

Abstract
Purpose.
Loss-of-function mutations in hemojuvelin (HJV) cause juvenile hemochromatosis, an iron-overload disease. Deletion of Hjv in mice results in excessive iron accumulation and morphologic changes in the retina. Here, we studied the retinal vasculature in Hjv−/− mice.
Methods.
Age-matched wild-type and Hjv−/− mice were used for fluorescein angiography and preparation of retinal cryosections, flat-mounts, and trypsin-digested blood vessels. Retinal angiogenesis was monitored by immunofluorescent detection of isolectin-B4, endoglin, and VEGF. Retinal vasculogenesis was monitored by immunofluorescent detection of collagen IV. Reactive gliosis was assessed based on the expression of glial fibrillary acidic protein and vimentin and CD11b/c as markers for Müller cells and microglia.
Results.
Between 18 and 24 months of age, retinas of Hjv−/− mice displayed marked disruptions in angiogenesis and vasculogenesis. Blood vessels in Hjv−/− mice were tortuous and dilated, with a decrease in the tight-junction protein occludin. There was also evidence of neovascularization in Hjv−/− mice with blood vessels appearing in the vitreous, which were leaky. There was reactive gliosis in these mice involving both Müller cells and microglia. Such changes were not detected at 2 weeks of age. Even at the age of 4 months, retinas of Hjv−/− mice were almost normal with changes just beginning to appear. Thus, the vascular changes in Hjv−/− mouse retinas represent an age-dependent phenomenon.
Conclusions.
Deletion of Hjv in mice leads to abnormal retinal angiogenesis/vasculogenesis, with proliferation of new, leaky blood vessels in the vitreous. These changes are accompanied with reactive gliosis involving Müller cells and microglia.
Keywords: iron, angiogenesis, retinal vasculature, gliosis
Deletion of Hemojuvelin (HJV) in mice is associated with excessive iron accumulation and morphologic changes in the retina. In this study, we find that HJV knockout mice have abnormal retinal angiogenesis and vasculogenesis along with robust reactive gliosis involving Müller cells and microglia.
Introduction
Hereditary hemochromatosis (HHC) is the most common autosomal recessive disorder in the United States, affecting approximately 1.5 million people.1–4 The prevalence of homozygous genotype is estimated to be 1 in 250 persons. The disease is characterized by increased iron absorption in the intestine all through life, consequently leading to accumulation of iron to toxic levels in multiple organs (liver, heart, pancreas, kidney, joints, etc.).1–4 Because of the lack of appreciation for the extremely high prevalence of the disease and the fact that multiple organs are affected with the involvement of a specific organ varying from person to person, most of the patients are treated for the symptoms without ever being diagnosed with HHC.
Until recently, it was believed that the retina was spared in hemochromatosis on the assumption that the blood–retinal barrier protects the retina from the systemic iron overload. Quite contrary to this belief, our studies with mouse models of hemochromatosis have shown unequivocally that the retina is indeed a target organ in this disease.5–9 In two different mouse models of hemochromatosis, iron accumulates in the retina to abnormal levels and causes oxidative stress and significant morphologic and structural damage. The involvement of the retina in hemochromatosis has now been confirmed in other laboratories.10–12 Interestingly, excessive iron in the retina leads to structural and morphologic changes similar to those found in AMD,13–15 identifying hemochromatosis as an important modifier of the progression and pathology of AMD. Curiously, just like AMD, hemochromatosis is also an age-related disease because, though genetic in origin, the clinical symptoms appear only at 50 to 60 years of age, thus underlining a key similarity between hemochromatosis and AMD. Furthermore, there is overwhelming evidence for iron accumulation in the retina in patients with AMD,16–18 again pinpointing the pivotal interrelationship among iron, hemochromatosis, and AMD. Another important disease in which hemochromatosis may play a contributing role is diabetes. Hemochromatosis causes diabetes due to iron-induced damage in pancreas; in addition, excessive iron has been implicated as a potential causative factor in diabetic retinopathy.19–21
One of the common features in diabetic retinopathy and the wet form of AMD is abnormal retinal angiogenesis with evidence of neovascularization.22–25 Because excessive iron is found in the retina in AMD, diabetes, and HHC, we asked whether HHC is associated with changes in retinal vasculature. In the present study, we addressed this question using hemojuvelin (Hjv−/−) mouse as an animal model of HHC. We have two mouse models of HHC: Hfe−/− mouse represents the prevalent adult form of the disease (Hfe: high Fe or histocompatibility complex associated with Fe regulation), whereas Hjv−/− mouse the less frequent, juvenile form of the disease. Both HFE and HJV promote the expression of the iron-regulatory hormone hepcidin in the liver and retina. Loss-of-function mutations in HJV lead to iron overload in tissues at a much younger age than loss-of-function mutations in HFE. Preliminary comparison of retinal vasculature in Hfe−/− mice and Hjv−/− mice indicated significant alterations in retinal vasculature only in the juvenile form of the disease (Hjv−/−) when examined at 18 to 24 months of age of the animals; this prompted us to conduct the present investigation with Hjv−/− mice.
Methods
Animals
Breeding pairs of Hjv± mice on a 129/SvEvTac (129/S) background, kindly provided by Nancy Andrews (Duke University School of Medicine, Durham, NC, USA), were used to establish our colony of Hjv+/+ and Hjv−/− mice. Genotyping, husbandry, and housing conditions for the mice have been described in one of our previous publications.7 For analysis of retinal vasculature, wild-type (Hjv+/+, n = 34) and knockout (Hjv−/−, n = 35) mice were used at different ages (2 weeks, 4, and 18–24 months). Experiments were approved by the Institutional Animal Care and Use Committee of Georgia Regents University (Augusta, GA, USA) and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Flat-Mounted Retinal Preparations
To visualize retinal vasculature and analyze retinas for new blood vessel formation, evidence of gliosis, and integrity of the blood–retinal barrier, a battery of markers were used. The sources and concentrations of primary and secondary antibodies are provided in the Table. Eyes were enucleated, fixed in 4% paraformaldehyde overnight and transferred to PBS. Retinas were dissected, washed with PBS, and incubated with Power Block (BioGenex, San Ramon, CA, USA). The retinas were incubated overnight at 4°C with antibodies specific for the markers for vasculature (isolectin-B4, collagen IV), new blood vessel formation (endoglin, also known as CD105, and VEGF), gliosis (glial fibrillary acidic protein [GFAP]), and blood–retinal barrier (occludin), followed by incubation with the appropriate secondary antibody for 1 hour at 37°C (Table). Retinas were cut partially at four places to allow the tissue to be flattened upon Superfrost microscope slides (Fisher Scientific, Pittsburgh, PA, USA). Immunofluorescent signals in these retinal flat mounts were visualized using an Axioplan-2 fluorescent microscope (Carl Zeiss, Göttingen, Germany) equipped with a high resolution microscope (HRM) camera (Carl Zeiss AG, Göttingen, Germany). Images were captured and processed using Zeiss Axiovision digital image processing software (version 4.7; Carl Zeiss). The number of capillary tufts per a given area of the retina was counted. The area of the capillary tufts was determined using ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA) and the tortuosity of the blood vessels was assessed using the FIJI program, an open-source platform for biological-image analysis.
