

Abstract

The genome of Mycobacterium tuberculosis contains a gene, blaC, which encodes a highly active β-lactamase (BlaC). We have previously shown that BlaC has an extremely broad spectrum of activity against penicillins and cephalosporins but weak activity against newer carbapenems. We have shown that carbapenems such as meropenem, doripenem, and ertapenem react with the enzyme to form enzyme–drug covalent complexes that are hydrolyzed extremely slowly. In the current study, we have determined apparent Km and kcat values of 0.8 μM and 0.03 min–1, respectively, for tebipenem, a novel carbapenem whose prodrug form, the pivalyl ester, is orally available. Tebipenem exhibits slow tight-binding inhibition at low micromolar concentrations versus the chromogenic substrate nitrocefin. FT-ICR mass spectrometry demonstrated that the tebipenem acyl–enzyme complex remains stable for greater than 90 min and exists as mixture of the covalently bound drug and the bound retro-aldol cleavage product. We have also determined the high-resolution crystal structures of the BlaC–tebipenem covalent acylated adduct (1.9 Å) with wild-type BlaC and the BlaC–tebipenem Michaelis–Menten complex (1.75 Å) with the K73A BlaC variant. These structures are compared to each other and to other carbapenem–BlaC structures.
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis that results in over a million deaths annually.1,2 According to a report published by the WHO, one-third of the world’s population is latently infected with Mtb, with new infections occurring at a rate of about one per second.3,4 Global tuberculosis control is facing major challenges today.5 Current treatments require long-term adherence (at least 6 months) and are not effective against nonreplicating forms of TB.6 The emergence of multidrug-resistant (MDR) and extensive drug-resistant (XDR) strains of Mtb is also a major public health concern. In addition, coinfection with TB and HIV, especially in Africa, makes control activities more complex and limited. So, development of novel and more effective therapeutics is an immediate and urgent necessity.7
Starting with the first report of penicillin in 1929, the β-lactam family is the most often prescribed antibacterial family and includes penicillin derivatives, cephalosporins, monobactams, carbapenems, and others.8,9 Extremely high specificity and low toxicity has made them essential for the treatment of both Gram-positive and Gram-negative bacterial infections.10 This class of compounds has been rarely used against Mtb, and the lack of efficacy is caused by the expression of a chromosomally encoded gene (blaC) that provides resistance to most classes of β-lactams.11 BlaC is a class A β-lactamase (EC 3.5.2.6).12 It catalyzes the opening of the β-lactam ring via nucleophilic attack by an active site serine residue (Ser70) to generate the acyl enzyme followed by hydrolysis of the ester bond to generate the inactive ring-opened product.13−17 Carbapenems, like all β-lactam antibacterials that bind to and inhibit the peptidoglycan cross-linking transpeptidases, were specifically designed to resist the action of β-lactamases with activity against penicillins and cephalosporins. All approved carbapenems that are used clinically for the treatment of bacterial infections (imipenem, meropenem, ertapenem, doripenem, and biapenem) contain the 6-α-1R-hydroxyethyl substituent on the β-lactam ring that sterically blocks binding to other class A β-lactamases.18
Tebipenem pivoxil (TBPM-PI, ME1211) has been under development as the first orally available carbapenem for the treatment of otolaryngological and respiratory infections caused by drug-resistant Streptococcus pneumoniae in pediatric patients.19 It has a 1-(1,3-thiazolin-2-yl)azetidin-3-ylthio group at the C-2 position (Scheme 1) and was developed by Wyeth Lederle Japan in 1994.20 TBPM-PI is a prodrug that is quickly hydrolyzed to tebipenem (TEBI), and the absorption rate of TBPM-PI is higher than that of other prodrug-type β-lactam antibiotics.21 Tebipenem is approved in Japan for treating children, as these oral antibiotics are often better tolerated than infusions.22
Scheme 1. Structure of Tebipenem.

