

Abstract
Background: A slight increase in albuminuria (urinary albumin excretion [UAE] ≥30 mg/d) is associated with hypertension, type 2 diabetes mellitus, dyslipidemia (high triglyceride [TG] and low high-density lipoprotein cholesterol [HDL-C] concentrations), and hyperuricemia. Although antihypertensive and antidiabetic therapies have been reported to reduce UAE, an association between improvement in dyslipidemia and/or hyperuricemia and a reduction in UAE has not been reported.
Objective: The aim of this study was to investigate the efficacy and tolerability of fenofibrate on albuminuria in patients with hypertriglyceridemia and/or hyperuricemia.
Methods: Patients with hypertriglyceridemia and/or hyperuricemia were recruited from general clinics and lipid clinics in Japan; they received fenofibrate (300 mg once daily) in this randomized, double-blind, placebo-controlled, crossover study. Patients in group A received fenofibrate for 8 weeks followed by placebo for an additional 8 weeks, whereas those in group B received placebo for 8 weeks followed by fenofibrate for 8 additional weeks. UAE was measured at baseline and at the end of each 8-week period. Blood tests were performed at baseline and every 4 weeks until study end. Each physician who participated in the study was to record adverse events at each study visit.
Results: A total of 43 patients entered this study (38 men, 5 women; mean [SE] age, 57.1 [1.4] years; mean [SE] body mass index, 24.3 [0.4] kg/m2). Twenty-one patients (18 men, 3 women) were randomly assigned to group A and 22 (20 men, 2 women) to group B. In group A, serum TG (P<0.001) and apolipoprotein (apo) C2, C3, and E (all P<0.01) concentrations decreased significantly with fenofibrate, and HDL-C and apo A1 and A2 increased significantly (all P<0.001). All of these parameters returned to near-baseline levels after placebo administration. In group B, serum TG, HDL-C, or apo A1, A2, B, C2, C3, and E concentrations did not change significantly with placebo, but TG (P<0.01), apo C3 (P<0.05), and apo E (P<0.05) were significantly decreased with fenofibrate. In addition, HDL-C (P<0.05), apo A1 (P<0.001), and apo A2 (P<0.01) were significantly increased with fenofibrate. Serum concentrations of TG (group A, P<0.001; group B, P<0.001); apo C2 (group A, P<0.01), C3 (group A, P<0.01; group B, P<0.05), and E (group A, P<0.01; group B, P<0.05); and uric acid (group A, P<0.001; group B, P<0.01) were significantly decreased with fenofibrate compared with placebo. HDL-C and apo A1 and A2 were significantly increased with fenofibrate compared with placebo (all P<0.001 in both groups). Fenofibrate treatment was associated with significant reductions in UAE (group A, P<0.05; group B, P<0.01). Spearman rank correlation analysis showed that changes in UAE were associated with changes in apo C2 (ρ = 0.43; P = 0.02) and apo C3 (ρ = 0.49; P = 0.01) concentrations. Multiple regression analysis revealed that a decrease in apo C3 concentration was independently and significantly associated with reductions in albuminuria (ρ = 0.48; P = 0.01). At the end of the study, neither drug-related nor clinical adverse events were evident in any of the patients, except for an increase in serum creatinine concentration above the upper limit of normal (1.40 mg/dL) in 3 patients (14.3%) in group A and 1 patient (4.5%) in group B.
Conclusions: In our study population of patients with hypertriglyceridemia and/or hyperuricemia, fenofibrate-induced ameliorations of impaired TG-rich lipoprotein metabolism were associated with reductions in albuminuria.
Keywords: fenofibrate, apolipoprotein C3, low level of high-density lipoprotein cholesterol, hyperuricemia, albuminuria
Introduction
Microalbuminuria (urinary albumin excretion [UAE] ≥30 mg/d) is a strong risk factor for cardiovascular disease in diabetic and hypertensive patients.1–3 The exact mechanisms of albuminuria and subsequent organ damage are debatable. One hypothesis states that generalized endothelial dysfunction underlies microalbuminuria.4 Strong support for this theory comes from studies in both diabetic and hypertensive patients showing that circulating levels of von Willebrand's factor are related to microalbuminuria.5,6
Microalbuminuria has been reported to be associated with hypertension, type 2 diabetes mellitus (DM), high serum triglyceride (TG) and uric acid concentrations, and low serum high-density lipoprotein-cholesterol (HDL-C) concentration.7–10 We recently found that patients with type 2 DM whose condition progressed from normoalbuminuria to microalbuminuria had increased serum TG and apolipoprotein (apo) B concentrations.11 Although intervention studies revealed that antidiabetic and antihypertensive treatment reduced albuminuria,12,13 studies in which low-density lipoprotein (LDL) cholesterol concentration was lowered showed conflicting results.14,15 No other studies have examined the effect on albuminuria of correcting dyslipidemia (high TG and low HDL-C concentrations) or high uric acid concentration, both of which were improved with fenofibrate.16
The aim of this study was to investigate the efficacy and tolerability of fenofibrate treatment on albuminuria in patients with hypertriglyceridemia and/or hyperuricemia.