Table.
Details of the Primary and Secondary Antibodies Used in Immunofluorescence Studies
|
Primary Antibody and Catalog Number |
Vendor |
Concentration |
Use as Marker |
Secondary Antibody |
| G. simplicifolia isolectin-B4 (B-1105) | Vector Labs, Burlingame, CA, USA | 1:133 | Marker for blood vessels | Texas Red avidin; Vector Labs |
| Collagen IV, rabbit polyclonal (SAB4500369) | Sigma-Aldrich, St. Louis, MO, USA | 1:200 | Marker for blood vessels | Alexafluor 555 (donkey anti-rabbit); Invitrogen, Eugene, OR, USA |
| CD105 (endoglin) rat anti-mouse (Lot 75619) | BD Biosciences, San Jose, CA, USA | 1:250 | Marker for angiogenesis | Alexafluor 555 (donkey anti-rat); Invitrogen |
| VEGF mouse monoclonal IgM (AB 38909) | Abcam, Cambridge, MA, USA | 1:250 | Marker for angiogenesis | Alexafluor 488 (donkey anti-mouse); Invitrogen |
| GFAP rabbit polyclonal (Lot# 00019620) | DakoCytomation, Carpenteria, CA, USA | 1:100 | Marker for stressed Müller cells and astrocytes | Alexafluor 488 (goat anti-rabbit); Invitrogen |
| CD11b/c equivalent antibody mouse monoclonal (Ab 33827) | Abcam | 1:200 | Marker for microglia | Alexafluor 555 (donkey anti-mouse); Invitrogen |
| Vimentin goat anti-human (AB1620) | Millipore, Billerica, MA, USA | 1:250 | Marker for Müller cells | Alexafluor 555 (mouse anti-goat); Invitrogen |
| Mouse monoclonal(AB 18401) | Abcam | 1:200 | Isotype control (VEGF study) | Alexafluor 488 (donkey anti-mouse); Invitrogen |
Detection of Vascular Markers in Retinal Cryosections by Immunofluorescence
Retinal cryosections were prepared from Hjv+/+ and Hjv−/− mice per our published method.7 Sections were fixed with 4% paraformaldehyde, washed with PBS-Triton X-100, incubated with Power Block and then incubated with primary antibody for either 3 hours at 37°C or overnight at 4°C; the sections were then washed thrice with PBS-Triton X-100 followed by incubation with secondary antibody for 1 hour at 37°C. Sections were again washed with PBS-Triton X-100 and coverslipped with Fluoroshield with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO, USA) to label nuclei. Sections were examined by epifluorescence as described above for the flat-mount preparations. The intensity of fluorescence was analyzed using Metamorph Image Analysis software (version 6.3; Molecular Devices, Sunnyvale, CA, USA). Similar exposure levels were used to compare the fluorescence intensity between images.
Isolation of Retinal Vasculature by Trypsin Digestion
Retinal vessels were isolated per the method of Stitt et al.26 In brief, freshly enucleated eyes were fixed with 2% paraformaldehyde overnight. Retinas were dissected, washed in PBS and incubated with 3% crude trypsin in 20 mM Tris buffer, pH 8, at 37°C (with shaking) for 2 hours until vitreous separated from retina. To separate vasculature from the remaining retinal tissue, retina was soaked in several washes of 5% and 2% Triton X-100. After the transparent vasculature was visualized, it was gently flattened onto the microscope slide, using a stereomicroscope, and the preparation was allowed to dry for several days. Vasculature was immunostained for occludin and isolectin-B4 and examined by fluorescent microscope as described above.
Fluorescein Angiography
To evaluate the changes in retinal vasculature and permeability in vivo, fluorescein angiography was preformed following our published methods.27 Briefly, mice were anesthetized using 20-μL intramuscular injection of rodent anesthesia cocktail (Ketamine 100 mg/mL, Xylazine 30 mg/mL, Acepromazine 10 mg/mL). Pupils were dilated using 1% tropicamide (Bausch & Lomb, Rochester, NY, USA). The mice were then placed on the imaging platform of the Phoenix Micron III retinal imaging microscope (Phoenix Research Laboratories, Pleasanton, CA, USA) and Goniovisc 2.5% (hypromellose; Sigma Pharmaceuticals, LLC, Monticello, IA, USA) was applied liberally to keep the eye moist during imaging. Mice were administered 10 to 20 μL fluorescein sodium (10% Lite; Apollo Ophthalmics, Newport Beach, CA, USA), while also receiving Gioniovisc 2.5%, and rapid acquisition of fluorescent images ensued for approximately 5 minutes. Fluorescein leakage was compared between wild-type and Hjv−/− mouse retinas by quantifying the fluorescence intensities collected after 1, 2, and 3 minutes following fluorescein injection. ImageJ software was used for this purpose. Statistical analysis was performed using the GraphPad software (La Jolla, CA, USA).
Results
Evaluation of the Vasculature
Previous studies of the retinas of Hjv−/− mice used retinal cryosections to investigate retinal architecture.7 Gross disruption was observed in several retinal layers of the Hjv−/− mice at the age of 18 months and older. This disruption corresponded with markedly increased iron accumulation compared with age-matched controls. Such changes were not observed in younger mice. These studies however did not focus on retinal vasculature. Therefore, mice in the age range of 18 to 24 months were used in the present study to evaluate the retinal vasculature in wild-type and Hjv−/− mice (age-matched). We also examined the retinal vasculature in some mice at younger ages (2 and 16 weeks) to determine if there are any vascular changes during the time of retinal vascular development (2 weeks) and at young age after retinal vascular development (16 weeks).
