

Abstract
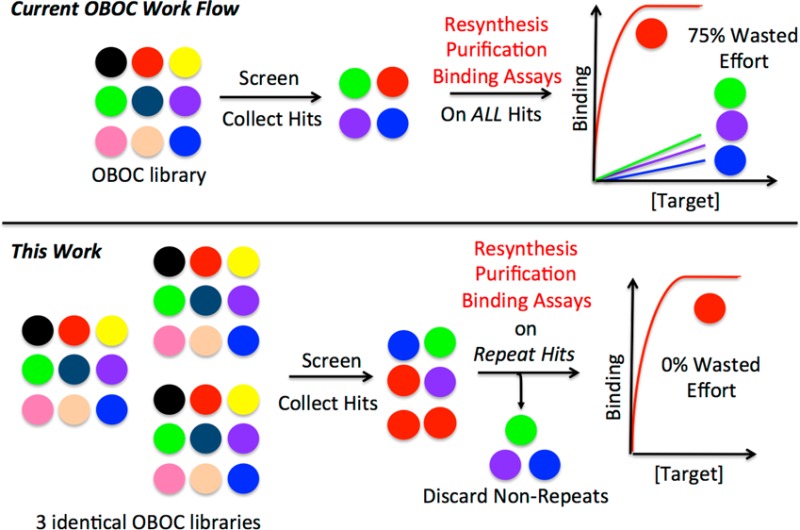
Large one-bead one-compound (OBOC) combinatorial libraries can be constructed relatively easily by solid-phase split and pool synthesis. The use of resins with hydrophilic surfaces, such as TentaGel, allows the beads to be used directly in screens for compounds that bind selectively to labeled proteins, nucleic acids, or other biomolecules. However, we have found that this method, while useful, has a high false positive rate. In other words, beads that are scored as hits often display compounds that prove to be poor ligands for the target of interest when they are resynthesized and carried through validation trials. This results in a significant waste of time and resources in cases where putative hits cannot be validated without resynthesis. Here, we report that this problem can be largely eliminated through the use of redundant OBOC libraries, where more than one bead displaying the same compound is present in the screen. We show that compounds isolated more than once are likely to be high quality ligands for the target of interest, whereas compounds isolated only once have a much higher likelihood of being poor ligands. While the use of redundant libraries does limit the number of unique compounds that can be screened at one time in this format, the overall savings in time, effort, and materials makes this a more efficient route to the isolation of useful ligands for biomolecules.
Keywords: OBOC library, peptoids, redundant library, nonspecific binding, antibody screen, antigen surrogate, serum screen
Introduction
A major goal of chemical biology is to identify small molecules with high affinity and selectivity for a variety of biological targets, including proteins and nucleic acids. Most such compounds are identified through some sort of high-throughput screen. While the most common technologies today employ various types of functional screens using compounds formatted in the wells of microtiter plates, an alternative and far more economical approach is to carry out binding screens with one bead one compound (OBOC) libraries created by solid-phase split and pool synthesis. This approach was first developed for the synthesis of peptide libraries1,2 and continues to be used most frequently for the creation of libraries of oligomers that can be sequenced by Edman degradation or mass spectrometry,3−6 since one cannot keep track of what compound is on what bead during the split and pool process. However, the use of encoding strategies7−10 has allowed this technology to be expanded to the creation of many different types of small molecule libraries. OBOC libraries created on beads with a hydrophilic surface, such as TentaGel, can easily be screened for binding to a labeled target.3 For example, a common approach is to directly or indirectly tag the target protein or nucleic acid with a fluorescent label and then monitor the bead population for those that have strong surface fluorescence after exposure to the target.11 An advantage of this kind of screen is that conditions can be adjusted to demand high selectivity12,13 by including a large excess of competitor proteins or nucleic acids.
However, the utility of these binding screens is compromised by several technical difficulties. One of the most problematic is the isolation of false positives. These are bead-displayed compounds that score as robust hits in the screening experiment, but fail to bind the target with reasonable affinity when resynthesized and tested in a variety of different formats. A striking example was published recently by Pei and co-workers,14 in which a TentaGel-displayed library of bicyclic peptides was screened against tumor necrosis factor-α (TNF-α). Despite the fact that several different methods were employed to score binding of the target protein to the beads,15 of the ≈400 hits originally isolated, only two proved to be ligands for TNF-α and one of these was a nonselective binder.
Clearly, if putative hits at the bead level must be resynthesized and purified to proceed to validation studies, a huge amount of effort would be wasted on compounds that ultimately prove to be of little value. Fortunately, there exist protocols for the validation of putative hits that do not require resynthesis. The most powerful of these, developed by Auer and co-workers,16 is to carry out on-bead labeling of the putative hits with a fluorescent tag using functionality in the invariant linker connecting the library compound to the bead. After cleavage from the bead, there is enough material on the 90–160 μm TentaGel beads used commonly for screening that the affinity of the compound for the protein of interest can be determined roughly using a fluorescence polarization (FP) assay. Indeed, this strategy was critical in allowing Pei and co-workers to distinguish the one useful TNF-α ligand from the large number of false positives isolated from the screen.14 Unfortunately, this strategy cannot be used when the target is difficult to produce in sufficient quantities for FP, is a minor component of a complex mixture, or is an integral membrane protein. For example, we have reported a study in which an OBOC peptoid library was screened against human serum samples to identify ligands that bind anti-Aquaporin 4 (AQP4) autoantibodies17 found commonly in patients with neuromyelitis optica (NMO).18 From a library of 100 000 beads, 10 were identified that appeared to bind high levels of antibodies from NMO patients that were not present in normal control sera. However, upon further validation, only one of these compounds proved to be a high quality anti-AQP4 ligand. Three were very weak ligands and the remaining compounds did not bind detectably to NMO antibodies at all when assayed in a microarray format.17 We have carried out several other screens using sera from patients with other diseases where the false positive rate was even worse (unpublished results). Unfortunately, the FP assay described above is not applicable to analysis of binding of the hits to low abundance antibodies in a serum sample. An alternative is to cleave the hits from the beads and spot them onto chemically modified glass slides and use these arrays for postscreening validation on individual serum samples.19 However, we have found that there is a high degree of variability in these arrays constructed from the material on a single bead (unpublished results). To acquire high quality array-based data, hit resynthesis and purification is required. Thus, a strategy to distinguish high from low quality bead screening hits without the need for resynthesis would have a major impact on the utility of this technology.