Patients and methods
Study design
This was a multicenter, randomized, double-blind, placebo-controlled, crossover study. Male and female patients with hyperlipidemia (fasting serum concentrations of total cholesterol ≥220 mg/dL and/or TG ≥150 mg/dL17) and/or hyperuricemia (uric acid concentration ≥7.0 mg/dL) were randomly selected from 3 lipid clinics and 4 general clinics in Japan and were invited to participate in the study if they fulfilled the following criteria: age 17 to <70; no sign of primary renal, hepatic, or cardiac disease; and no sign of insufficiently treated DM (hemoglobin A1c [HbA1c] >9.0%) or hypertension (blood pressure [BP] ≥160/95 mm Hg). Female patients who were pregnant, possibly pregnant, or lactating were excluded.
After providing written informed consent, patients were asked to revisit the clinic 4 weeks later, for biochemical screening and initiation of the treatment. The study protocol was approved by the local ethics committee of each study site and was in accordance with the principles of the Declaration of Helsinki II.
The patients were instructed to maintain their dietary habits throughout the study. Diet and exercise were not assessed at study visits.
Patients
Patients were randomly assigned to 1 of 2 treatment groups. In group A, patients received fenofibrate (300 mg once daily) for 8 weeks, followed by placebo for an additional 8 weeks, whereas group B received placebo for 8 weeks, followed by fenofibrate (300 mg once daily) for an additional 8 weeks. All patients received placebo for 4 weeks before the study began.
UAE, creatinine clearance rate (CCR), uric acid excretion, uric acid clearance, and fractional excretion of uric acid (FEUA) in a 24-hour pooled urine sample were measured at baseline and at the end of both treatment periods. Blood tests after an overnight fast and BP measurements were performed at baseline and every 4 weeks until study end.
Laboratory tests
Serum cholesterol, TG, creatinine, free fatty acid (FFA), and uric acid concentrations were measured enzymatically using a Hitachi 7350® Autoanalyzer (Hitachi Ltd., Mito, Ibaraki, Japan). HbA1c (reference interval, 4.5%–5.8%) and fasting plasma glucose (FPG) concentration were assayed by affinity chromatography and a glucose oxidase method, respectively. Insulin was measured using enzyme immunoassay. HDL-C and apo concentrations were measured by a precipitation method using phosphotungstate and manganese chloride18 and an immunoturbidimetric assay,19 respectively. UAE was measured using a commercially available latex immunoassay20 kit (Eiken Alb-II®, Eiken Chemicals, Tokyo, Japan) with a detection limit of 0.4 mg/mL. Intra- and interassay coefficients of variation were ≤2.5% and ≤1.8%, respectively. Urinary creatinine concentration was determined by Jaffe's reaction using a Hitachi 7450® Autoanalyzer (Hitachi Ltd.). All blood and urine samples were analyzed at the SRL Laboratory (Tokyo, Japan).
Each physician who participated in the study was to record adverse events at each study visit.
Statistical analysis
SAS statistical software (SAS Institute Inc., Cary, North Carolina) was used to perform statistical analyses. The results for continuous variables are presented as mean (SE). Wilcoxon signed rank test and the Mann-Whitney U test were used to assess the significance of differences within and between group means, respectively. The Fisher exact test was used to assess differences in proportions between groups. Associations of changes in UAE with those in other variables were assessed by Spearman rank correlation analysis and then multiple regression analysis. P<0.05 was considered significant. To improve skew and kurtosis of the distribution, log-transformations were made when appropriate.
Results
A total of 43 patients (38 men, 5 women; mean [SE] age, 57.1 [1.4] years; mean [SE] body mass index [BMI], 24.3 [0.4] kg/m2) entered this study. Twenty-one patients (18 men, 3 women) were allocated to group A and 22 (20 men, 2 women) to group B. Of these, 9 (42.9%) and 11 (50.0%) patients in groups A and B, respectively, were selected from general clinics and 12 (57.1%) and 11 (50.0%) patients, respectively, were selected from lipid clinics.