In our current study, we have performed an in vitro biochemical characterization of tebipenem (TEBI) with BlaC. We have also solved the high-resolution crystal structure of the Michaelis–Menten complex (1.7 Å) of TEBI and the covalent BlaC acylated adduct (1.9 Å). These are compared to covalent acyl-adducts of other carbapenems with BlaC.
Experimental Procedures
Materials
Tebipenem was kindly provided by Anacor Pharmaceuticals (Palo Alto, CA). Buffer reagents for crystallography were purchased from Hampton Research (Aliso Viejo, CA). Unless noted, other chemicals were from Sigma-Aldrich (St. Louis, MO).
Cloning and Purification of WT and Mutant BlaC
The blaC gene was amplified from genomic M. tuberculosis H37Rv DNA and cloned into pET28 using NdeI and HindIII. BlaC was expressed as an N-terminally truncated form, lacking the first 40 amino acids, as previously described.15 The same pET28-based plasmid was used as a template for site-directed mutagenesis to generate the K73A mutant form of BlaC.23 After both of the constructs were confirmed by sequencing, the plasmids expressing N-terminal His6-tagged BlaC WT and mutant proteins were transformed into Escherichia coli BL21/DE3 cells and cultured in LB broth at 37 °C. When the culture OD600 reached 0.6, the cultures were cooled to 16 °C, and protein expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). After continued shaking for 18 h, cells were harvested, resuspended in 25 mM Tris-HCl, pH 7.5, containing 300 mM NaCl, and disrupted by sonication. After centrifugation, the soluble extract was loaded onto a Ni-NTA agarose column (Qiagen) and eluted with 200 mM imidazole in 25 mm Tris-HCl, pH 7.5, containing 300 mM NaCl. The eluted fractions were dialyzed against the same buffer without imidazole. To remove the His-tag, the eluted protein was incubated with thrombin (Novagen Madison, WI) overnight at 4 °C (1.6 units/mg of protein). The cleaved protein was separated from the His6-tag peptide by size-exclusion chromatography using a HiLoad 26/60 Superdex 200 column (GE Healthcare Life Science, Uppsala, Sweden).
Inhibition Assays
The steady-state hydrolysis of tebipenem by BlaC was monitored at 297 nm (Δε = 9550 M–1 cm–1). For the inhibition assays, the hydrolysis of 100 μM nitrocefin was monitored at 486 nm (Δε = 20 500 M–1 cm–1) with 0.6 nM BlaC in the presence of 1 to 160 μM tebipenem. All assays were performed in 100 mM MES, pH 6.5, and reactions were initiated by the addition of BlaC.
The progress curves from the inhibition assays17 were fitted to the following equation
| 1 |
where [P] is the product concentration, vi and vs correspond to the initial and steady-state velocities, respectively, and kiso is the apparent first-order rate constant.
The general inhibition mechanism is modeled as
| 2 |
where k1 and k–1 represent the rate constants of the binding and dissociation of tebipenem and BlaC, and k2 and k3 represent the rate constants for the acylation and deacylation steps, respectively. kiso, determined from eq 1, was plotted against tebipenem concentration (eq 3) to obtain the values of k2 and k3 as well as K, which is equal to k–1/k1.
| 3 |
Km and kcat can then be calculated with the eqs 4 and 5
| 4 |
| 5 |
Mass Spectrometric Analysis
BlaC (50 μM) was incubated with 50 μM tebipenem at room temperature as previously described.15,17 Mass spectra were determined after 0, 30, 60, and 90 min incubation times using a 9.6 T Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Ionspec, Lake Forest, CA). The molecular weight of the protein species was calculated for the +25 charge state using the equation m = (m/z × 25) – 25 on the isotopic centroid.
Crystallization
Hanging-drop vapor diffusion was used for crystallization of both the WT and K73A BlaC variants. The composition of the well consisted of 0.1 M HEPES, pH 7.5, and 2 M NH4H2PO4, which makes the final pH of the well solution 4.1. Protein at a concentration of 12 mg/mL was mixed 1:1 with the well solution and incubated at 10 °C. The mutant was initially seeded with the native enzyme crystals, and then after iterative crystal growth, the pure mutant crystals were obtained. Repeated microseeding resulted in efficient crystal growth as well as improved morphology to produce diffraction-quality crystals of the mutant enzyme.