In this study, we explore the hypothesis that there is significant inhomogeneity in a given bead population with respect to compound density at the surface20 and that most of these false positives represent very weak ligands for the protein of interest that happen to be displayed at extremely high local concentrations on a particular bead.15 Evidence consistent with this idea is presented. With this model in mind, we further hypothesized that these low quality hits could be distinguished from high quality hits without tedious postscreening efforts if one employs a redundant library in which each compound is represented by several beads. The logic is that the chances that a given compound in the library will bind to the target protein, however poorly, are low, and that the fraction of beads in the population that have these unusually high surface densities is also low. Therefore, the odds of isolating a particular false positive are long and it is extremely unlikely that this would happen more than once when screening a redundant library. This leads to the idea that one could ignore hits isolated only once from a redundant library and focus all postscreening efforts only on compounds isolated multiple times. We show here that this is indeed the case. In a model serum antibody screening experiment, the hits identified multiple times were, without exception, high quality ligands while compounds isolated only once were far more likely to be of low affinity. We anticipate that this insight will allow studies using OBOC libraries to proceed more efficiently by eliminating unproductive postscreening resynthesis and analysis of what eventually prove to be poor quality compounds.
Results
Illustration of Bead Heterogeneity in a Hit Validation Experiment
Figure 1 shows an example of the unusual behavior manifested by many hits in a serum screen of the type discussed above for NMO.17 In this case, an OBOC peptoid library was screened against a pool of serum samples from lung cancer patients after having first cleared the library of beads that retain antibodies in the serum of control subjects. Retention of primary antibodies to each bead was visualized by incubation with a quantum dot-conjugated secondary antibody. The beads that displayed significant fluorescence were picked as hits. One of the brightest beads in this experiment was picked and mass spectrometric analysis showed its structure to be that shown in Figure 1A. This sequence was resynthesized on new TentaGel beads of the same type employed for the screen and approximately 100 of these beads were incubated with the same lung cancer serum pool employed in the screen. As shown in Figure 1B, a wide range of fluorescence intensities was observed on these beads, even though they all displayed the same peptoid. Only a few of the beads in the population displayed a florescence intensity similar to that observed during the primary screen. This perplexing observation could be explained, as suggested in the introduction, by postulating that the peptoid is an exceedingly weak ligand for antibodies in the pool and that retention of IgGs is only readily visible on relatively rare beads that happen to display dense clusters of the compound. Indeed, subsequent experiments in which this peptoid was used as a ligand in ELISA experiments with serum samples from many different lung cancer patients revealed that it is not a high quality ligand for lung cancer-specific antibodies (data not shown). This is exactly the type of hit that ideally would be disposed of prior to resynthesis and hit validation for the sake of efficiency, but which consumes large amounts of resources in the current work flow for the discovery of synthetic ligands for diagnostically useful antibodies.17,21
Figure 1.

Examples of heterogeneity between TentaGel macrobeads. (A) Chemical structure of a lung cancer antibody ligand derived from a OBOC screen. (B) Photomicrograph depicting the binding of lung cancer serum antibodies to beads containing only the compound shown in panel A. Bead hetereogeneity can be visualized by observing the variable brightness of red halos that result from binding of anti-IgG conjugated to red Qdots655 to primary serum antibodies. (C) Chemical structure of ADP3, which is synthesized with a C-terminal cysteine, and Met-ADP3, which is synthesized with a C-terminal methionine to facilitate cleavage from TentaGel macrobeads. Residues that are critical for IgY binding are highlighted in yellow. (D) Relative ligand density determined by synthesizing Met-ADP3 on a pool of 160 μm TentaGel beads and, after cleaving each bead, comparing the ADP3 parent ion intensity (Iparent) with respect to the internal standard ion intensity (Istandard).
TentaGel Macrobeads Display Varying Ligand Densities
To test the idea that there may be a wide variation in the loading capacity of different TentaGel beads in a population, we examined the composition of individual beads using mass spectrometry. The methionine derivative of an eight-residue peptoid called ADP321 (Figure 1C) was synthesized on 160 μm TentaGel resin. 96 individual beads were picked randomly from the population and the beads were placed into the wells of a microtiter plate. After liberating the peptoid from each bead, the relative intensity of the parent ion peak relative to an internal standard was used as a measure of individual bead loading. The ratio of these two numbers—parent ion m/z (ADP3) vs parent ion m/z (standard)—provides a relative indication of ligand loading for each bead (Supporting Information Figure S1). The smallest ratio was arbitrarily taken as 1.0 and the remainder were normalized to this value. As shown in Figure 1D, a 19-fold range of ligand densities with a broad distribution was observed. Note that this measurement is of total ligand capacity, whereas the relevant issue for protein binding is ligand density at the surface, since large proteins, such as IgG antibodies, cannot access the interior of TentaGel beads.20 Nonetheless, these data illustrate the general point that there is a great deal of heterogeneity in the bead population.
Isolation of Antibody Ligands from a Redundant Library
As mentioned in the introduction, a possible solution to this vexing problem of bead heterogeneity is to employ redundant libraries and only invest effort at the postscreening stage in hits that are observed multiple times. Again, the logic is that high affinity ligands for antibodies will be much less dependent than weak ligands on bead architecture to retain antibody.
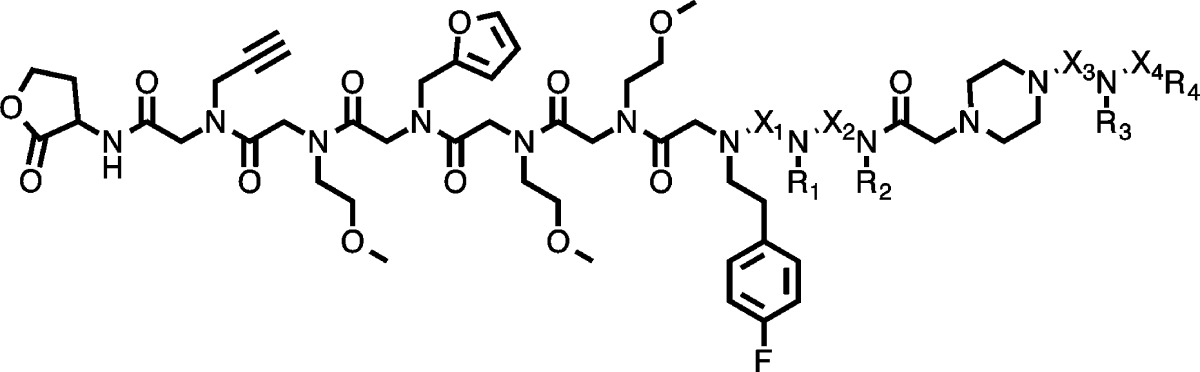
To test this idea, a screen was conducted to identify ligands for IgY antibodies from chickens immunized with ADP3 peptoid (Figure 1C) using the library shown in Figure 2A. Previous work showed that the side chains at positions 2 and 7 in ADP3 (highlighted in Figure 1C) are most critical for antibody binding.22 Therefore, the library that was employed in this experiment included these so-called Nlys and Npip residues as submonomers (Figure 2B) with the expectation that this somewhat biased library might contain high affinity ligands. Moreover, the library was composed of both peptoid and peptide tertiary amide (PTA) units (Figure 2C). PTA units combine a chiral center at the α-carbon, like peptides, with N-substitution. This results in greater conformational constraints, which we anticipate will lead to higher affinity binding.23 Specifically, bromoacetic acid and both enantiomers of 2-bromopropionic acid (with one stereoisomer encoded by deuteration (Figure 2B)) were employed as variable submonomers in the library synthesis. Note that this library does not contain the native antigen ADP3 nor compounds in which the critical Npip and Nlys side chains are spaced in the same way as ADP3.