Thirty patients (69.8%) had hypertension (BP >140/>90 mm Hg),21 8 (18.6%) had type 2 DM (FPG >126 mg/dL),22 and 4 (9.3%) (2 each in groups A [9.5%] and B [9.1%]) had both conditions (Table I). The number of patients with clinical disorders at baseline were similar in the 2 groups. The number of patients using antihypertensives or angiotensin-converting enzyme (ACE) inhibitors also were similar in the 2 groups.
Table I.
| Disorder | Group A (n = 21) | Group B (n = 22) |
|---|---|---|
| Hypertriglyceridemia | 16 (76.2) | 18 (81.8) |
| Hyperuricemia | 16 (76.2) | 15 (68.2) |
| Hypertension† | 15 (71.4) | 15 (68.2) |
| Hypercholesterolemia | 14 (66.7) | 15 (68.2) |
| Microalbuminuria | 3 (14.3) | 8 (36.4) |
| Type 2 diabetes mellitus‡ | 2 (9.5) | 6 (27.3) |
| Impaired fasting glycemia | 1 (4.8) | 3 (13.6) |
No significant differences were found between the 2 groups.
Some patients had >1 disorder.
Two patients in each group (9.5% in group A, 9.1% in group B) had both hypertension and type 2 diabetes mellitus.
At the end of the study, neither drug-related nor clinical adverse events were evident in any of the patients, except for an increase in serum creatinine concentration above the upper limit of normal (1.40 mg/dL) in 3 patients (14.3%) in group A and 1 patient (4.5%) in group B. Although the mean CCR decreased significantly from baseline with active treatment in group B (from 11517 mL/min to 8914 mL/min; P<0.01), this rate remained within the normal range. No significant changes were found in BMI, BP, FPG, HbA1c, fasting serum insulin, or FFA concentrations in either group (Table II). In both groups, fenofibrate treatment was associated with a significant decrease in mean (SE) serum uric acid concentration (group A, P<0.001; group B, P<0.01). This treatment also was associated with a concomitant significant increase in the FEUA (both P<0.05), demonstrating a uricosuric effect of fenofibrate.
Table II.
Clinical and biochemical values in study patients. (Data are expressed as mean [SE].)∗
| Parameter | Normal Value | Baseline | 8 Weeks | 16 Weeks |
|---|---|---|---|---|
| BMI, kg/m2 | <25 | |||
| Group A | 25.1 (0.9) | 25.2 (1.0) | 25.1 (1.1) | |
| Group B | 25.2 (1.0) | 24.3 (0.6) | 23.9 (0.6) | |
| SBP, mm Hg | ≤140 | |||
| Group A | 137 (4) | 136 (3) | 134 (4) | |
| Group B | 138 (2) | 135 (3) | 140 (4) | |
| DBP, mm Hg | ≤90 | |||
| Group A | 82 (3) | 81 (3) | 79 (2) | |
| Group B | 80 (1) | 79 (1) | 81 (2) | |
| FPG, mg/dL | <126 | |||
| Group A | 107 (10) | 102 (7) | 118 (15) | |
| Group B | 131 (12) | 128 (11) | 122 (13) | |
| HbA1c, % | 4–7 | |||
| Group A | 5.7 (0.2) | 6.1 (0.4) | 5.4 (0.3) | |
| Group B | 6.3 (0.5) | 6.1 (0.4) | 6.2 (0.5) | |
| Fasting insulin, μU/mL | 3.1–16.9 | |||
| Group A | 11.2 (1.9) | 11.3 (1.9) | 13.5 (2.7) | |
| Group B | 10.8 (1.4) | 11.1 (1.2) | 11.2 (1.2) | |
| FFA, mEq/L | 0.14–0.85 | |||
| Group A | 0.70 (0.11) | 0.56 (0.16) | 0.58 (0.13) | |
| Group B | 0.71 (0.11) | 0.57 (0.04) | 0.76 (0.29) | |
| Serum uric acid, mg/dL | 3.0–7.0 | |||
| Group A | 7.8 (0.2) | 5.9 (0.3)† | 7.6 (0.2) | |
| Group B | 8.1 (0.2) | 6.9 (0.5)‡ | 5.7 (0.3)§‖ | |
| FEUA, % | ≤5 | |||
| Group A | 6.6 (0.5) | 9.5 (1.4)¶ | 6.5 (0.6) | |
| Group B | 5.4 (0.5) | 7.8 (1.0)¶ | 10.6 (1.0)¶# | |
| Serum creatinine, mg/dL | 0.70–1.40 | |||
| Group A | 1.09 (0.07) | 1.26 (0.09)† | 1.11 (0.08) | |
| Group B | 0.99 (0.04) | 1.03 (0.05)‡ | 1.13 (0.05)† | |
| CCR, mL/min | 90–140 | |||
| Group A | 82 (10) | 68 (8) | 79 (12) | |
| Group B | 115 (17) | 124 (29) | 89 (14)§ |
BMI = body mass index; SBP = systolic blood pressure; DBP = diastolic blood pressure; FPG = fasting plasma glucose; HbA1c = hemoglobin A1c; FFA = free fatty acid; FEUA = fractional excretion of uric acid; CCR = creatinine clearance rate.