The retinas were incubated with isolectin-B4 to label endothelial cells in frozen sections and flat-mount preparations. Wild-type mouse retinas labeled with isolectin-B4 with DAPI stain to label nuclei showed normal retinal structure with evenly distributed endothelial cells within the retina. Blood vessels were observed in the ganglion cell layer (GCL), inner nuclear layer (INL), and inner and outer plexiform layers. In contrast, the retinas of the Hjv−/− mice displayed marked disruptions, an observation that confirms earlier findings.7 Isolectin-B4–stained blood vessels were observed not only in the retina but also within the vitreous of Hjv−/− mice, indicating formation of aberrant blood vessels (Figs. 1A, 1B). Isolectin-B4 staining was also performed with retinal flat-mount preparations (Fig. 1C). Compared with the normal vascular architecture observed in wild-type mouse retina, the vasculature in Hjv−/− mouse retinas were tortuous, dilated, and thickened. In addition, berry-like vascular masses were also observed in Hjv−/− mouse retinas. Most of the Hjv−/− mice (>90%) revealed these vascular alterations, which were evident in the central as well as the peripheral parts of the retina, while the vascular masses were observed more in the superficial plexus of the retina.
Figure 1.

Evidence of neovascularization in Hjv−/− mouse retinas as detected by isolectin-B4 staining. (A, B) Retinal cryosections from Hjv+/+ and Hjv−/− mice were incubated with biotinylated isolectin-B4, a specific endothelial cell marker, to label blood vessels. Blood vessels growing into the vitreous were detected in Hjv−/− mouse retina; arrows point to the same blood vessels observed in the vitreous of these mutant mice at low and high magnification. Calibration bar: 50 μm. (C) Retinal flat-mounts were stained with biotinylated isolectin-B4 to label retinal vasculature. Neovascular tufts and vascular masses (arrows) were observed in retinas of Hjv−/− mice compared with normal vasculature in retinas of wild-type mice. Calibration bar: 50 μm. (D) Quantification of the data obtained from metamorphic analysis of the color intensity of the retinas stained with isolectin-B4 (**P < 0.01, n = 3 mice for each genotype). ONL, outer nuclear layer.
We also examined the retinal vasculature with collagen IV staining. The berry-like masses observed with isolectin-B4 staining also stained with collagen IV. These masses were found attached to multiple branching blood vessels, a feature observed exclusively in Hjv−/− mouse retinas (Figs. 2A, 2B). Taken together, these data show abnormal proliferation of blood vessels involving both the endothelium (isolectin-B4) and the basal lamina (collagen IV), suggesting angiomatous proliferation in Hjv−/− mouse retinas. We counted the number of tufts in blood vessels per a given retinal area (13 mm2) and found the number to be much higher in Hjv−/− mice than in wild-type mice (Figs. 2C, 2D). Further, the blood vessels in the knockout mouse retinas were tortuous; the tortuosity index, measured as the branch length or Euclidean distance, was significantly greater for the blood vessels in Hjv−/− mouse retinas than for those in wild-type mice (Fig. 2D).
Figure 2.

Evidence of neovascularization in Hjv−/− mouse retina. Retinal flat-mounts were stained with collagen IV to label retinal vasculature. (A) Superficial vascular mass was noted at the periphery of the retina in Hjv−/− mice. (B) Blood vessels in Hjv−/− mice showed marked tortuosity with vascular masses (arrows) extending out of the vessel wall compared with normal appearing vasculature in wild-type mouse retina. (C, D) Isolectin-B4–stained retinal flat-mounts were used to count capillary tufts per a given retinal area and to quantify the vessel tortuosity. Scale bar: 20 μm. Data are representative of the experiments performed in three wild-type mice and three Hjv−/− mice.
Assessment of Angiogenesis
Endoglin (CD105) is a proliferation-associated and hypoxia-inducible protein abundantly expressed in angiogenic endothelial cells.28,29 It is used as a marker for new blood vessels. To investigate new blood vessel formation in the Hjv−/− mouse retina, endoglin expression was analyzed by immunofluorescence in retinal cryosections and retinal flat-mounts. At the exposure at which endoglin was not detected in the neural retina of wild-type mice, the angiogenic marker was detectable in multiple retinal layers of the Hjv−/− mice, including the ganglion cell and INLs, providing evidence of neovascularization in Hjv−/− mouse retinas (Figs. 3A, 3B). Endoglin-stained retinal flat-mounts at low (Fig. 3C) and high (Fig. 3D) magnification revealed markedly branching, tortuous, and dilated blood vessels. There was a marked increase in endoglin immunoreactivty in the Hjv−/− mice compared with age-matched wild-type mice (Fig. 3E).
Figure 3.

Increased expression of endoglin (CD105) in Hjv−/− mouse retinas. (A, B) Fluorescent immunodetection of endoglin (red), a marker of neovascularization, was performed in retinal cryosections of Hjv+/+ and Hjv−/− mice; DAPI (blue) was used to label nuclei. There was minimal detection of endoglin in Hjv+/+ mouse retinas whereas endoglin levels were markedly increased in Hjv−/− mouse retinas, particularly where the blood vessels are predominant (GCL, inner plexiform layer, and INL). Scale bar: 50 μm. (C, D) Retinal flat-mounts were stained for endoglin and assessed at low magnification (C) and high magnification (D). The retinas from Hjv−/− mice showed markedly branching, tortuous, dilated blood vessels with increased endoglin staining compared with normal appearing and less endoglin-stained retinas from Hjv+/+ mice. Scale bar: 50 μm (C) and 20 μm (D). (E) Quantification of the data obtained from metamorphic analysis of the color intensity of the retinas stained with CD105 in (C) (*P < 0.05, n = 3 mice for each genotype).
To confirm neovascularization in Hjv−/− mouse retinas, experiments to detect VEGF were performed. Vascular endothelial growth factor is a potent endothelial cell mitogen. It stimulates proliferation, migration, and tube formation resulting in new blood vessels.30,31 Retinal cryosections of wild-type and Hjv−/− mice were immunostained for VEGF and isolectin-B4 (Fig. 4). Vascular endothelial growth factor expression in Hjv−/− mouse retina was markedly increased and was localized in blood vessels as evident from the colocalization of the marker with isolectin-B4. The data were obtained using a fluorescence microscope in studies in which IgG1 was used instead of the primary antibody (isotype control) or when the primary antibody was omitted (negative control) and the exposure time was held constant. There was no signal detected in these negative controls; data are shown in Figure 4 for the isotype control. Metamorph analysis of the sections indicated approximately 2-fold increase in VEGF levels in the Hjv−/− retinas (Fig. 4B).