Figure 2.

PTA-peptoid hybrid library design and IgY screening schematic. (A) PTA-peptoid library design depicting the invariant region (red) and diversity region (black). The scaffold contains a piperazine linker to stiffen the backbone and ease synthetic coupling of consecutive PTA units. (B) Primary amines used in the diversity positions (R) of the library. Amines that are important for IgY binding are highlighted in yellow in Figure 1C. (C) Cα substituents at position X. The R stereochemistry was encoded by (S)-2-bropropionic acid-d4. (D) Screening was performed on three copies of a 200 000 bead library (triply redundant) by first removing hits to nonspecific IgY. The remainder of the library was incubated with total IgY from chickens immunized with ADP3 and hits were visualized using anti-IgY Qdots. The hit beads were stripped in acetonitrile and resubmitted to the identical screening procedure to identify true hits.
The screen employed serum from chickens not exposed to ADP3 (200 μg/mL total protein) into which was spiked 250 nM total IgY from a chicken that either had or had not been immunized with ADP3. We estimate that the anti-ADP3 antibodies comprise approximately 1% of the total IgY fraction (Supporting Information Figure S2), meaning that the concentration of the anti-ADP3 antibodies in the serum sample was approximately 2.5 nM.
The beads were first exposed to the control serum and, after washing, beads binding IgY were visualized by the addition of an anti-IgY secondary antibody conjugated to a red quantum dot (Qdot655). Brightly fluorescent beads were removed from the population using a micropipet under a low power fluorescence microscope. The remaining beads were incubated with the serum sample spiked with anti-ADP3 antibodies. Again, after washing, the hits were visualized using Qdot655-conjugated anti-IgY and manually picked out of the library for further analysis. To ensure that the beads isolated at this step represented true hits, they were stripped of antibody by incubation in acetonitrile/water and re-equilibrated with buffer. The beads were then re-exposed to the anti-ADP3 IgY-containing serum and processed as described. Thirty-five beads displaying a bright red halo were isolated (Figure 3). After cleavage of the compounds from each bead using CNBr, the compounds were sequenced using MALDI-TOF MS/MS (Table 1).
Figure 3.

(A) Photomicrographs of all 35 hits that exhibited a red halo after stripping and rebinding. (B and C) Magnified images of hits shown in panel A.
Table 1. Sequences of Hit Compounds from IgY Screena.
| compound | X1b | R1c | X2b | R2c | X3b | R3c | X4b | R4c |
|---|---|---|---|---|---|---|---|---|
| 1 | S | Nmea | N | Nleu | N | Npip | N | Nlys |
| 1 | S | Nmea | N | Nleu | N | Npip | N | Nlys |
| 1 | S | Nmea | N | Nleu | N | Npip | N | Nlys |
| 2 | S | Nmea | N | Napp | N | Npip | N | Nlys |
| 2 | S | Nmea | N | Napp | N | Npip | N | Nlys |
| 3 | S | Napp | N | Npip | N | Npip | N | Nlys |
| 3 | S | Napp | N | Npip | N | Npip | N | Nlys |
| 4 | S | Npip | N | Nlys | N | Npip | N | Nlys |
| 4 | S | Npip | N | Nlys | N | Npip | N | Nlys |
| 5 | S | Nmea | N | Npip | N | Npip | N | Napp |
| 6 | N | Nlys | N | Napp | N | Npip | N | Nleu |
| 7 | N | Napp | N | Nmea | N | Npip | N | Nlys |
| 8 | N | Napp | N | Npip | N | Npip | N | Nlys |
| 9 | S | Nlys | N | Nleu | N | Npip | N | Nleu |
| 10 | S | Nlys | N | Napp | N | Npip | N | Nlys |
| 11 | S | Nlys | N | Nleu | N | Npip | N | Nlys |
| 12 | N | Nleu | N | Nlys | N | Npip | N | Nlys |
| 13 | N | Npip | N | Nlys | N | Npip | N | Nlys |
| 14 | S | Nmea | S | Nmea | N | Npip | N | Nlys |
| 15 | S | Npip | S | Nmea | N | Npip | N | Nlys |
| 16 | S | Nmea | N | Nleu | N | Npip | N | Nlys |
| 17 | N | Napp | R | Nmorph | N | Npip | N | Nlys |
| 18 | N | Nlys | N | Napp | N | Napp | N | Nlys |
| 19 | N | Nleu | N | Napp | N | Npip | N | Napp |
| 20 | S | Nmea | N | Npip | S | Nmea | N | Napp |
| 21 | N | Nlys | N | Npip | N | Npip | S | Nmoph |
| 22 | N | Napp | S | Nmea | N | Npip | N | Nlys |
| 23 | N | Nlys | N | Nleu | S | Nmorph | R | Nmorph |
Four compounds were isolated at least two times from the library (Figure 4A). Moreover, they all shared significant sequence homology. Each contained a terminal piperonylamine and diaminobutane dyad (Figure 4A), units that were also found in many of the 35 hits (Table 1). This was not surprising, since the same dyad is present in ADP3, though the N-terminal Nlys residue is not essential for binding of the peptoid antigen to the antibodies. Furthermore, each replicate hit also contained a chiral methyl group with the S configuration at the first variable position (Figure 4A).
Figure 4.

Characterization of anti-ADP3 IgY ligands. (A) Chemical structures of hit and control compounds with the number of times each hit was isolated from the screen. The structure of the linker of the cleaved compounds is shown in Table 1 in gray. Homologous side chains in the repeat hits are highlighted in orange and identical Cα stereochemistry is highlighted in blue. (B) Saturation binding curves generated for each of the four redundant hits using fluorescence polarization (FP) against anti-ADP3 IgY (solid line) or control IgY (dotted line). (C) Competition FP of fluorescein-conjugated ADP3 and 1 using ADP3 (solid line) or a control peptoid (dotted line) as a competitor. (D) Binding saturation curves for compounds that were isolated only one time during the screen against anti-ADP3 IgY (solid line) and control IgY for ADP3 and 5 (dotted line).