Data Collection and Refinement
Mineral oil was added to the solution as a cryo-protectant. Diffraction data were collected from a single frozen crystal using a RAXIS-IV++ detector mounted on a Rigaku RH-200 rotating anode (copper anode) X-ray generator. The data were processed using HKL2000.24,25 The previous structure of BlaC with bound NXL104 (PDB entry 4HFX)14 was used to phase the data using the CCP4 software suite.26 Multiple rounds of structural refinement and model building were performed in Refmac5,27 Phenix,28 and Coot.29 Structure figures were generated using PyMOL.30 Atomic coordinates and experimental structure factors have been deposited in the Protein Data Bank (Table 1). Table 1 lists the data collection statistics for the structures as well as the final refinement statistics.
Table 1. Summary of Data Collection and Refinement Statistic for the K73A BlaC–Tebipenem and WT BlaC–Tebipenem Complexes.
| data collection statistics | K73A BlaC–tebipenem complex | WT BlaC–tebipenem complex | |
|---|---|---|---|
| X-ray source | rotating anode | rotating anode | |
| wavelength (Å) | 1.5418 (Cu anode) | 1.5418 (Cu anode) | |
| temperature (K) | 100 | 100 | |
| resolution range (Å) | 30.00–1.75 | 30.00–1.90 | |
| reflection | 25 499 | 20 497 | |
| completeness (%) | 97.3 | 97.3 | |
| redundancy | 8.6 | 3.4 | |
| space group | P212121 | P212121 | |
| unit cell (Å) | |||
| a | 49.57 | 49.93 | |
| b | 67.56 | 68.03 | |
| c | 75.11 | 75.55 | |
| α = β = γ | 90.0° | 90.0° | |
| molecules per a.u. | 1 | 1 | |
| Refinement statistics | |||
| Rwork (%) | 15.27 | 16.73 | |
| Rfree (%) | 17.83 | 22.51 | |
| number of atoms | |||
| protein (chain A) | 2000 | 2019 | |
| phosphate (chain B) | 20 | 25 | |
| tebipenem (chain T) | 25 | 25 | |
| water (chain W) | 323 | 366 | |
| rms deviation | |||
| bond length (Å) | 0.008 | 0.006 | |
| bond angles (deg) | 1.363 | 1.210 | |
| average B-factors (Å2) | |||
| protein main chain | 10.21 | 16.27 | |
| protein side chain | 12.42 | 15.22 | |
| protein whole chain | 11.24 | 17.45 | |
| phosphate | 32.37 | 37.91 | |
| tebipenem | 25.59 | 33.06 | |
| water | 24.74 | 31.60 | |
| PDB accession code | 4QB8 | 4Q8I | |
Result and Discussion
Inhibition Studies
We attempted to directly monitor the steady-state hydrolysis of tebipenem by BlaC at 297 nm. The reaction rate remained unchanged when the tebipenem concentration was varied from 2 to 20 μM, suggesting that Km is smaller than 2 μM. The kcat value was estimated to be 0.04 min–1.