Figure 4.

Increased expression of VEGF in Hjv−/− mouse retinas. (A) Retinal cryosections from Hjv+/+ and Hjv−/− mice were immunostained for VEGF (green), a marker of neovascularization, and isolectin-B4 (red), a marker of blood vessels. The levels of VEGF were markedly increased in the Hjv−/− mouse retinas compared with wild-type (Hjv+/+) mouse retinas. The far left panel is a negative control in which an isotype antibody (IgG1 was used instead of the primary antibody, but with the secondary antibody). Scale bar: 50 μm. (B) Quantification of the intensity levels of VEGF immunofluorescence. **P < 0.01, n = 3 mice for each genotype; Scale bar: 50 μm.
Evidence of Reactive Gliosis in Hjv−/− Mouse Retina
We investigated whether these new blood vessels seen in Hjv−/− mouse retinas were associated with activation of glial cells, including macroglia (astrocytes and Müller) and microglia. This was done by the immunofluorescent detection of the GFAP, an intermediate filament protein that is a major constituent of astrocytes, in retinal cryosections and flat-mount preparations from wild-type and Hjv−/− mouse retinas. Under normal conditions, Müller cells contain low levels of GFAP; however, GFAP expression is strongly upregulated under a variety of conditions including retinal degeneration, retinal detachment, and ischemia.32 Significantly higher levels of GFAP were detected in retinal cryosections of Hjv−/− mice compared with wild-type mice (Figs. 5A, 5B). The astrocytes in the vicinity of the GCL were positive in wild-type mouse retinas and a low level GFAP was present in the outer plexiform layer; in contrast, GFAP levels were increased dramatically in Hjv−/− mouse retinas. Labeling of the Müller cell radial fibers was quite prominent in Hjv−/− mice compared with wild-type mice. The low level of Müller cell activation seen in wild-type mouse retinas was expected at old age as the mice used in the study were 18- to 24-months old. Glial fibrillary acidic protein labeling was also investigated in flat-mount preparations along with isolectin-B4 staining to detect retinal blood vessels (Figs. 5C, 5D). In wild-type mouse retinas, the presence of GFAP-positive astrocytes was evident; these astrocytes had a normal star-shaped appearance. A few of these cells were found to ensheath blood vessels. In contrast, there were more GFAP-labeled glia in Hjv−/− mouse retinas; the cells however had a different morphologic appearance. They exhibited more diffuse branching patterns and a ragged appearance. In addition, many of these glial cells were found in association with isolectin B4-labeled vessels, and the vessels themselves showed abnormal masses growing out of the blood vessels. These findings suggest presence of angiomas in Hjv−/− mouse retinas.
Figure 5.

Increased expression of GFAP in Hjv−/− mouse retinas. Glial fibrillary acidic protein levels were detected by immunofluorescence in retinal cryosections (A, B) and flat-mount preparations (C, D). Retinal cryosections of Hjv+/+ and Hjv−/− mice were incubated with an antibody against GFAP followed by incubation with a secondary antibody (green). Glial fibrillary acid protein immunoreactivity in both astrocytes and Müller cells was much higher in Hjv−/− retinas than in Hjv+/+ retinas. Retinal flat-mount preparations from Hjv+/+ and Hjv−/− mice were immunostained for GFAP (green) and isolectin-B4 (red) to visualize the vasculature. Altered vasculature with vascular masses extending out of the blood vessels and hypertrophied astrocytes with thickened and enlarged processes and increased GFAP expression (ragged appearance) were evident in Hjv−/− retinas, suggesting reactive astrocytes. Such changes were not evident in wild-type retinas. Data are representative of the experiments performed in three wild-type mice and three Hjv−/− mice. Scale bar: 50 μm.
Retinal frozen sections and flat-mount preparations were examined for microglia reactivity using a specific antibody, CD11b/c (Fig. 6). Microglia localization was more prominent in the blood vessels of the Hjv−/− mouse retinas than of the wild-type mouse retinas. Metamorph analysis of the sections indicated a significant increase in GFAP, CD11b/c, and vimentin staining in the Hjv−/− retinas (Figs. 6E–G). The retinal flat-mount evaluation with collagen IV revealed increased staining of astrocytes in Hjv−/− mouse retinas than in wild-type mouse retinas (Fig. 7A). These data agree with literature reports on the positive expression of collagen IV in reactive astrocytes.31,32 CD11b/c staining of retinal flat-mounts revealed elongated vascular microglia (amoeboid) in Hjv−/− mouse retinas (Fig. 7B). In contrast, normal, nonactive, resting microglia were found in wild-type mouse retinas.
Figure 6.

Evidence of reactive gliosis in Hjv−/− mouse retinas. Retinal cryosections from Hjv+/+ and Hjv−/− mice were immunostained for the astrocyte marker GFAP (A, C), microglia marker CD11b/c (B, C) and Müller cell marker vimentin (D). Hjv−/− retinas showed activated astrocytes and microglia, mainly in the GCL and INL where the blood vessels are predominant; in addition, the Müller cell radiating fibers were more extensively stained with vimentin in Hjv−/− retinas compared with Hjv+/+ mice retinas. Quantification of the data obtained from metamorphic analysis of the color intensity of the retinas stained with GFAP (E) CD11b/c (F) and vimentin (G). **P < 0.01; ***P < 0.001; n = 3 mice for each genotype. Calibration bar: 50 μm.
Figure 7.

Additional evidence of reactive gliosis in Hjv−/− mouse retinas. Retinal flat-mounts from Hjv+/+ and Hjv−/− mice were immunostained for collagen IV, another marker for reactive astrocytes (A) and CD11b/c, a marker for microglia (B). Hjv+/+ retinas showed small rounded, resting microglia, which are normally found encased within the wall of the blood vessels. In contrast, Hjv−/− retinas showed amoeboid, rod-shaped reactive microglia. In addition, collagen IV staining became evident in astrocytes in Hjv−/− mouse retinas while the staining was much less in astrocytes in wild-type mouse retinas. Data are representative of the experiments performed in two wild-type mice and three Hjv−/− mice. Scale bar: 20 μm.