Characterization of the Binding Properties of the Repeat Hits
To see how well these redundant hits performed in a binding assay, they were resynthesized and labeled with maleimide-fluorescein on a cysteine included at the C-terminus. Binding to increasing amounts of total IgY isolated from the ADP3-immunized, or control, chickens was then monitored by fluorescence polarization. The four repeat hits exhibited strong binding to IgY, with Kd values similar to the native antigen, ADP3. When corrected for the ≈1% abundance of anti-ADP3 antibodies in the total IgY sample, the data indicate a dissociation constant of approximately 25–30 nM. All four compounds, as well as ADP3, exhibited a much lower affinity for control IgY (Figure 4B). A negative control compound, 24, that did not exhibit binding during the primary screen, yet shares some sequence homology with the four redundant hits, failed to show binding to the anti-ADP3 IgY. These data show that the repeat hits are, as hoped, high affinity, selective ligands for the anti-ADP3 antibodies.
To ensure that the ligands bind in the antigen-binding pocket of the antibody, as assumed, a competition experiment was carried out in which the binding of fluorescein-labeled ADP3 or fluorescein-labeled 1 to the anti-ADP3 antibodies were challenged with unlabeled ADP3, which was monitored by fluorescence polarization. If the unlabeled molecule binds to the same site as the labeled molecule, then the polarization observed should decrease as the concentration of the unlabeled competitor increases. As expected, when ADP3 was used to compete the fluorescein-labeled ADP3 probe, a dose-dependent decrease in polarization was observed (Figure 4C). When ADP3 was used to compete labeled 1, a similar dose-dependent decrease in binding was observed. Neither the labeled ADP3 nor 1 were competed significantly by a control peptoid 25. We conclude that 1 indeed binds to the antigen binding site of the IgY antibodies. While this experiment has not been carried out for all of the hits, we assume that they also bind the same surface based on their similar structures and the fact that compounds that bind outside of the antigen-binding pocket should have been removed from the population in the prescreen in which the beads were exposed to an IgY population that did not contain anti-ADP3 antibodies.
Characterization of the Binding Properties of the Single Hits
To determine if the hits isolated only once in the library screen were of comparable quality or inferior to those isolated multiple times, their binding to the anti-ADP3 antibodies was also analyzed using fluorescence polarization. These compounds displayed some degree of similarity to the repeat hits. One had a chiral center in the same position as the four repeat hits (5), whereas compounds 6–8 were devoid of PTA units but shared some side chains with 1–4. All of them contained an Npip residue at the second to last position. Two of them display an N-terminal Nlys residue. As shown in Figure 4D, none of these compounds exhibited the high affinity displayed by the four repeat hits. Indeed, the affinity of the peptoids 5–8 for the anti-ADP3 antibodies more closely resembled the binding of the four repeat hits to the control antibodies (compare Figure 4B and 4D). Compound 5, which contained the chiral center was better than the other three, but still clearly inferior to ADP3 and the repeat hits.
Identification of Critical Residues in the Repeat Hits
In addition to the fact that compounds 1-4 were isolated more than once from the library, there is a great deal of structural similarity between them. Are these similarities indicative of the units in the molecule most critical for binding the antibodies? To determine this a “methyl scan” of one of the redundant hits, 1, was performed. Specifically, a series of compounds was made that were identical to 1, except that one of the side chains was replaced by a methyl group. This analysis included the most N-terminal residue in the invariant linker as well as all of the library-encoded positions. Each derivative was fluorescently labeled and their affinities for anti-ADP3 IgY, or control antibodies, were determined using an FP assay (Figure 5). The data show that Npip was critical for binding to the IgY population (Figure 5). Additionally, replacement of the Nlys residue at the N-terminus with methylamine decreased affinity by >10-fold (Table 2). This is interesting, given that the N-terminal Nlys residue in ADP3 was not critical for IgY binding, possibly suggesting that the screening hits bind in a different fashion to the antibodies than does ADP3. Finally, substitution of the fluoroaromatic side chain at the last position of the invariant linker with a methyl group also reduced binding affinity somewhat, indicating it also plays a modest role in antibody recognition.
Figure 5.
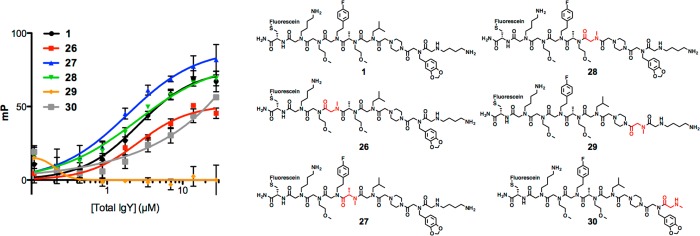
FP saturation curves for 26–30 to study the importance of each residue on binding to IgY. The position that contains the substitution is displayed in red for clarity.
Table 2. IgY Binding Affinities for Side Chain and Stereochemical Analogues of Repeat Antigen Surrogates.
Reported Kd is an average Kd for all polyclonal anti-ADP3 IgY.
If at least one curve failed to saturate during the experiment, its Kd is reported as >10 μM
Next a series of compounds was created to test the importance of the chiral methyl group present at the first variable position of all of the repeat hits. Fluorescein-tagged derivatives of 1 and 2 were created that eliminated the methyl group at this position. As shown in Figure 6A, this essentially abolished binding of the compound to the anti-ADP3 antibodies. This is interesting given that ADP3 has no chiral centers and is consistent with the idea that the PTA unit strongly stabilizes a particular conformation of the molecule favorable for binding, though it is also possible that the methyl group itself makes important contacts. To probe this further, the fluorescein-labeled enantiomer of 1 was synthesized and tested (compound 33). As shown in Figure 6B, its affinity for anti-ADP3 IgY was much lower than that of the enantiomer 1, though not as low as the des-methyl compound. These data provide yet another striking example of the superiority of protein ligands containing conformationally constrained units relative to simple peptoids.23,24 In general, the data show clearly that, as suspected, the structural units shared by each of the four repeat hits are important for antibody binding.
Figure 6.

Importance of Cα in IgY ligands. A. FP binding curves for 1, 2, and analogues lacking a Cα methyl substituent, 31 and 32; the Cα is highlighted in yellow for clarity. B. FP binding curve for 1 and its enantiomer, 33. For reference, the des-methyl derivative (32) was used as an experimental negative control.