Carbapenems such as meropenem,16 doripenem, and ertapenem17 have been demonstrated to be poor substrates of BlaC. The deacylation step is extremely slow, thus trapping BlaC as the covalent acyl intermediate. Because of the tight binding of tebipenem to BlaC, the very slow rate of hydrolysis, and the modest extinction coefficient, we chose a second assay to determine the kinetic parameters and rate constants of acylation and deacylation for the hydrolysis of tebipenem. We performed an inhibition assay, where nitrocefin, a good substrate of BlaC (kcat ∼ 1800 min–1), was used as a reporter for the conversion of BlaC into the stable acyl adduct. As shown in Figure 1A, the hydrolysis of nitrocefin by BlaC was inhibited in the presence of tebipenem in a time- and concentration-dependent manner. The first-order rate constant kiso was determined after fitting the progress curves to eq 1 and then plotted against tebipenem concentration (Figure 1B). The values of k2 and k3 were determined to be 3.9 and 0.03 min–1, respectively, whereas the dissociation constant K was 122 μM. We then calculated the values of kcat (0.03 min–1) and Km (0.8 μM) using eqs 4 and 5. In Scheme 2, we compare the kinetic constants for tebipenem with meropenem, doripenem, and ertapenem and show the different C2 substitutents. Tebipenem is most similar to doripenem and ertapenem, with kcat and Km values that are below those of meropenem. Of the C2 substituents, only the meta-aminobenzoic acid substituent of doripenem makes contact with the enzyme after isomerization17 (see below).
Figure 1.
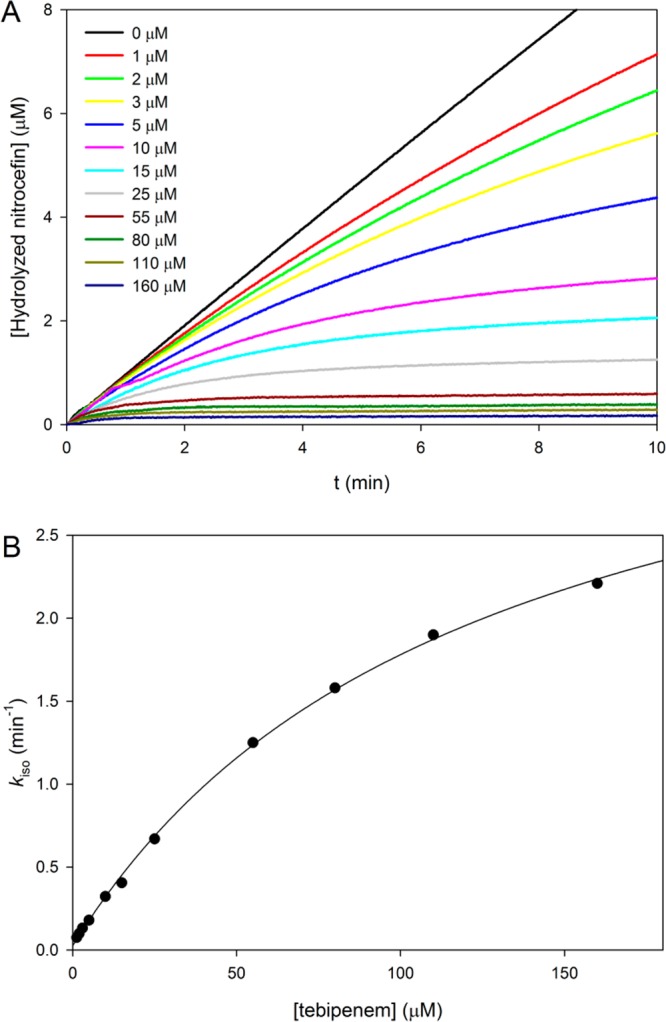
Determination of the inhibition rate constants. (A) Time-dependent hydrolysis of nitrocefin in the presence of various concentrations of tebipenem. The apparent first-order rate constants (kiso) were calculated by fitting the curves to eq 1. (B) Plot of kiso vs tebipenem concentration. The data were fit to eq 4 to obtain k2/K and k3 values.
Scheme 2. Kinetic Parameters of Carbapenem Hydrolysis by BlaC and Structure of the C2 Substituent.