Patients in group A received fenofibrate for 8 weeks followed by placebo for 8 weeks, and group B received placebo followed by fenofibrate.
P<0.001 versus baseline.
P<0.05 versus group A (Mann-Whitney U test).
P<0.01 versus baseline.
P<0.001 versus group A (Mann-Whitney U test).
P<0.05 versus baseline.
P<0.01 versus group A (Mann-Whitney U test).
As shown in Table III, serum concentrations of TG (P<0.001) and apo C2, C3, and E (all P<0.01) decreased significantly in group A after 8 weeks of fenofibrate therapy, and HDL-C and apo A1 and A2 significantly increased (all P<0.001). All of these parameters returned to near-baseline levels after 8 weeks of placebo administration.
Table III.
Serum concentrations of lipids and apolipoproteins during treatment in study patients. (Data are expressed as mean [SE].)∗
| Component | Normal Value | Baseline | 8 Weeks | 16 Weeks |
|---|---|---|---|---|
| TC, mg/dL | <220 | |||
| Group A | 238 (10) | 225 (10) | 232 (9) | |
| Group B | 245 (11) | 231 (8) | 220 (7)† | |
| TG, mg/dL | <150 | |||
| Group A | 352 (60) | 205 (47)‡ | 339 (110) | |
| Group B | 342 (65) | 262 (33)§ | 220 (36)† | |
| HDL-C, mg/dL | <40 | |||
| Group A | 40.0 (2.2) | 49.5 (2.5)‡ | 43.1 (2.5) | |
| Group B | 49.4 (5.1) | 47.4 (5.1) | 54.5 (6.4)‖ | |
| Apo A1, mg/dL | 119–155 (men) | |||
| 126–165 (women) | ||||
| Group A | 148.8 (5.3) | 172.0 (4.7)‡ | 152.2 (6.6) | |
| Group B | 163.3 (6.5) | 157.3 (8.4)¶ | 180.1 (7.3)‡§ | |
| Apo A2, mg/dL | 25.9–35.7 (men) | |||
| 24.6–33.3 (women) | ||||
| Group A | 34.2 (1.0) | 42.1 (1.1)‡ | 33.6 (1.3) | |
| Group B | 35.4 (1.4) | 34.5 (1.6)# | 43.0 (1.8)†# | |
| Apo B, mg/dL | 73–109 (men) | |||
| 66–101 (women) | ||||
| Group A | 140.8 (7.5) | 129.1 (8.4) | 130.5 (5.3)‖ | |
| Group B | 138.7 (7.5) | 137.1 (4.3) | 122.8 (6.0)† | |
| Apo C2, mg/dL | 1.8–4.6 (men) | |||
| 1.5–3.8 (women) | ||||
| Group A | 7.5 (0.7) | 6.2 (0.6)† | 7.4 (1.1) | |
| Group B | 7.5 (0.8) | 7.0 (0.8) | 6.6 (0.7) | |
| Apo C3, mg/dL | 5.8–10.0 (men) | |||
| 5.4–9.0 (women) | ||||
| Group A | 17.9 (1.8) | 14.0 (1.6)† | 18.7 (3.4) | |
| Group B | 20.0 (2.5) | 18.0 (2.0) | 16.5 (2.1)‖ | |
| Apo E, mg/dL | 2.7–4.3 (men) | |||
| 2.8–4.6 (women) | ||||
| Group A | 8.1 (0.8) | 6.3 (0.8)† | 9.1 (1.6) | |
| Group B | 8.6 (1.2) | 7.2 (0.8) | 7.1 (0.9)‖ |
TC = total cholesterol; TG = triglycerides; HDL-C = high-density lipoprotein cholesterol; apo = apolipoprotein.
Patients in group A received fenofibrate for 8 weeks followed by placebo for 8 weeks, and group B received placebo followed by fenofibrate.
P<0.01 versus baseline.
P<0.001 versus baseline.
P<0.01 versus group A.
P<0.05 versus baseline.
P<0.05 versus group A.
P<0.001 versus group A.