Assessment of the Blood–Retinal Barrier
Newly formed blood vessels are often immature and leaky.33–35 The data showing new blood vessel formation in retinas of Hjv−/− mice prompted investigation of the tight-junction protein occludin, a marker for an intact blood–retinal barrier. Maintenance of the blood-retinal barrier is essential for retinal homeostasis. The tight-junction protein occludin contributes to the retinal permeability barrier and is present in endothelial cells. Occludin levels were investigated by immunofluorescence. Retinas were digested with trypsin to permit visualization of blood vessels, and were subsequently subjected to dual-labeling immunofluorescence methods to detect occludin and isolectin-B4. The isolectin-B4 clearly labeled blood vessels in wild-type and Hjv−/− mouse retinas (Fig. 8). There was robust expression of occludin in wild-type mouse retinal blood vessels; however, the expression was markedly reduced in Hjv−/− mouse retinal blood vessels (Figs. 8A, 8D), indicating compromised inner blood–retinal barrier in Hjv−/− mice. There were, however, vessel structures in Hjv−/− mouse retinas with intense labeling for occludin; these structures represent angiomas and capillary tufts with increased density of endothelial cells. Such structures were less numerous in wild-type mouse retinas.
Figure 8.

Decreased expression of occludin in Hjv−/− mouse retinas. (A–C) Trypsin-digested retinal blood vessel preparations were immunostained for isolectin-B4 (red) and occludin (green). (D) Quantification of the data obtained from metamorphic analysis of the color intensity of the retinas stained with occluding. *P < 0.05; n = 3 mice for each genotype; Scale bar: 50 μm.
Live Hjv−/− mice were also subjected to fluorescein angiography to evaluate the retinal vasculature and to determine whether permeability was altered. In wild-type mice, there was normal filling of retinal blood vessels with fluorescein and there was no leakage of fluorescein into the surrounding area (Fig. 9A), reflecting the intactness of the blood–retinal barrier. In contrast, when the age-matched Hjv−/− mice were subjected to fluorescein angiography, neovascularization and fluorescein leakage were observed. There was a significant increase in fluorescence intensity resulting from the leakage in Hjv−/− mouse retinas compared with wild-type mouse retinas. The presence of leaky blood vessels was observed in all four Hjv−/− mice examined (age, 18–24 months), but with the magnitude of leakage varying from mouse to mouse. The fluorescein staining intensity, monitored as a measure of the leakage, was significantly higher in Hjv−/− mouse retinas than in wild-type mouse retinas (Fig. 9B). Furthermore, the large blood vessels in Hjv−/− mouse retinas were more dilated than those in wild-type mouse retinas.
Figure 9.

Evidence of leaky blood vessels in Hjv−/− mouse retinas. (A) Fluorescein angiography was performed in age-matched and live wild-type and Hjv−/− mice to evaluate the retinal vasculature and to determine whether permeability was altered. Retinas of the Hjv−/− mice showed neovascularization (small white arrows) as well as increased perivascular fluorescein leakage (large white arrows). Wild-type mouse retinas showed normal blood vessels with no evidence of leakiness. Data are representative of the experiments performed in four mice for each genotype. (B) Relative fluorescence intensity in wild-type and Hjv−/− mouse retinas.
Since hemochromatosis is an age-dependent disease in terms of detectable phenotypical changes in tissues and in retina, the present study was carried out primarily to assess the disease-associated changes in retinal vasculature in Hjv−/− mice at old age (18–24 months). To confirm that the observed changes in retinal vasculature in these mice are indeed age-dependent, we also examined the retinas of wild-type and knockout mice at younger ages (2 weeks and 4 months; three mice each). There were no detectable changes in retinal vasculature in the knockout mice at 2 weeks of age (Fig. 10A). At the age of 4 months also, the architecture of the retinal vasculature was almost normal in the knockout mice, with angiomas and vascular tufts just beginning to appear (Figs. 10B, 10C). These results demonstrate that the vascular changes observed in hemochromatosis mouse retinas represent an age-dependent phenomenon.
Figure 10.
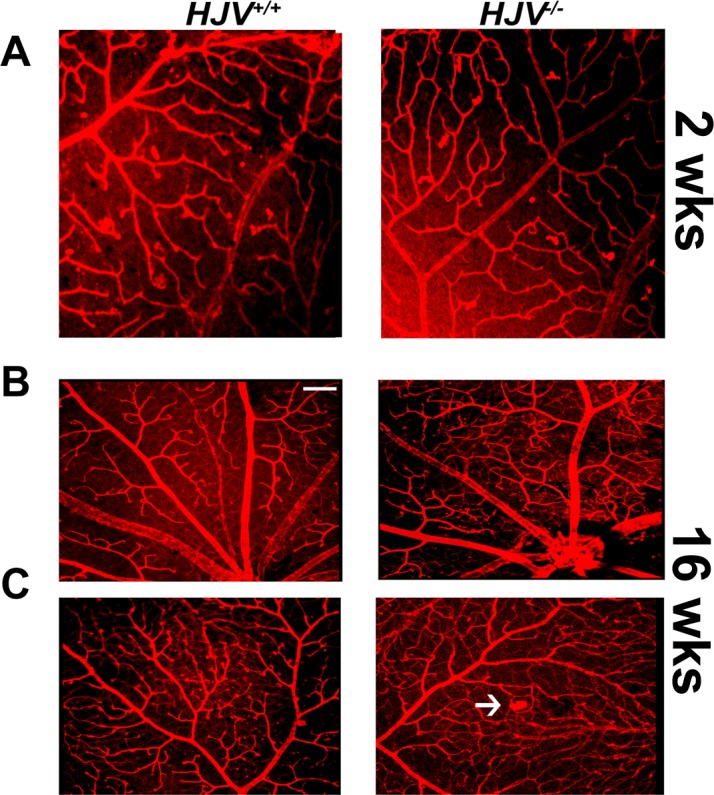
Retinal vasculature in Hjv−/− mice at 2 and 16 weeks of age. Flat-mount preparations of the retinas from wild type and Hjv−/− mice at 2-weeks of age (A) and 16-weeks of age (B, C) were labeled with isolectin-B4 for endothelial cells. The appearance of capillary tufts, marked by white arrows, was evident in Hjv−/− mouse retinas at 16-weeks of age.