Lower Affinity Ligands Reveal More Heterogeneous Antibody Binding in Mock Bead Screening Experiments
As discussed above, our hypothesis is that the weaker the ligand, the more dependent it will be on being displayed on a high density bead in order to be scored as a hit. To further test this idea, one high affinity and one low affinity ligand for anti-ADP3 antibodies (1 and 7, respectively; see Figure 4A) were resynthesized on 160 μm TentaGel beads and approximately 250 of the beads were assayed for binding at different IgY concentrations. After incubating the two pools of resin with different concentrations of anti-ADP3 IgY, followed by washing and incubation with a Qdot655-conjugated secondary antibody, the beads displaying an obvious red halo were counted. As shown in Figure 7, 18% and 25% of the beads displaying the low affinity ligand 7 were scored as hits at low antibody concentrations (10 and 25 nM, respectively). Increasing the antibody concentration to 75 or 200 nM resulted in >90% the beads being scored as hits. In comparison, the bead population displaying the high affinity ligand 1 also showed heterogeneous behavior at 10 nM antibodies (∼40% scored as hits), but at 25 nM IgY, > 90% showed clear antibody binding. These data are consistent with our hypothesis that weaker ligands are more dependent on bead architecture to bind detectable amounts of antibody.
Figure 7.

Comparison of 1 and 7 binding on TentaGel macrobeads in a mock IgY screen. (A) 1 and 7 were resynthesized on 160 μm TentaGel macrobeads to test how well IgY binds to the hit in a population of heterogeneous macrobeads—the linker in the sequences shown is represented in Figure 1A (red). Approximately 250 beads from each population were subjected to a mock screen for anti-ADP3 IgY ligands at varying concentrations of target. The percentage of beads visually scored as a hit were recorded at each population of target. (B) Photomicrographs of beads containing 1 after screening at 25 nM anti-ADP3 IgY and Qdot655-conjugated secondary antibody. (C) Photomicrographs of beads containing 7 after screening at 25 nM anti-ADP3 IgY followed by red Qdot655 anti-IgY.
Discussion
OBOC library screens are a potentially powerful approach to the identification of ligands for a variety of interesting biomolecules. We have been particularly interested in a novel application of this technology, which is the identification of “antigen surrogates”, that is, synthetic compounds that bind to the antigen-binding sites of antibodies.17,21 Specifically, we have described a screening protocol designed to identify IgG antibodies in human serum samples that are linked to a given disease state. This involves incubation of an OBOC library with control sera from patients that do not have the disease of interest, followed by a labeled anti-IgG secondary antibody to denude the population of ligands to uninteresting IgGs. The remainder of the library is then exposed to a pool of sera from patients that share the disease of interest. The hits from this round of screening are further evaluated as potentially interesting antigen surrogates (Figure 2). These compounds, when immobilized on an appropriate surface, could then be used as “capture agents” in ELISA-like assays designed to monitor the level of the biomarker antibody in the blood.17
As illustrated by the peptoid isolated in a screen using sera collected from lung cancer patients (Figure 1), a major problem with the current workflow is that “false positives” are exceedingly common, often comprising the majority of the hits from bead screens of this type. These are beads that “light up” brightly at the screening stage, yet the compounds they display fail to show useful antibody-binding properties in subsequent assays. If postscreening analysis requires resynthesis of the hits, then these false positives consume considerable time and resources.
In this study, we explore the hypothesis that at least one explanation for these false positives is that they represent poor ligands for the antibodies of interest but happen to be displayed on rare beads in a heterogeneous population with unusually high surface density. This can result in the antibody being effectively trapped on the bead. This is especially true for screens targeting antibodies, since these are bivalent molecules and binding to surface-immobilized molecules can involve large avidity effects. If this idea is correct, then a simple solution would be to employ redundant libraries in which several different beads display the same compound and focus postscreening efforts solely on the hits isolated more than once.
As shown in Figure 1D, quantitative analysis of a batch of TentaGel beads revealed an approximately 20-fold range of compound loading from bead to bead. While this measurement may or may not reflect the density of compound displayed at the protein-accessible20 surface of each bead, it shows clearly that there is considerable heterogeneity in the bead population with respect to compound loading. This is at least consistent with our hypothesis for the nature of the false positives.
To test the utility of redundant libraries as a solution to this problem, approximately three copies of a TentaGel-displayed library of 200 000 compounds constructed from peptoid and PTA building blocks (Figure 2) was screened against an antipeptoid IgY antibody doped into chicken serum. Four compounds were isolated more than once. Moreover, there was significant structural similarity between all four (Figure 4). Because of the high levels of anti-ADP3 antibodies in the total IgY purified from the immunized chicken, we found that we could employ FP assays to quantify binding of the compounds to the target antibodies. The results (Figure 4) showed clearly that all four of the repeat hits were high affinity ligands for the anti-ADP3 antibodies. Indeed, the binding curves were quite similar to that exhibited by the ADP3 antigen itself. Binding was selective, as control IgY antibodies exhibited much lower affinities (>100-fold) for these compounds (Figure 4). In contrast, almost all of the hits isolated only once recognized the anti-ADP3 antibodies with far lower affinities that more closely resembled the binding of the repeat hits to the control antibodies. These data strongly support the idea that in future serum screens of this type, it would indeed be wise to focus postscreening validation efforts solely on the repeat hits. This is important, because in most biomarker discovery efforts using this technology, the levels of interesting antibodies are likely to be too low to allow for FP analysis of the quality of the screening hits without resynthesis. Instead, it is far more likely that hits of interest will have to be resynthesized, purified and mounted onto some type of ELISA-like analytical platform.22 Thus, we believe that the strategy of using redundant libraries and focusing only on hits isolated multiple times will greatly increase the utility of this screening technology for the discovery of diagnostically useful antibodies. It should also prove to be useful in screening projects where the properties of the target protein make the high-throughput FP-based validation technique16 of all of the hits impractical, for example if it is difficult to purify in quantity or if it is an integral membrane protein.
While the main point of this study was to evaluate the wisdom of focusing solely on repeat hits in postscreening efforts, the analysis of the determinants of binding of the repeat hits to the anti-ADP3 antibodies was also carried out and provided interesting data. Not surprisingly, the structural features shared by all four compounds proved to be important for binding. Thus, one can rely on the appearance of a consensus sequence as another indication that a given compound is a high quality hit, as has been seen many times in screens of biologically encoded libraries of peptides or aptamers. These include the two N-terminal side chains as well as the chiral methyl group at the α carbon of the most C-terminal variable unit. Of particular interest to us was that the peptoid analogue of 1 lacking a chiral center essentially failed to bind to the anti-ADP3 antibodies and that the enantiomer of 1 was a much weaker ligand (Figure 5). This provides yet another striking example in which conformational restriction contributes greatly to high affinity binding of peptoid-like molecules to proteins23,24 and strongly suggests that libraries constructed from these types of building blocks will be a source of superior protein ligands.