Mass Spectrometry
Because of the slow deacylation reaction, the acyl BlaC intermediate could be detected by FT-ICR mass spectrometry. After incubating the enzyme with an equal amount of tebipenem for 10 min, peak A, corresponding to free BlaC, disappeared, and two new peaks were observed (Figure 2). Peak C corresponds to the intact tebipenem–BlaC adduct, whereas peak B corresponds to the adduct minus mass 44, which comes from the covalent intermediate after a retro-aldol fragmentation to release the C6-hydroxyethyl side chain as acetaldehyde (Scheme 3), as we proposed originally for meropenem.16 This fragmentation also occurs with both doripenem and ertapenem.17 Interestingly, no breakdown of the covalent intermediate was seen after 90 min, in contrast to that for the meropenem–BlaC adduct,16 suggesting an even slower deacylation of tebipenem compared to that of meropenem.
Figure 2.

FT-ICR mass spectra of BlaC and the (BlaC–tebipenem)25+ covalent adduct.
Scheme 3. Mechanism of Acylation, Fragmentation, and Hydrolysis of Tebipenem by BlaC.

Structures of the Covalent and Noncovalent Tebipenem Complexes
Lys73 is involved in the activation of the acylating nucleophile, Ser70 (Ambler notation). A mutation of Lys73 to alanine abolishes the acylation reaction,23 thus allowing us to trap the Michaelis–Menten complex by soaking the K73A BlaC mutant crystal with tebipenem. The three-dimensional structure was solved at a resolution of 1.75 Å, with Rwork 15.3 and Rfree 17.8 (Table 1). In this structure of the tebipenem Michaelis complex (Figure 3), there is clear electron density corresponding to the intact β-lactam ring, and Ser70 adopts two different conformations (Figure 3B). The C2 atom of the pyrroline ring is sp2 hybridized, again confirming that tebipenem is bound, but it is not covalently attached via Ser70. There are four hydrogen bonds to the tebipenem carboxylate (Figure 3C). The side chain hydroxyls of Thr237 and Thr239 contribute three, whereas a conserved active site water molecule (W1) contributes the fourth. The hydroxyl group of Ser70 interacts with the carbonyl of the β-lactam ring, as does a second active site water molecule (W2) that is involved in hydrolysis of the acylated enzyme. This same water molecule interacts with the C6 hydroxyl group. There are an additional two hydrogen bonds to the C6 hydroxyl group from Glu166 and Ser130. There are no interactions with the azetidine and thiazoline rings of the C2 substituent that differentiates tebipenem from other carbapenems.
Figure 3.

Crystal structure of BlaC K73A–tebipenem Michaelis–Menten complex. (A) Overall backbone structure of BlaC with the bound tebipenem molecule displayed by atom color (resolution 1.75 Å). (B) Fo – Fc omit map of the BlaC–tebipenem Michaelis–Menten complex contoured at 2.0σ. (C) Active site of BlaC–tebipenem Michaelis–Menten complex showing interactions and interatomic distances between tebipenem and active site residues.
The extremely slow deacylation, via hydrolysis, of the BlaC–tebipenem covalent adduct allowed us to structurally characterize the covalent tebipenem acyl intermediate by soaking the apo WT BlaC crystals with tebipenem. The three-dimensional structure was solved at a resolution of 1.9 Å, with Rwork 16.7 and Rfree 22.5 (Table 1). In this structure, Ser70 is covalently linked to the β-lactam ring-opened tebipenem. The electron density of the covalently bound tebipenem is shown under the calculated Fo – Fc omit map (Figure 4B), revealing that the β-lactam ring has been opened and that the initially formed Δ2 pyrroline has now isomerized to generate the S-Δ1 pyrroline in which C2 is sp3 hybridized (Scheme 2). The S stereochemistry is sterically preferred, with the C1 methyl group and the C2 substituent in a trans orientation on opposite sides of the pyrroline ring. This ring-opening and isomerization have been previously observed for carbapenems reacting with BlaC.16,17 The differences in the orientation of the linked azetidine and thiazoline rings are the result of the change in the hybridization of the C2 position of the pyrroline ring. The hydrogen-bonding pattern to the tebipenem carboxylate and C6 hydroxyethyl group are remarkably similar in the covalent complex (Figure S2). The one exception is that the hydrogen bonding between Ser130 and the C6 hydroxyethyl group observed in the tebipenem–K73A BlaC Michaelis complex is replaced in the covalent complex with an interaction between the hydroxyl group and the ε-amino group of K73, and Ser130 now hydrogen bonds exclusively to the pyrroline ring nitrogen. Most importantly, the conserved hydrolytic water (W2) is hydrogen-bonded to the C6 hydroxyl group, but it has been displaced from its position in the Michaelis complex and no longer interacts with the carbonyl group of the ester. This accounts for the stability of the tebipenem covalent complex to hydrolysis. There are again no significant interactions between the enzyme and the heteroatoms of either the azetidine or thiazoline rings. An overlay of the tebipenem Michaelis and covalent complexes is shown in Figure 5A.