In group B, placebo administration for 8 weeks did not produce significant changes from baseline in any parameters. However, fenofibrate treatment for 8 weeks resulted in significant decreases in TG (P<0.01), apo C3 (P<0.05), and apo E (P<0.05). In addition, HDL-C (P<0.05), apo A1 (P<0.001), and apo A2 (P<0.01) were significantly increased by fenofibrate (Table III).
As shown in Table IV, serum concentrations of TG (group A, P<0.001; group B, P<0.001); apo C2 (group A, P<0.01), C3 (group A, P<0.01; group B, P<0.05), and E (group A, P<0.01; group B, P<0.05); and uric acid (group A, P<0.001; group B, P<0.01) were significantly decreased with fenofibrate compared with placebo. HDL-C and apo A1 and A2 were significantly increased with fenofibrate compared with placebo (all P<0.001 in both groups).
Table IV.
Mean (SE) changes in serum lipids, apolipoproteins, and uric acid concentrations.∗
| Component | 8 Weeks | 16 Weeks |
|---|---|---|
| TC, mg/dL | ||
| Group A | −13 (11) | +14 (10) |
| Group B | −14 (8) | −12 (9) |
| TG, mg/dL | ||
| Group A | −146 (55)† | +203 (75) |
| Group B | −80 (47) | −35 (27)† |
| HDL-C, mg/dL | ||
| Group A | +9.5 (1.5)† | −6.6 (1.8) |
| Group B | −2.0 (1.7) | +7.8 (2.1)† |
| Apo A1, mg/dL | ||
| Group A | +23.2 (4.7)† | −18.7 (5.6) |
| Group B | −5.9 (4.8) | +23.6 (5.8)† |
| Apo A2, mg/dL | ||
| Group A | +7.9 (1.1)† | −8.2 (1.6) |
| Group B | −0.9 (0.8) | +9.1 (1.9)† |
| Apo B, mg/dL | ||
| Group A | −11.7 (6.3) | +7.0 (5.2) |
| Group B | −1.6 (3.6) | −14.5 (6.2)§ |
| Apo C2, mg/dL | ||
| Group A | −1.2 (0.6)‡ | +1.2 (0.8) |
| Group B | −0.8 (0.5) | −0.3 (0.6) |
| Apo C3, mg/dL | ||
| Group A | −3.9 (1.8)‡ | +5.2 (2.1) |
| Group B | −1.9 (1.4) | −1.4 (1.6) |
| Apo E, mg/dL | ||
| Group A | −1.8 (0.7)‡ | +2.8 (1.2) |
| Group B | −1.3 (0.8) | −0.3 (0.8)§ |
| Uric acid, mg/dL | ||
| Group A | −2.0 (0.3)† | +1.7 (0.4) |
| Group B | −0.6 (1.4) | −1.6 (0.5)‡ |
TC = total cholesterol; TG = triglycerides; HDL-C = high-density lipoprotein cholesterol; apo = apolipoprotein.
Patients in group A received fenofibrate for 8 weeks followed by placebo for 8 weeks, and group B received placebo followed by fenofibrate.
P<0.001 versus placebo.
P<0.01 versus placebo.
P<0.05 versus placebo.
As shown in the figure, fenofibrate treatment in group A was associated with a significant reduction in UAE (P<0.05 vs baseline), which remained after 8 weeks of placebo administration (P<0.05). In group B, UAE was not changed significantly after placebo administration, but it was significantly decreased after fenofibrate therapy (P<0.01 vs baseline). Baseline UAE was similar in both groups; however, after 8 weeks, the difference in UAE was statistically significant between the 2 groups (P<0.05).
Figure.
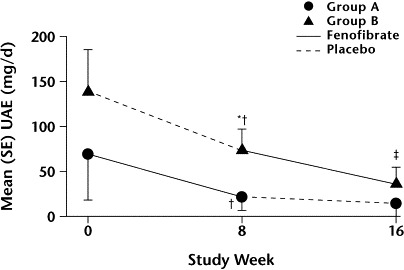
Mean (SE) urinary albumin excretion (UAE) with fenofibrate and placebo. ∗P<0.05 versus group A. †P<0.05 versus baseline. ‡P<0.01 versus baseline.
Using Spearman rank correlation analysis, changes in UAE were associated with significant changes in apo C2 (ρ = 0.43; P = 0.02) and apo C3 (ρ = 0.49; P = 0.01). However, no significant correlation was found in changes in serum levels of TG; HDL-C; apo A1, A2, or E; or uric acid.