Experimental Procedures
General Information
Deuterated (S)-(−)-2-bromopropionic acid was prepared as described previously.23 TentaGel R resin was purchased from Rapp Polymere. All the Fmoc-protected amino acids and Knorr Amide MBHA resin were purchased from Novabiochem. All reagents were purchased from Sigma-Aldrich or Alfa Aesar, unless otherwise specified. All steps involving water utilized distilled water filtered through a Barnstead Nanopure filtration system (Thermo Scientific)
Peptoid/PTA and Azapeptoid Oligomer Synthesis
Oligomers were synthesized on Rink Amide resin (0.44 mmol/g) using previously described protocols.3,25−27 Resin (0.1 g, 0.032 mmol) was swelled in DMF for 2 h prior to use. 9-Fluorenylmethoxycarbonyl (Fmoc) was removed by 20% piperidine and washed thoroughly in DMF. N-α-Fmoc-S-p-methoxytrityl-l-cysteine, (0.1 g, 0.16 mmol) was coupled to the resin using O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU, 0.061 g, 0.16 mmol) and diisopropylethylamine (DIPEA, 0.06 mL, 0.32 mmol) for 3 h at room temperature (RT). For compounds synthesized on TentaGel macrobeads, methionine was the first amino acid loaded onto the resin. The beads were washed (3 × 3 mL dimethylformamide (DMF)), and Fmoc was deprotected with 20% piperidine and washed (3 × 3 mL DMF). To install peptoid and azapeptoid subunits, the growing chain was bromoacetylated using 1 mL 2 M 2-bromoacetic acid (BAA) and 1 mL 2.5 M diisopropylcarbodiimide (DIC). The mixture was shaken at 37 °C for 10 min and washed thoroughly. Primary amines and carbazides were added to the bromoacetylated resin as 1 M solutions in DMF and shaken at 37 °C for 1 h. 1-(t-Butoxycarbonyl)-diaminobutane and glycine tert-butyl ester were used for aminating with the Nlys amine. For compounds containing PTA monomers, (R)-2-bromopropionic acid (R)-BPA) or (S)-2-bromopropionic acid ((S)-BPA) was coupled in place of bromoacetic acid using previously described conditions with slight modification.23 Briefly, (R)- or (S)-BPA (20 μL, 0.22 mmol) was dissolved in a solution of 20 mg mL–1 bis(trichloromethyl) carbonate (BTC, 0.022 g, 0.075 mmol) in anyhydrous tetrahydrofuran (THF) and precooled to −20 °C in a freezer for 15 min. The resin was washed with DMF (5 × 3 mL), dichloromethane (DCM, 5 × 3 mL) and anhydrous tetrahydofuran (5 × 3 mL). DIPEA (61 μL, 0.35 mmol) in 1 mL anhydrous THF was added to the beads. 2,4,6-Trimethylpyridine (60 μL,0.44 mmol) was added to the cooled BPA/BTC solution and quickly added to the resin slurry, vented, and shaken for 2 h at RT. The solution remained a pale yellow solution throughout the incubation period. A darker color is an indication that excessive heat accompanied the transformation, and can be avoided by further cooling the bead and/or the acyl chloride solution. After completion of the reaction, the beads were washed successively (3 × 3 mL) in THF, DCM and DMF, respectively. Amination reactions were performed using 1 M of the primary amine solution at 60 °C overnight, followed by thorough washing in DMF (3 × 3 mL). Once oligomer synthesis was complete, the beads were washed in DCM (3 × 3 mL) followed by incubation with 2% TFA in DCM (8 × 2 min) to remove the MMT protecting group and yield the free sulfhydryl. The resin was neutralized with 10% DIPEA, washed (5 × 2 mL DMF), and incubated with a 5 mM solution of fluorescein-5-maleimide in DMF for 3 h at room temperature. The beads were washed with DMF (3 × 3 mL) and DCM (5 × 3 mL) and then cooled to 4 °C. Oligomers were simultaneously deprotected and liberated from the resin by incubating in a precooled cocktail of TFA/DCM/TIS (49.5:49.5:1) for 1 h at 4 °C. The TFA/DCM solution was evaporated under nitrogen and the oligomer was precipitated in cold ether and harvested by centrifugation. Cleaved compounds were purified on a Vydac reverse-phase C18 column (Grace), freeze-dried, and stored. Compound identity was confirmed by MALDI-TOF MS using a 4800 Plus MALDI TOF/TOF Analyzer (Applied Biosystems) using α-cyano-4-hydroxycinnamic acid (CHCA) as matrix.
Mock Screen with Lung Cancer Hit
The hit peptoid (Figure 1A) was resynthesized on a small batch of ∼100 160 μm TentaGel beads as described above. The beads were washed in water (10 × 1 mL) and TBS-T (3 × 1 mL). Beads were incubated with serum from an adenocarcinoma patient diluted to 80 μg/mL in 50% PBS Starting Block in TBS-T overnight at 4 °C. The beads were washed in TBS-T (3 × 1 mL) and hybridized with Qdot655 goat antihuman IgG as a 1:200 dilution in 50% Starting Block in TBS-T for 1 h at RT. The beads were washed in TBS-T (3 × 1 mL) and imaged under an inverted fluorescent microscope using a DAPI filter.
Mass Spectrometric Determination of Bead Heterogeneity
Met-ADP3 was synthesized on 160 μm TentaGel Macrobeads (100 mg) as described. Following synthesis, the beads were washed with DCM (3 × 3 mL) and protecting groups were removed by incubating in TFA/H2O/TIS (95:2.5:2.5) for 2 h at RT. The beads were washed in DCM (3 × 3 mL) and 50% acetonitrile/water (3 × 3 mL). 96 beads were separated into the wells of a 96-well plate and the acetonitrile/water was removed by vacuum centrifugation. Compound was cleaved from the bead by incubating each bead in 20 μL of a 50 mg mL–1 solution of cyanogen bromide (CNBr) dissolved in acetic acid:acetonitrile:water (5:4:1) overnight at RT. The next day, the CNBr solution was evaporated by vacuum centrifugation and the dry residue in each well was dissolved in 20 μL of a 75:25 mixture of water:acetonitrile containing 0.1% TFA. To avoid evaporation of this solution during the experiment, the solution was only added to 12 wells at a time. To aid in dissolution and homogenizing the compound solution, each well was aspirated 5–6 times with a pipet immediately prior to spotting onto a MALDI-TOF MS plate. 0.7 μL from each well was cospotted with a 10 mg/mL solution of CHCA dissolved in 25% acetonitrile in water containing 0.1% TFA and 500 ng/mL of ADP3 lacking the C-terminal methionine as an internal standard. MALDI-TOF MS spectra were obtained on a 4800 Plus MALDI TOF/TOF Analyzer (Applied Biosystems). Intensities for ADP3 (IMet-ADP3) and the internal standard (IStandard) were recorded and the loading on each bead was determined as
and normalized to the lowest ratio, which was given a value of 1.