Figure 4.

Crystal structure of BlaC–tebipenem covalent adduct. (A) Overall structure of BlaC with the tebipenem covalent adduct displayed by atom color (resolution 1.9 Å). (B) Fo – Fc omit map of the BlaC–tebipenem covalent adduct formed at the active site residue Ser70 contoured at 2.0σ. (C) Active site of BlaC–tebipenem covalent adduct showing interactions and interatomic distances between tebipenem and active site residues.
Figure 5.
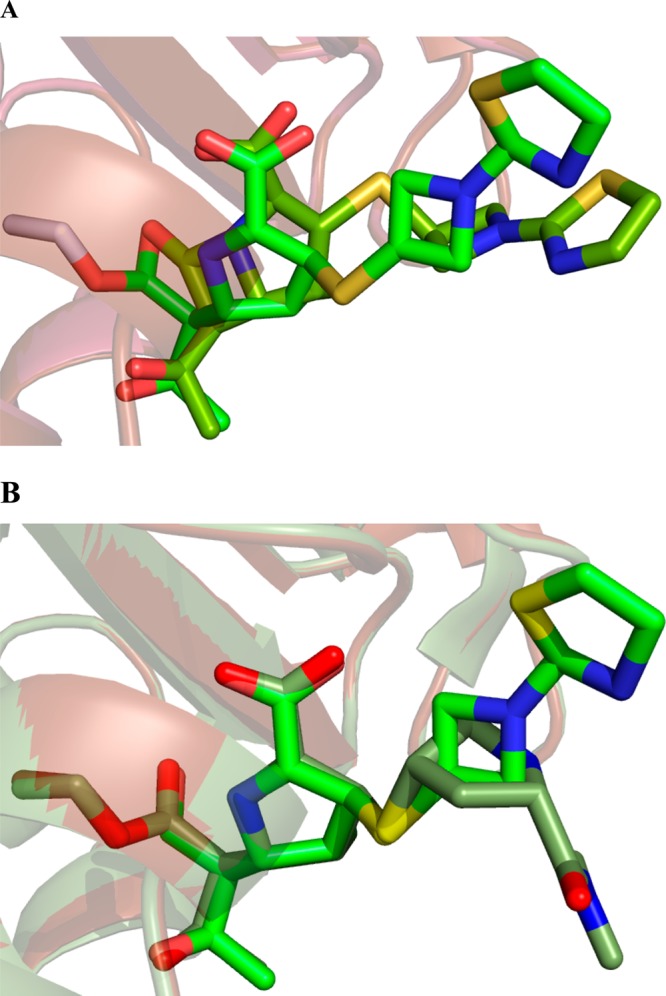
Overlay of the BlaC–tebipenem covalent adduct with the BlaC–tebipenem Michaelis–Menten complex (A) and the BlaC–tebipenem covalent adduct with the BlaC–meropenem covalent adduct (B) to show the significant difference in the orientation of the C2 substituent.