Using multiple regression analysis, changes in serum TG, apo A1 and C3, and uric acid concentrations emerged as determinants of changes in UAE. These changes explained 49% of UAE reductions. Of these variables, the only association that was statistically significant was apo C3 (P = 0.01).
Discussion
In this study, in patients with hypertriglyceridemia and/or hyperuricemia, fenofibrate decreased mean serum concentrations of TG by 30% and uric acid by 27% and increased HDL-C by 20%. Fenofibrate-induced improvements in hypertriglyceridemia and hyperuricemia were associated with reductions in albuminuria (as assessed by UAE), despite no change in blood pressure or glycemia (as assessed by HbA1c). Multiple regression analysis showed that reductions in UAE were independently and significantly associated with decreases in serum apo C3 concentration. Although instructed not to do so, some patients may have changed their diet or exercise during the study, which may have contributed to the nonsignificant reduction in UAE during placebo administration. Apo C3 has been shown to inhibit the activity of lipoprotein lipase and the rate of uptake of very-low-density lipoprotein remnants by the liver,23 both of which result in prolonged residence time of TG-rich lipoproteins in the circulation. Hence, we suggest that fenofibrate-induced improvements in impaired metabolism of TG-rich lipoproteins may be associated with reductions in albuminuria in patients with hypertriglyceridemia and/or hyperuricemia.
Several possible explanations exist for these findings, and these explanations are not necessarily mutually exclusive. First, fenofibrate-induced reduction in albuminuria, which reflects widespread endothelial dysfunction,4 may represent an improvement in impaired endothelial function, as was recently demonstrated in chronic but not in acute hypertriglyceridemia.24–26 De Man et al26 reported that patients with chronic hypertriglyceridemia had impaired endothelium-dependent vasodilation mediated by the nitric oxide pathway, which was reversed with lipid-lowering therapy using atorvastatin. They suggested that lipids did not interfere directly with nitric oxide availability because they did not find changes in endothelial vasodilation in acute hypertriglyceridemia. However, other investigators27,28 have found small LDL particles to be associated with impaired in vivo endothelial function independent of TG concentration. As fenofibrate has been shown to increase small LDL size,29 fenofibrate-induced albuminuria reduction found in the present study may be associated with an increase in small LDL size, although we did not measure LDL size. In this context, it should be noted that ciprofibrate therapy improved endothelial function and reduced postprandial lipidemia in type 2 DM.30
Second, reductions in albuminuria found in fenofibrate-treated patients with hypertriglyceridemia and/or hyperuricemia may be associated with improved insulin resistance, because fenofibrate has been shown to reduce insulin resistance.31 Indeed, in the present study, fenofibrate treatment improved not only hypertriglyceridemia and low HDL-C but also hyperuricemia and microalbuminuria, all of which are features of the insulin resistance syndrome.32 Although no change in fasting serum insulin concentrations was found in the present study, fasting serum insulin concentration may not be an accurate estimate of insulin sensitivity.
Several limitations of this study deserve mention. The sample size was very small. Also, many patients were diabetic and hypertensive; consequently, they received other drugs in addition to fenofibrate or placebo.
The results of the present study show that fenofibrate-induced improvements in impaired TG-rich lipoprotein metabolism were associated with reductions in albuminuria. Long-term studies are needed to confirm our observations and to investigate putative mechanisms involved in reductions in albuminuria.
Conclusions
In our study population of patients with hypertriglyceridemia and/or hyperuricemia, fenofibrate-induced ameliorations of impaired TG-rich lipoprotein metabolism were associated with reductions in albuminuria. Further studies of patients who are not receiving ACE inhibitors should be performed.
Acknowledgements
This study was funded by a grant from Grelan Pharmaceutical Co., Ltd., Tokyo, Japan.
Members of the Fenofibrate Study Group: Tsutomu Kazumi, MD, PhD, Department of Food Sciences and Nutrition, School of Human Environmental Sciences, Mukogawa Women's University, Hyogo; Tsutomu Hirano, MD, PhD, First Department of Internal Medicine, Showa University School of Medicine, Tokyo; Gen Yoshino, MD, PhD, Department of Laboratory Medicine, Toho University School of Medicine, Tokyo; Masahiko Amano, MD, Department of Internal Medicine, Nishiwaki Municipal Hospital, Kobe; Tadanobu Chinzei, MD, Department of Internal Medicine, Kakogawa Municipal Hospital, Kobe; Takeo Goto, MD, and Shozo Miki, MD, Department of Internal Medicine, Takasago Municipal Hospital, Kobe; Joji Hari, MD, Department of Internal Medicine, Hyogo Prefectural Kakogawa Hospital, Kobe; Toshiki Hozumi, MD, and Yoshihiko Ishida, MD, Department of Internal Medicine, Hyogo Medical Center for Adults, Kobe; Yoshiki Nishizawa, MD, Second Department of Internal Medicine, Osaka City University School of Medicine, Osaka, Japan.