Discussion
Hemojuvelin is a critical regulator of iron homeostasis, and it is expressed in mammalian retina in all cell types.1–4 Inactivating mutations in HJV lead to excessive accumulation of iron in systemic circulation, consequently leading to iron overload in multiple organs, including the retina. HFE is another important regulator of iron homeostasis, and mutations in HFE also lead to excessive iron accumulation in systemic circulation, thus resulting in iron overload in the retina and other organs.1–4 Both proteins regulate iron by controlling the expression of the iron-regulatory hormone hepcidin.36 It is generally believed that hepcidin is secreted only by the liver, but our studies have shown that this hormone is also made in the retina, most likely for the local control of iron homeostasis.37 While loss-of-function mutations in HFE are the cause of the adult form of the iron-overload disease hemochromatosis, loss-of-function mutations in HJV cause juvenile hemochromatosis, highlighting the difference in the time-dependent accumulation of iron in organs and the resultant failure of organ function between the two forms of the disease. It is therefore apparent that loss of HJV function leads to a more rapidly progressing disease than the one caused by the loss of HFE function.
The present study was undertaken to evaluate the influence of iron overload on retinal angiogenesis in the mouse model of juvenile hemochromatosis (Hjv−/− mouse). We also examined the potential changes in retinal angiogenesis in Hfe−/− mice and found that alterations in retinal vasculature are more severe in Hjv−/− mice than in Hfe−/− mice. This suggests that the retina as a target organ for iron-induced damage is affected much more rapidly in juvenile hemochromatosis than in adult hemochromatosis. The more accelerated accumulation of iron in systemic blood and in tissues in juvenile hemochromatosis than in adult hemochromatosis is the most likely reason for this phenomenon.
We made three important observations in the Hjv−/− mouse model of juvenile hemochromatosis that have not been reported previously: abnormal neovascularization and angiomas, reactive gliosis, and disruption of the blood–retinal barrier. The increased angiogenesis and neovascularization were evident in in Hjv−/− mice not only in the retinal layers but also in the vitreous. Analysis of collagen IV as a marker of blood vessels demonstrated abnormality also in vasculogenesis in these mice. These changes were accompanied with increased expression of VEGF. The blood vessels were thicker, tortuous, and abnormal with evidence of angiomas at multiple sites in the retina. There was also strong evidence of reactive gliosis, involving both Müller cells and microglia. Retinal astrocytes were not only more numerous but also abnormal in Hjv−/− mouse retinas compared with those in wild-type mouse retinas. Similarly, the shape and appearance of microglia were also different in Hjv−/− mouse retinas from those in wild-type mouse retinas. These abnormalities in retinal blood vessels in Hjv−/− mice were associated with barrier dysfunction as evident from decreased expression of the tight-junction protein occludin. These vascular changes were not evident in 2-week-old Hjv−/− mouse retinas. Even at the age of 4 months, the retinas in the knockout mice were almost normal. Iron accumulation and resultant oxidative stress in Hjv−/− mouse retinas is an age-dependent characteristic. The observations that the changes in retinal vasculature in these mice are also age-dependent suggest that excessive iron is the cause of these changes.
Potentially there are multiple pathways and mechanisms that might be involved in the observed abnormal angiogenesis and vasculogenesis in Hjv−/− mouse retina. The most fundamental factor is the increased accumulation of free iron in the retina in Hjv−/− mice. This initiates a cascade of events that promote angiogenesis and vasculogenesis in an uncontrolled manner leading to abnormality in the newly-formed blood vessels. First, excess Fe2+ in the retina would generate reactive oxygen species (ROS) via Fenton reaction in which H2O2 is converted into highly reactive hydroxyl radical and at the same time Fe2+ is oxidized to Fe3+. The increase in ROS would lead to elevated levels of hypoxia inducible factor 1α (HIF-1α) through activation of the inflammatory transcription factor nuclear factor–kappa B (NF-κB) and the protein kinase Akt(also known as Protein Kinase B or PKB).38 Another mechanism by which ROS can increase HIF-1α levels is through depletion of ascorbic acid by oxidizing the vitamin into dehydroascorbic acid. The levels of HIF-1α are regulated by proteosomal degradation, a process involving specific prolyl hydroxylases, which use Fe2+, O2, and α-ketoglutarate as cofactors.39 Ascorbic acid plays a key role in the function of prolyl hydroxylases by maintaining iron in the biologically active Fe2+ form.40 Therefore, depletion of ascorbic acid in the retina due to iron-induced generation of ROS would interfere with the function of prolyl hydroxylases. In addition, the conversion of Fe2+ to Fe3+ during the generation of ROS would increase the Fe3+/Fe2+ ratio, which is unfavorable for the function of prolyl hydroxylases. The resultant decrease in the activity of prolyl hydroxylases would prevent the proteosomal degradation of HIF-1α, thus leading to an increase in HIF-1α levels. Since HIF-1α is a potent inducer of VEGF synthesis, it is reasonable to expect accelerated angiogenesis. We have already documented the increase in iron levels and ROS production in the retinas of Hjv−/− mice,7 and in the present study we have shown that the expression of VEGF is elevated in the retinas of these mice. These findings support the hypothesis that the Fe2+/ROS/HIF-1α/VEGF pathway drives the accelerated angiogenesis observed in the retinas of Hjv−/− mice. Second, the antioxidant transcription factor Nrf2 (nuclear factor [erythroid-derived 2]-like 2) may also play a role in the abnormal angiogenesis in Hjv−/− mouse retinas. We have shown that deletion of Hjv in mice increases Nrf2 signaling as evident from the increased expression of several Nrf2-target genes, including the amino acid transporter Slc7a11 and the enzyme heme oxygenase-1 (HO-1).7 Nrf2 and HO-1 have been implicated as potent promoters of angiogenesis.41-43 Heme oxygenase-1 is involved in the catabolism of heme, which generates CO, a potent vasoactive and pro-angiogenic gaseous molecule. We have demonstrated previously that hemochromatosis is likely to be associated with increased intracellular levels of heme because of the alterations in the expression of various heme transporters.9 The increase in heme levels in Hjv−/− mouse retinas is directly relevant to the pro-angiogenic role of HO-1. The third potential mechanism underlying the accelerated angiogenesis in Hjv−/− mouse retina involves ferritin. Elevation of intracellular levels of iron leads to an increase in ferritin levels, and we have already shown that retinal levels of ferritin are higher in mouse models of hemochromatosis than in wild-type mice.5,7 Ferritin consists of a light chain (ferritin-L) and a heavy chain (ferritin-H). Ferritin-H is a potent modulator of angiogenesis via its ability to bind high molecular weight kininogen fragment HKa with high affinity and consequently block its antiangiogenic effect.44,45 Therefore, the increase in ferritin levels observed in Hjv−/− mouse retinas would also be potentially involved in promoting angiogenesis. The fourth possible mechanism underlying the accelerated angiogenesis in Hjv−/− mouse retina is related to the G-protein-coupled receptor GPR91. This receptor is activated by succinate, and is known to have a profound stimulatory effect on retinal angiogenesis.46 We have already shown that the expression and function of this receptor are upregulated in retina in a mouse model of hemochromatosis,6 indicating that GPR91 might contribute to the changes in retinal angiogenesis observed in Hjv−/− mice.