The structures of the covalent acyl intermediates of BlaC structures with bound tebipenem and bound meropenem were compared (Figure 5B). The two structures are extremely similar, with rmsd values of 0.11 (for all 1669 atoms). The pyrroline rings, carboxylate, and C6 hydroxyethyl group are superimposable, whereas the C2 side chains are found in quite different orientations because of their different chemical composition, and neither makes any contacts with the protein. The conserved active site hydrolytic water molecule is also in the same place and shares the same hydrogen-bonding partners in both structures. This water molecule interacts with the C6 hydroxyl group and is unable to make a close approach to the carbonyl carbon of the ester linkage (Table S1), ruling out the possibility that the very slow hydrolysis of bound carbapenems is due to the potential exclusion of the hydrolytic water molecule from the active site. The C6 hydroxyethyl substituent common to all the carbapenems is sterically accommodated at the active site of BlaC in both the Michaelis complex and in the covalent complex, with few changes in the interactions observed between the two (Figures S1 and S2).
These biochemical and structural studies reveal several potential advantages for the use of tebipenem in the treatment of MDR- and XDR-TB. The prodrug pivalyl ester is orally available, as opposed to meropenem, doripenem, and ertapenem. It is rapidly absorbed into intestinal cells21 and has high bioavailabilty with favorable pharmacokinetics.22 Tebipenem is an extremely poor substrate for the sole M. tuberculosis β-lactamase, BlaC, and hydrolyzes very slowly from the enzyme. The use of meropenem in combination with the β-lactamase inhibitor clavulanate has been shown to be effective in the treatment of MDR-TB infected patients,31 suggesting that tebipenem might also be useful in the treatment of the disease.
Acknowledgments
We thank the Almo lab staff for their support and Dr. Hui Xiao at Albert Einstein College of Medicine for his assistance in the FT-ICR analysis.
Supporting Information Available
Numbering of tebipenem and meropenem common atoms used in Table S1; side chain of meropenem; interactions of tebipenem and meropenem covalent adducts with M. tuberculosis β-lactamase; active site molecular description of BlaC–tebipenem Michaelis–Menten complex; and active site molecular description of BlaC–tebipenem covalent adduct. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The Protein Data Bank entry for the WT BlaC–tebipenem adduct is 4Q8I, and that for the mutant K73A BlaC–tebipenem Michaelis–Menten complex is 4QB8.
This work was supported by NIH grant AI60899 (to J.S.B.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- (2007) Robbins Basic Pathology, 8th ed. (Kumar V., Abbas A. K., Fausto N., and Mitchell R. N., Eds.) pp 516–522, Saunders Elsevier, Philadelphia, PA. [Google Scholar]
- Konstantinos A. (2010) Testing for tuberculosis. Australian Prescriber 33, 12–18. [Google Scholar]
- (2011) The sixteenth global report on tuberculosis, World Health Organization, Geneva, Switzerland, http://www.who.int/tb/publications/global_report/2011/gtbr11_full.pdf.
- (2010) Multidrug and extensively drug-resistant TB (M/XDR-TB): 2010 global report on surveillance and response, World Health Organization, Geneva, Switzerland, http://whqlibdoc.who.int/publications/2010/9789241599191_eng.pdf.
- Netto E. M.; Dye C.; Raviglione M. C. (1999) Progress in global tuberculosis control 1995–1996, with emphasis on 22 high-incidence countries. Global Monitoring and Surveillance Project. Int. J. Tuberc. Lung Dis. 3, 310–320. [PubMed] [Google Scholar]
- Fact sheet: a global perspective on tuberculosis, CDC, Atlanta, GA, http://www.cdc.gov/tb/publications/factsheets/default.htm.
