticles using phase-sensitive optical coherence tomography,” Opt. Express 16(7), 4376–4393 (2008). 10.1364/OE.16.004376 [DOI] [PubMed] [Google Scholar]
- 29.Jung Y., Reif R., Zeng Y. G., Wang R. K., “Three-Dimensional High-Resolution Imaging of Gold Nanorods Uptake in Sentinel Lymph Nodes,” Nano Lett. 11(7), 2938–2943 (2011). 10.1021/nl2014394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tucker-Schwartz J. M., Hong T., Colvin D. C., Xu Y. Q., Skala M. C., “Dual-modality photothermal optical coherence tomography and magnetic-resonance imaging of carbon nanotubes,” Opt. Lett. 37(5), 872–874 (2012). 10.1364/OL.37.000872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paranjape A. S., Kuranov R., Baranov S., Ma L. L., Villard J. W., Wang T. Y., Sokolov K. V., Feldman M. D., Johnston K. P., Milner T. E., “Depth resolved photothermal OCT detection of macrophages in tissue using nanorose,” Biomed. Opt. Express 1(1), 2–16 (2010). 10.1364/BOE.1.000002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hobbs S. K., Monsky W. L., Yuan F., Roberts W. G., Griffith L., Torchilin V. P., Jain R. K., “Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment,” Proc. Natl. Acad. Sci. U.S.A. 95(8), 4607–4612 (1998). 10.1073/pnas.95.8.4607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi H. S., Liu W., Misra P., Tanaka E., Zimmer J. P., Itty Ipe B., Bawendi M. G., Frangioni J. V., “Renal clearance of quantum dots,” Nat. Biotechnol. 25(10), 1165–1170 (2007). 10.1038/nbt1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyer A. K., Khaled G., Fang J., Maeda H., “Exploiting the enhanced permeability and retention effect for tumor targeting,” Drug Discov. Today 11(17-18), 812–818 (2006). 10.1016/j.drudis.2006.07.005 [DOI] [PubMed] [Google Scholar]
- 35.Murphy C. J., Sau T. K., Gole A. M., Orendorff C. J., Gao J. X., Gou L., Hunyadi S. E., Li T., “Anisotropic metal nanoparticles: Synthesis, assembly, and optical applications,” J. Phys. Chem. B 109(29), 13857–13870 (2005). 10.1021/jp0516846 [DOI] [PubMed] [Google Scholar]
- 36.Wojtkowski M. D., Ko T. H., Fujimoto J. G., Bajraszewski T., Gorczynska I., Targowski P., Kowalczyk A., Schuman J. S., Duker J. S., “Ultrahigh speed, ultrahigh resolution optical coherence tomography using spectral domain detection,” Invest. Ophthalmol. Vis. Sci. 45, U50 (2004). [Google Scholar]
- 37.Moon S., Lee S. W., Chen Z. P., “Reference spectrum extraction and fixed-pattern noise removal in optical coherence tomography,” Opt. Express 18(24), 24395–24404 (2010). 10.1364/OE.18.024395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akkin T., Joo C., de Boer J. F., “Depth-resolved measurement of transient structural changes during action potential propagation,” Biophys. J. 93(4), 1347–1353 (2007). 10.1529/biophysj.106.091298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hendargo H. C., Estrada R., Chiu S. J., Tomasi C., Farsiu S., Izatt J. A., “Automated non-rigid registration and mosaicing for robust imaging of distinct retinal capillary beds using speckle variance optical coherence tomography,” Biomed. Opt. Express 4(6), 803–821 (2013). 10.1364/BOE.4.000803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perrault S. D., Walkey C., Jennings T., Fischer H. C., Chan W. C. W., “Mediating Tumor Targeting Efficiency of Nanoparticles Through Design,” Nano Lett. 9(5), 1909–1915 (2009). 10.1021/nl900031y [DOI] [PubMed] [Google Scholar]
- 41.Dreher M. R., Liu W. G., Michelich C. R., Dewhirst M. W., Yuan F., Chilkoti A., “Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers,” J. Natl. Cancer Inst. 98(5), 335–344 (2006). 10.1093/jnci/djj070 [DOI] [PubMed] [Google Scholar]
- 42.Wang T. Y., Halaney D., Ho D., Feldman M. D., Milner T. E., “Two-photon luminescence properties of gold nanorods,” Biomed. Opt. Express 4(4), 584–595 (2013). 10.1364/BOE.4.000584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnida, M. M. Janat-Amsbury, A. Ray, C. M. Peterson, and H. Ghandehari, “Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages,” European journal of pharmaceutics and biopharmaceutics: official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e. V 77, 417-423 (2011). [DOI] [PMC free article] [PubMed]
- 44.Manzoor A. A., Lindner L. H., Landon C. D., Park J. Y., Simnick A. J., Dreher M. R., Das S., Hanna G., Park W., Chilkoti A., Koning G. A., ten Hagen T. L. M., Needham D., Dewhirst M. W., “Overcoming Limitations in Nanoparticle Drug Delivery: Triggered, Intravascular Release to Improve Drug Penetration into Tumors,” Cancer Res. 72(21), 5566–5575 (2012). 10.1158/0008-5472.CAN-12-1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subhash H. M., Xie H., Smith J. W., McCarty O. J. T., “Optical detection of indocyanine green encapsulated biocompatible poly (lactic-co-glycolic) acid nanoparticles with photothermal optical coherence tomography,” Opt. Lett. 37(5), 981–983 (2012). 10.1364/OL.37.000981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones S. W., Roberts R. A., Robbins G. R., Perry J. L., Kai M. P., Chen K., Bo T., Napier M. E., Ting J. P. Y., Desimone J. M., Bear J. E., “Nanoparticle clearance is governed by Th1/Th2 immunity and strain background,” J. Clin. Invest. 123(7), 3061–3073 (2013). 10.1172/JCI66895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guan G. Y., Reif R., Huang Z. H., Wang R. K. K., “Depth profiling of photothermal compound concentrations using phase sensitive optical coherence tomography,” J. Biomed. Opt. 16(12), 126003 (2011). 10.1117/1.3659211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pache C., Bocchio N. L., Bouwens A., Villiger M., Berclaz C., Goulley J., Gibson M. I., Santschi C., Lasser T., “Fast three-dimensional imaging of gold nanoparticles in living cells with photothermal optical lock-in Optical Coherence Microscopy,” Opt. Express 20(19), 21385–21399 (2012). 10.1364/OE.20.021385 [DOI] [PubMed] [Google Scholar]