The mechanisms described above as potential players in abnormal angiogenesis in the retina are common in hemochromatosis irrespective of whether the disease is caused by deletion of Hfe or Hjv. Increased accumulation of Fe2+, excessive generation of ROS, increased Nrf2 signaling, elevated levels of ferritin, and upregulation of Gpr91 are observed in Hfe−/− mouse retinas as well as in Hjv−/− mouse retinas. There may be a molecular mechanism for promotion of retinal angiogenesis that is unique to Hjv−/− mice. This involves the signaling via the bone morphogenetic proteins (BMPs). These proteins are potent promoters of angiogenesis and vasculogenesis.47,48 Bone morphogenetic proteins signal through BMP receptors, and HJV is an important modifier of BMP signaling.49 Hemojuvelin exists in two forms, a membrane-anchored form and a soluble form. The membrane-bound form facilitates BMP signaling by serving as a coreceptor for BMPs, whereas the soluble form interferes with BMP signaling by binding BMPs and consequently preventing their interaction with their receptors. Therefore, the relative levels of the two forms of HJV would determine the magnitude of BMP signaling, and hence, the potential of this pathway in retinal angiogenesis. Deletion of Hjv in mice will deplete the membrane-bound form as well as the soluble form of Hjv. Which of these two forms predominate in wild-type mouse retina is not known. If the soluble form of Hjv is much higher than the membrane-anchored form in wild-type mouse retina, increased BMP signaling would be predicted in Hjv−/− mouse retina, which might contribute to the enhanced angiogenesis and vasculogenesis observed in Hjv−/− mouse retina. Additional research is needed to interrogate each of the four potential pathways to determine their relative contribution to the abnormal retinal angiogenesis in Hjv−/− mice. Two other mouse models of excessive iron accumulation in the retina have been reported to exhibit neovascularization: ceruloplasmin/hephaestin double-knockout mouse and hepcidin-knockout mouse.10,50 Since iron accumulation in the retina at abnormally high levels in these mouse models is a feature that is common to Hjv−/− mice, it is possible that excessive iron is the cause of neovascularization in the retina. The pro-angiogenic receptor Gpr91 and/or altered BMP signaling might mediate the effects of excessive iron on the retinal vasculature.
Acknowledgments
Supported by a grant from the National Eye Institute (EY019672; Bethesda, MD, USA).
Disclosure: A. Tawfik, None; J.P. Gnana-Prakasam, None; S.B. Smith, None; V. Ganapathy, None
References
- 1. Babitt JL, Lin HY. The molecular pathogenesis of hereditary hemochromatosis. Semin Liver Dis. 2011; 31: 280–292 [DOI] [PubMed] [Google Scholar]
- 2. Pietrangelo A, Caleffi A, Corradini E. Non-HFE hepatic iron overload. Semin Liver Dis. 2011; 31: 302–318 [DOI] [PubMed] [Google Scholar]
- 3. Kaplan J, Ward DM, De Domenico I. The molecular basis of iron overload disorders and iron-linked anemias. Int J Hematol. 2011; 93: 14–20 [DOI] [PubMed] [Google Scholar]
- 4. Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012; 1823: 1434–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gnana-Prakasam JP, Thangaraju M, Liu K, et al. Absence of iron-regulatory protein Hfe results in hyperproliferation of retinal pigment epithelium: role of cystine/glutamate exchanger. Biochem J. 2009; 424: 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gnana-Prakasam JP, Ananth S, Prasad PD, et al. Expression and iron-dependent regulation of succinate receptor GPR91 in retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2011; 52: 3751–3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gnana-Prakasam J, Tawfik A, Romej M, et al. Iron-mediated retinal degeneration in hemojuvelin knockout mice. Biochem J. 2012; 441: 599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gnana-Prakasam JP, Veeranan-Karmegam R, Coothankandaswamy V, et al. Loss of Hfe leads to progression of tumor phenotype in primary retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2013; 54: 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gnana-Prakasam JP, Reddy SK, Veeranan-Karmegam R, Smith SB, Martin PM, Ganapathy V. Polarized distribution of heme transporters in retinal pigment epithelium and their regulation in the iron-overload disease hemochromatosis. Invest Ophthalmol Vis Sci. 2011; 52: 9279–9286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hadziahmetovic M, Song Y, Ponnuru P, et al. Age-dependent retinal iron accumulation and degeneration in hepcidin knockout mice. Invest Ophthalmol Vis Sci. 2011; 52: 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hadziahmetovic M, Song Y, Wolkow N, et al. The oral iron chelator deferiprone protects against iron overload-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2011; 52: 959–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hadziahmetovic M, Song Y, Wolkow N, et al. Bmp6 regulates retinal iron homeostasis and has altered expression in age-related macular degeneration. Am J Pathol. 2011; 179: 335–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gnana-Prakasam JP, Martin PM, Simth SB, Ganapathy V. Expression and function of iron-regulatory proteins in retina. IUBMB Life. 2010; 62: 363–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. He X, Hahn P, Iacovelli J, et al. Iron homeostasis and toxicity in retinal degeneration. Prog Retin Eye Res. 2007; 26: 649–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song D, Dunaief JL. Retinal iron homeostasis in health and disease. Front Aging Neurosci. 2013; 5: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dunaief JL. Iron induced oxidative damage as a potential factor in age-related macular degeneration: the Cogan Lecture. Invest Ophthalmol Vis Sci. 2006; 47: 4660–4664 [DOI] [PubMed] [Google Scholar]
- 17. Wong RW, Richa DC, Hahn P, Green WR, Dunaief JL. Iron toxicity as a potential factor in AMD. Retina. 2007; 27: 997–1003 [DOI] [PubMed] [Google Scholar]
- 18. Blasiak J, Szaflik J, Szaflik JP. Implications of altered iron homeostasis for age-related macular degeneration. Front Biosci. 2011; 16: 1551–1559 [DOI] [PubMed] [Google Scholar]