- Barry C. E. 3rd; Blanchard J. S. (2010) Chemical biology of new drugs in development for tuberculosis. Curr. Opin. Chem. Biol. 14, 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin C.; Ghuysen J. M. (1998) Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol Mol. Biol. Rev. 62, 1079–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holten K. B.; Onusko E. M. (2000) Appropriate prescribing of oral beta-lactam antibiotics. Am. Fam. Physician 62, 611–620. [PubMed] [Google Scholar]
- Neu H. C. (1969) Effect of beta-lactamase location in Escherichia coli on penicillin synergy. Appl. Microbiol. 17, 783–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores A. R.; Parsons L. M.; Pavelka M. S. Jr. (2005) Genetic analysis of the beta-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to β-lactam antibiotics. Microbiology 151, 521–532. [DOI] [PubMed] [Google Scholar]
- Hall B. G.; Barlow M. (2005) Revised Ambler classification of β-lactamases. J. Antimicrob. Chemother. 55, 1050–1051. [DOI] [PubMed] [Google Scholar]
- Fisher J. F.; Meroueh S. O.; Mobashery S. (2005) Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105, 395–424. [DOI] [PubMed] [Google Scholar]
- Xu H.; Hazra S.; Blanchard J. S. (2012) NXL104 irreversibly inhibits the β-lactamase from Mycobacterium tuberculosis. Biochemistry 51, 4551–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugonnet J. E.; Blanchard J. S. (2007) Irreversible inhibition of the Mycobacterium tuberculosis β-lactamase by clavulanate. Biochemistry 46, 11998–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugonnet J. E.; Tremblay L. W.; Boshoff H. I.; Barry C. E. 3rd; Blanchard J. S. (2009) Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323, 1215–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay L. W.; Fan F.; Blanchard J. S. (2010) Biochemical and structural characterization of Mycobacterium tuberculosis β-lactamase with the carbapenems ertapenem and doripenem. Biochemistry 49, 3766–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassetti M.; Nicolini L.; Esposito S.; Righi E.; Viscoli C. (2009) Current status of newer carbapenems. Curr. Med. Chem. 16, 564–575. [DOI] [PubMed] [Google Scholar]
- Sato N.; Kijima K.; Koresawa T.; Mitomi N.; Morita J.; Suzuki H.; Hayashi H.; Shibasaki S.; Kurosawa T.; Totsuka K. (2008) Population pharmacokinetics of tebipenem pivoxil (ME1211), a novel oral carbapenem antibiotic, in pediatric patients with otolaryngological infection or pneumonia. Drug Metab. Pharmacokinet. 23, 434–446. [DOI] [PubMed] [Google Scholar]
- Hikida M.; Itahashi K.; Igarashi A.; Shiba T.; Kitamura M. (1999) In vitro antibacterial activity of LJC 11,036, an active metabolite of L-084, a new oral carbapenem antibiotic with potent antipneumococcal activity. Antimicrob. Agents Chemother. 43, 2010–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K.; Shirasaka Y.; Kuraoka E.; Kikuchi A.; Iguchi M.; Suzuki H.; Shibasaki S.; Kurosawa T.; Tamai I. (2010) Intestinal absorption mechanism of tebipenem pivoxil, a novel oral carbapenem: involvement of human OATP family in apical membrane transport. Mol. Pharmaceutics 7, 1747–1756. [DOI] [PubMed] [Google Scholar]
- Kijima K.; Morita J.; Suzuki K.; Aoki M.; Kato K.; Hayashi H.; Shibasaki S.; Kurosawa T. (2009) Pharmacokinetics of tebipenem pivoxil, a novel oral carbapenem antibiotic, in experimental animals. Jpn. J. Antibiot. 62, 214–240. [PubMed] [Google Scholar]
- Tremblay L. W.; Xu H.; Blanchard J. S. (2010) Structures of the Michaelis complex (1.2 Å) and the covalent acyl intermediate (2.0 Å) of cefamandole bound to the active site of the Mycobacterium tuberculosis β-lactamase K73A and E166A mutants. Biochemistry 49, 9685–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Potterton E.; Briggs P.; Turkenburg M.; Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr., Sect. D: Biol. Crystallogr. 59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Pannu N. S.; Murshudov G. N.; Dodson E. J.; Read R. J. (1998) Incorporation of prior phase information strengthens maximum-likelihood structure refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 54, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.3, Schrödinger, LLC.
- Payen M. C.; De Wit S.; Martin C.; Segsels R.; Muylle I.; Van Laethem Y.; Clumeck N. (2012) Clinical use of the meropenem–clavulanate combination for extensively drug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 16, 558–560. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.