References
- 1.Mattock M.B, Morrish N.J, Viberti G. Prospective study of microalbuminuria as predictor of mortality in NIDDM. Diabetes. 1992;41:736–741. doi: 10.2337/diab.41.6.736. [DOI] [PubMed] [Google Scholar]
- 2.Yudkin J.S, Forrest R.D, Jackson C.A. Microalbuminuria as predictor of vascular disease in non-diabetic subjects. Islington Diabetes Survey. Lancet. 1988;2:530–533. doi: 10.1016/s0140-6736(88)92657-8. [DOI] [PubMed] [Google Scholar]
- 3.Jensen J.K, Feldt-Rasmussen B, Strandgaard S. Arterial hypertension, microalbuminuria, and risk of ischemic heart disease. Hypertension. 2000;35:898–903. doi: 10.1161/01.hyp.35.4.898. [DOI] [PubMed] [Google Scholar]
- 4.Deckert T, Feldt-Rasmussen B, Borch-Johnsen K. Albuminuria reflects widespread vascular damage. The Steno hypothesis. Diabetologia. 1989;32:219–226. doi: 10.1007/BF00285287. [DOI] [PubMed] [Google Scholar]
- 5.Stehouwer C.D, Stroes E.S, Hackeng W.H. von Willebrand factor and development of diabetic nephropathy in IDDM. Diabetes. 1991;40:971–976. doi: 10.2337/diab.40.8.971. [DOI] [PubMed] [Google Scholar]
- 6.Ferri C, Bellini C, Desideri G. Clustering of endothelial markers of vascular damage in human salt-sensitive hypertension: Influence of dietary sodium load and depletion. Hypertension. 1998;32:862–868. doi: 10.1161/01.hyp.32.5.862. [DOI] [PubMed] [Google Scholar]
- 7.Hirano T, Naito H, Kurokawa M. High prevalence of small LDL particles in non-insulin-dependent diabetic patients with nephropathy. Atherosclerosis. 1996;123:57–72. doi: 10.1016/0021-9150(95)05772-2. [DOI] [PubMed] [Google Scholar]
- 8.Bianchi S, Bigazzi R, Valtriani C. Elevated serum insulin levels in patients with essential hypertension and microalbuminuria. Hypertension. 1994;23:681–687. doi: 10.1161/01.hyp.23.6.681. [DOI] [PubMed] [Google Scholar]
- 9.Pedrinelli R, Giampietro O, Carmassi F. Microalbuminuria and endothelial dysfunction in essential hypertension. Lancet. 1994;344:14–18. doi: 10.1016/s0140-6736(94)91047-2. [DOI] [PubMed] [Google Scholar]
- 10.Pontremoli R, Sofia A, Ravera M. Prevalence and clinical correlates of microalbuminuria in essential hypertension: The MAGIC Study. Microalbuminuria: A Genoa Investigation on Complications. Hypertension. 1997;30:1135–1143. doi: 10.1161/01.hyp.30.5.1135. [DOI] [PubMed] [Google Scholar]
- 11.Kazumi T, Hozumi T, Ishida Y. Increased urinary transferrin excretion predicts microalbuminuria in patients with type 2 diabetes. Diabetes Care. 1999;22:1176–1180. doi: 10.2337/diacare.22.7.1176. [DOI] [PubMed] [Google Scholar]
- 12.Janssen W.M, de Jong P.E, de Zeeuw D. Hypertension and renal disease: Role of microalbuminuria. J Hypertens Suppl. 1996;14:S173–S177. [PubMed] [Google Scholar]
- 13.United Kingdom Prospective Diabetes Study (UKPDS) Group Intensive blood-glucose control with sulphonylurea or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 14.Nielsen S, Schmitz O, Møller N. Renal function and insulin sensitivity during simvastatin treatment in type 2 (non-insulin-dependent) diabetic patients with microalbuminuria. Diabetologia. 1993;36:1079–1086. doi: 10.1007/BF02374502. [DOI] [PubMed] [Google Scholar]
- 15.Tonolo G, Ciccarese M, Brizzi P. Reduction of albumin excretion rate in normotensive microalbuminuric type 2 diabetic patients during long-term simvastatin treatment. Diabetes Care. 1997;20:1891–1895. doi: 10.2337/diacare.20.12.1891. [DOI] [PubMed] [Google Scholar]