- 19. Ciudin A, Hernandez C, Simo R. Iron overload in diabetic retinopathy: a cause or a consequence of impaired mechanisms? Exp Diabetes Res. 2010; 2010. pii: 714108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rajpathak SN, Crandall JP, Wylie-Rosett J, Kabat GC, Rohan TE, Hu FB. The role of iron in type 2 diabetes in humans. Biochim Biophys Acta. 2009; 1790: 671–681 [DOI] [PubMed] [Google Scholar]
- 21. Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab. 2013; 17: 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crawford TN, Alfaro DV, Kerrison JB, Jablon EP. Diabetic retinopathy and angiogenesis. Curr Diabetes Rev. 2009; 5: 8–13 [DOI] [PubMed] [Google Scholar]
- 23. Zhang W, Liu H, Al-Shabrawey M, Caldwell RW, Caldwell RB. Inflammation and diabetic retinal microvascular complications. J Cardiovasc Dis Res. 2011; 2: 96–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chappelow AV, Kaiser PK. Neovascular age-related macular degeneration: potential therapies. Drugs. 2008; 68: 1029–1036 [DOI] [PubMed] [Google Scholar]
- 25. Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol Aspects Med. 2012; 33: 295–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stitt AW, Li YM, Gardiner TA, Bucala R, Archer DB, Vlassara H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am J Pathol. 1997; 150: 523–531 [PMC free article] [PubMed] [Google Scholar]
- 27. Tawfik A, Al-Shabrawey M, Roon P, et al. Alterations of retinal vasculature in cystathionine-β-synthase mutant mice, a model of hyperhomocysteinemia. Invest Ophthalmol Vis Sci. 2013; 54: 939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang J, Duh EJ, Caldwell RB, Behzadian MA. Antipermeability function of PEDF involves blockade of the MAP kinase/GSK/beta-catenin signaling pathway and uPAR expression. Invest Ophthalmol Vis Sci. 2010; 51: 3273–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fonsatti E, Sigalotti L, Arslan P, Altomonte M, Maio M. Emerging role of endoglin (CD105) as a marker of angiogenesis with clinical potential in human malignancies. Curr Cancer Drug Targets. 2003; 3: 427–432 [DOI] [PubMed] [Google Scholar]
- 30. Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, Hartnett ME. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008; 27: 331–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hernandez MR, Ye H, Roy S. Collagen type IV gene expression in human optic nerve heads with primary open angle glaucoma. Exp Eye Res. 1994; 59: 41–51 [DOI] [PubMed] [Google Scholar]
- 32. Anik I, Kokturk S, Genc H, et al. Immunohistochemical analysis of TIMP-2 and collagen types I and IV in experimental spinal cord ischemia-reperfusion injury in rats. Spinal Cord Med. 2011; 34: 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller JW, Le Couter J, Strauss EC, Ferrara N. Vascular endothelial growth factor a in intraocular vascular disease. Ophthalmology. 2013; 120: 106–114 [DOI] [PubMed] [Google Scholar]
- 34. Lewis GP, Fisher SK. Up-regulation of glial fibrillary acidic protein in response to retinal injury: its potential role in glial remodeling and a comparison to vimentin expression. Int Rev Cytol. 2003; 230: 263–290 [DOI] [PubMed] [Google Scholar]
- 35. Erickson KK, Sundstrom JM, Antonetti DA. Vascular permeability in ocular disease and the role of tight junctions. Angiogenesis. 2007; 10: 103–117 [DOI] [PubMed] [Google Scholar]
- 36. Meynard D, Babitt JL, Lin HY. The liver: conductor of systemic iron regulation. Blood. 2014; 123: 168–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gnana-Prakasam JP, Martin PM, Mysona BA, Roon P, Smith SB, Ganapathy V. Hepcidin expression in mouse retina and its regulation via lipopolysaccharide/Toll-like receptor-4 pathway independent of Hfe. Biochem J. 2008; 411: 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou Y, Yan H, Guo M, Zhu J, Xiao Q, Zhang L. Reactive oxygen species in vascular formation and development. Oxid Med Cell Longev. 2013; 2013: 374963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Myllyharju J. HIF prolyl 4-hydroxylases and their potential as drug targets. Curr Pharm Des. 2009; 15: 3878–3885 [DOI] [PubMed] [Google Scholar]
- 40. Salnikow K, Kasprzak KS. Ascorbate depletion: a critical step in nickel carcinogenesis? Environ Health Perspect. 2005; 113: 577–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim TH, Hur EG, Kang SJ, et al. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 2011; 71: 2260–2275 [DOI] [PubMed] [Google Scholar]
- 42. Grochot-Przeczek A, Kotlinowski J, Kozakowska M, et al. Heme oxygenase-1 is required for angiogenic function of bone marrow-derived progenitor cells: role in therapeutic revascularization. Antioxid Redox Signal. 2014; 20: 1677–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation. 2008; 117: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McCrae KR, Donate F, Merkulov S, Sun D, Qi X, Shaw DE. Inhibition of angiogenesis by cleaved high molecular weight kininogen (HKa) and HKa domain 5. Curr Cancer Drug Targets. 2005; 5: 519–528 [DOI] [PubMed] [Google Scholar]
- 45. Coffman LG, Parsonage D, D'Agostino R Jr, Torti FM, Torti SV. Regulatory effects of ferritin on angiogenesis. Proc Natl Acad Sci U S A. 2009; 106: 570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sapieha P, Sirinyan M, Hamel D, et al. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med. 2008; 14: 1067–1076 [DOI] [PubMed] [Google Scholar]
- 47. Cai J, Pardali E, Sanchez-Duffhues G, ten Dijke P. BMP signaling in vascular diseases. FEBS Lett. 2012; 586: 1993–2002 [DOI] [PubMed] [Google Scholar]
- 48. Beets K, Huylebroeck D, Moya IM, Umans L, Zwijsen A. Robustness in angiogenesis: notch and BMP shaping waves. Trends Genet. 2013; 29: 140–149 [DOI] [PubMed] [Google Scholar]
- 49. Stirnberg M, Gutschow M. Matriptase-2, a regulatory protease of iron homeostasis: possible substrates, cleavage sites, and inhibitors. Curr Pharm Des. 2013; 19: 1052–1061 [DOI] [PubMed] [Google Scholar]
- 50. Hahn P, Qian Y, Dentchev T, et al. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004; 101: 13850–13855 [DOI] [PMC free article] [PubMed] [Google Scholar]