Synthesis of a PTA-Peptoid Hybrid Library
The invariant linker was installed onto 90 μm Tentagel R macrobeads (1 g, 0.27 mmol/g, Rapp-Polymere) using standard BAA/DIC couplings described above using microwave conditions. For the first variable position, the beads were split into three equal portions, coupled with bromoacetic acid, (S)-2-bromoproprionic acid-d4 or (R)-2-bromopropionic acid, respectively. Bromoacetic acid couplings were activated with DIC and carried out using microwave conditions. For bromopropionic acids, bis(trichloromethyl) carbonate (BTC) was used as a coupling reagent. BTC (92.1 mg, 0.31 mmol) was dissolved in 5 mL anhydrous THF in a glass vial. Bromopropionic acid (89 μL, 0.95 mmol) was then added to the vial and the vial was cooled to −20 °C in the freezer for 15 min. Beads were washed using DMF, DCM and then THF, respectively, for 5 times each. A 2:1 THF/DIPEA (750 μL THF, 375 μL DIPEA, 2.2 mmol) solution was added to the beads and gently shaken. 2,4,6-Trimethylpyridine (356 μL, 2.7 mmol), was added to the cold solution of bromopropionic acid with BTC. A white precipitate formed quickly following the addition and the suspension was applied to the beads. The reaction vessel was put on a shaker for 2 h at room temperature. As stated previously, the solution in the vessel should be a pale yellow suspension during the entire course of the reaction. A darker color is an indication of excessive heat released during the initial addition of the acid chloride solution. If this problem persists, it can be ameliorated by further cooling down both the acid chloride and the bead solutions. The beads were washed five times with DCM followed by five times with DMF. A chloranil test was used to monitor the completion of the reaction. All three portions of the beads were then pooled together and the beads were incubated with a 2 M solution of the corresponding primary amine in DMF at 60 °C overnight. The completion of the reaction was monitored by the chloranil and silver acetate tests. Nlys was protected with MMT for synthesis, which was subsequently removed by washing the beads in 1% TFA in DCM at room temperature five times followed by washing in DCM (3 × 5 mL).
Screening for Anti-ADP3 IgY Ligands
The OBOC library was washed with 10 × 10 mL of water for 5 min and further equilibrated in water overnight. The beads were washed in 1× tris-buffered saline (20 mM tris, 137 mM NaCl, pH 7.6) containing 0.05% Tween-20 (TBS-T) for 1 h. Beads were blocked with 50% PBS Starting Block (Thermo) in 1× PBS (11.8 mM phosphates, 137 mM NaCl, 2.7 mM KCl, pH 7.4) for 1 h. The library was separated into three equal aliquots and screening was performed separately on each. First, 250 nM of normal chicken IgY (Santa Cruz) was doped into 100 μg/mL of chicken serum (Sigma-Aldrich). Three milliliters of this solution was applied to the OBOC library and incubated overnight at 4 °C. The library was washed (3 × 4 mL TBS-T) followed by addition of 3 mL of a 1:200 dilution of anti-IgY conjugated to red Qdots (Life Technologies) in 50% PBS Starting Block. The hybridization solution was incubated for 1 h at room temperature. The beads were washed 5 × 4 mL in TBS-T and visualized under an inverted fluorescent microscope using a DAPI excitation and emission filter. Beads that exhibited a red halo were manually removed from the library using a micropipet and discarded. The library was incubated with 3 mL of 250 nM anti-ADP3 IgY doped into 100 μg/mL of chicken serum overnight at 4 °C. The library was washed (3 × 4 mL TBS-T) and incubated with 3 mL of a 1:200 dilution of anti-IgY conjugated to red quantum dots for 1 h at room temperature. The library was washed 5 × 4 mL TBS-T and beads were visualized on an inverted fluorescent microscope fitted with a DAPI excitation and emission filter. Beads that displayed a red halo were removed. The screen was repeated for the remaining two aliquots of beads. ∼300 library beads were removed in total. These beads were collected and stripped of bound protein by incubating in 50% acetonitrile/water (3 × 2 mL) for 30 min at 37 °C followed by incubation in acetonitrile 2 × 2 mL) for 60 min at 37 °C.
Secondary Validation Screen
Hit beads were washed in water (10 × 2 mL) and re-equilibrated in water overnight. The beads were equilibrated in TBS-T for 1 h, followed by blocking in 50% Starting Block diluted into PBS. 100 nM of normal chicken IgY (Santa Cruz) was doped into 200 μg/mL of chicken serum (Sigma-Aldrich) and 500 μL of this solution was applied to the hit beads and incubated overnight at 4 °C. The library was washed (3 × 2 mL TBS-T), followed by addition of 500 μL of a 1:200 dilution of quantum dot-conjugated anti-IgY (Life Technologies) in 50% PBS Starting Block and incubated for 1 h at room temperature. The beads were washed (5 × 2 mL) in TBS-T and visualized under an inverted fluorescent microscope using a DAPI excitation and emission filter. Beads that exhibited a red halo were manually removed from the library and discarded. The beads were incubated overnight at 4 °C with 500 μL of 100 nM anti-ADP3 IgY doped into 200 μg/mL of chicken serum. The beads were washed (3 × 2 mL TBS-T) and hybridized with 500 μL of a 1:200 dilution of anti-IgY conjugated to red quantum dots for 1 h at room temperature. The beads were washed 5 × 2 mL TBS-T and visualized on an inverted fluorescent microscope fitted with a DAPI excitation and emission filter. Compounds exhibiting a red halo like those shown in Figure 3 were collected, stripped by incubating in acetonitrile at 37 °C for 1 h (2 × 1 mL). The beads were washed in ethanol to facilitate separation into a 96-well plate.
Sequence Identification of Validated Hits Using Mass Spectrometry
Individual beads were separated into individual wells of 96-well plates and the compounds were cleaved from the beads by incubation with 20 μL of 0.2 N HCl containing CNBr for 2 h at RT. The HCl solution was removed using a vacuum centrifuge and the cleaved compounds were dissolved in 10 μL of 50% acetonitrile/water. 0.7 μL of this solution was cospotted on a MALDI plate with 0.7 μL of 10 mg/mL CHCA in 50% acetonitrile/water containing 0.1% TFA. The spot was dried, and the mass spectra and tandem mass spectra of these compounds were collected using MALDI-TOF mass instrument. Compounds that were isolated more than one time from the screen were selected for postscreening validation (Supporting Information S3).