- 16.Elisaf M, Tsimichodimos V, Bairaktari E, Siamopoulos K.C. Effect of micronized fenofibrate and losartan combination on uric acid metabolism in hypertensive patients with hyperuricemia. J Cardiovasc Pharmacol. 1999;34:60–63. doi: 10.1097/00005344-199907000-00010. [DOI] [PubMed] [Google Scholar]
- 17.Saito Y. Prevention of coronary heart disease and lipid-lowering therapy in Japan. Eur Heart J Suppl. 2000;2(Suppl D):D49–D50. [Google Scholar]
- 18.Warnick G.R, Benderson J, Albers J.J. Dextran sulfate-Mg2+ precipitation procedure for quantitation of high-density-lipoprotein cholesterol. Clin Chem. 1982;28:1379–1388. [PubMed] [Google Scholar]
- 19.Ikeda T, Shibuya Y, Senba U. Automated immunoturbidimetric analysis of six plasma apolipoproteins: Correlation with radial immunodiffusion assays. J Clin Lab Anal. 1991;5:90–95. doi: 10.1002/jcla.1860050204. [DOI] [PubMed] [Google Scholar]
- 20.Bernard A.M, Lauwerys R.R. Continuous-flow system for automation of latex immunoassay by particle counting. Clin Chem. 1983;29:1007–1011. [PubMed] [Google Scholar]
- 21.The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med. 1997;157:2413–2446. doi: 10.1001/archinte.157.21.2413. [DOI] [PubMed] [Google Scholar]
- 22.Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183–1197. doi: 10.2337/diacare.20.7.1183. [DOI] [PubMed] [Google Scholar]
- 23.Jong M.C, Hofker M.K, Havekes L.M. Role of ApoCs in lipoprotein metabolism: Functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler Thromb Vasc Biol. 1999;19:472–484. doi: 10.1161/01.atv.19.3.472. [DOI] [PubMed] [Google Scholar]
- 24.Kusterer K, Pohl T, Fortmeyer H.P. Chronic selective hypertriglyceridemia impairs endothelium-dependent vasodilatation in rats. Cardiovasc Res. 1999;42:783–793. doi: 10.1016/s0008-6363(98)00331-9. [DOI] [PubMed] [Google Scholar]
- 25.Lewis T.V, Dart A.M, Chin-Dusting J.P. Endothelium-dependent relaxation by acetylcholine is impaired in hypertriglyceridemic humans with normal levels of plasma LDL cholesterol. J Am Coll Cardiol. 1999;33:805–812. doi: 10.1016/s0735-1097(98)00667-6. [DOI] [PubMed] [Google Scholar]
- 26.de Man F.H, Weverling-Rijnsburger A.W, van der Laarse A. Not acute but chronic hypertriglyceridemia is associated with impaired endothelium-dependent vasodilation: Reversal after lipid-lowering therapy by atorvastatin. Arterioscler Thromb Vasc Biol. 2000;20:744–750. doi: 10.1161/01.atv.20.3.744. [DOI] [PubMed] [Google Scholar]
- 27.Vakkilainen J, Mäkimattila S, Seppälä-Lindroos A. Endothelial dysfunction in men with small LDL particles. Circulation. 2000;102:716–721. doi: 10.1161/01.cir.102.7.716. [DOI] [PubMed] [Google Scholar]
- 28.Lupattelli G, Lombardini R, Schillaci G. Flow-mediated vasoactivity and circulating adhesion molecules in hypertriglyceridemia: Association with small, dense LDL cholesterol particles. Am Heart J. 2000;140:521–526. doi: 10.1067/mhj.2000.108508. [DOI] [PubMed] [Google Scholar]
- 29.Packard C.J. Overview of fenofibrate. Eur Heart J. 1998;19(Suppl A):A62–A65. [PubMed] [Google Scholar]
- 30.Evans M, Anderson R.A, Graham J. Ciprofibrate therapy improves endothelial function and reduces postprandial lipemia and oxidative stress in type 2 diabetes mellitus. Circulation. 2000;101:1773–1779. doi: 10.1161/01.cir.101.15.1773. [DOI] [PubMed] [Google Scholar]
- 31.Guerre-Millo M, Gervois P, Raspe E. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem. 2000;275:16638–16642. doi: 10.1074/jbc.275.22.16638. [DOI] [PubMed] [Google Scholar]
- 32.Hansen B.C. The metabolic syndrome X. Ann N Y Acad Sci. 1999;892:1–24. doi: 10.1111/j.1749-6632.1999.tb07782.x. [DOI] [PubMed] [Google Scholar]