Fluorescence Polarization Assay
Probe concentrations were determined using the absorbance of fluorescein at 495 nm (ε495 = 78 000 M–1 cm–1) using a Nanodrop UV–vis spectrophotometer (Thermo Scientific). FP binding saturation experiments were performed in 384-well half area, medium bind microtiter plates (Greiner Bio-One) by titrating serially diluted IgY (10 nM to 14 μM) into PBS containing a probe concentration of 10 nM. FP experiments were performed on a 2104 EnVision Multilabel Plate Reader (PerkinElmer) using 450 excitation and 515 nm emission filters. Kd values and fitted saturation curves were obtained using Prism (GraphPad Software, Inc.) with a one-site saturation with Hill slope model. The data shown are averages of 3 experiments ± the standard deviation. Competition experiments were performed by first incubating 10 μM anti-ADP3 IgY with 10 nM of fluorescein-containing probe. Fourteen microliters of this solution was aliquoted into a 384-well plate. One microliter of serially diluted competitor ligand was added to each well and incubated for 15 min in the dark. FP was measured as described above. Plots shown are representative of 2 independent experiments. Curves were fit using Prism (GraphPad Software, Inc.) using a one site–Fit log(IC50) model.
Mock Screen Using Resynthesized Ligands 1 and 7
Compounds 1 and 7 were each resynthesized on 100 mg of 160 μm TentaGel macrobeads as described previously. After cleavage of the protecting groups, the beads were neutralized in 10% DIPEA in DMF. After washing the beads thoroughly in DMF, they were washed in water (10 × 2 mL) and equilibrated in water overnight. The following day, the beads were incubated first in TBS-T and then in PBS starting block. Each of the bead populations was separated into 4 aliquots of ∼250 beads and incubated with 10 nM, 25 nM, 75 nM or 200 nM anti-ADP3 IgY diluted into 50% starting block overnight at 4 °C. The beads were washed in TBS-T (3 × 2 mL) and hybridized with QDot655 anti-IgY as a 1:200 dilution in 50% starting block for 1 h at RT. The beads were washed in TBS-T (5 × 2 mL) and viewed under an inverted fluorescent microscope using DAPI filters. 100 beads from each population containing either 1 or 7 were scored as a hit or nonhit. Beads were scored as a hit if they exhibited a distinct red halo. The results in Figure 7 are given as percentage of beads scored as a hit.
Glossary
Abbreviations
- AQP4
aquaporin 4
- BAA
bromoacetic acid
- BPA
2-bromopropionic acid
- CHCA
α-cyano-4-hydroxycinnamic acid
- CNBr
cyanogen bromide
- DCM
dichloromethane
- DIC
N,N′-diisopropylcarbodiimide
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- ELISA
enzyme-linked immunosorbent assay
- FP
fluorescence polarization
- HPLC
high-performance liquid chromatography
- IgG
Immunoglobulin G
- IgY
immunoglobulin Y
- MALDI-TOF MS
matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry
- MMT
monomethoxytrityl
- NMO
neuromyelitis optica
- OBOC
one bead one compound
- PTA
peptide tertiary amide
- RT
room temperature
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TNF-α
tumor necrosis factor-α
Supporting Information Available
Further experimental details, the chemical structures of all compounds reported in this study, full description of the characterization of the library employed. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by a grant from the National Institutes of Health (DP3 DK094309) and a contract from the NHLBI (N01-HV-00242).
T.K. discloses a significant financial stake in Opko Health Inc., which is attempting to commercialize aspects of the diagnostic technology described in this Research Article.
The authors declare the following competing financial interest(s): I have substantial stock options (>$10,000) in Opko Health Inc., which is attempting to commercialize some of the technology described in this paper. This competing interest is indicated at the appropriate point in the manuscript.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Houghten R. A.; Pinilla C.; Blondelle S. E.; Appel J. R.; Dooley C. T.; Cuervo J. H. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 1991, 354, 84–86. [DOI] [PubMed] [Google Scholar]
- Lam K. S.; Salmon S. E.; Hersh E. M.; Hruby V. J.; Kazmierski W. M.; Knapp R. J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [DOI] [PubMed] [Google Scholar]
- Alluri P. G.; Reddy M. M.; Bachhawat-Sikder K.; Olivos H. J.; Kodadek T. Isolation of protein ligands from large peptoid libraries. J. Am. Chem. Soc. 2003, 125, 13995–14004. [DOI] [PubMed] [Google Scholar]
- Paulick M. G.; Hart K. M.; Brinner K. M.; Tjandra M.; Charych D. H.; Zuckermann R. N. Cleavable hydrophilic linker for one-bead-one-compound sequencing of oligomer libraries by tandem mass spectrometry. J. Comb. Chem. 2006, 8, 417–426. [DOI] [PubMed] [Google Scholar]
- Joo S. H.; Xiao Q.; Ling Y.; Gopishetty B.; Pei D. High-throughput sequence determination of cyclic peptide library members by partial Edman degradation/mass spectrometry. J. Am. Chem. Soc. 2006, 128, 13000–13009. [DOI] [PubMed] [Google Scholar]
- Thakkar A.; Cohen A. S.; Connolly M. D.; Zuckermann R. N.; Pei D. High-throughput sequencing of peptoids and peptide–peptoid hybrids by partial Edman degradation and mass spectrometry. J. Comb. Chem. 2009, 112294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Marik J.; Lam K. S. A novel peptide-based encoding system for “one-bead one-compound” peptidomimetic and small molecule combinatorial libraries. J. Am. Chem. Soc. 2002, 124, 7678–7680. [DOI] [PubMed] [Google Scholar]
- Halpin D. R.; Harbury P. DNA display 1. Sequence-encoded routing of DNA populations. PLoS Biol. 2004, 2, 1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannocci L.; Zhang Y.; Scheuermann J.; Leimbacher M.; De Bellis G.; Rizzi E.; Dumelin C.; Melkko S.; Neri D. High-throughput sequencing allows the identification of binding molecules isolated from DNA-encoded chemical libraries. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17670–17675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M. A.; Acharya R. A.; Arico-Muendel C. C.; Belyanskaya S. L.; Benjamin D. R.; Carlson N. R.; Centrella P. A.; Chiu C. H.; Creaser S. P.; Cuozzo J. W.; Davie C. P.; Ding Y.; Franklin G. J.; Franzen K. D.; Gefter M. L.; Hale S. P.; Hansen N. J.; Israel D. I.; Jiang J.; Kavarana M. J.; Kelley M. S.; Kollmann C. S.; Li F.; Lind K.; Mataruse S.; Medeiros P. F.; Messer J. A.; Myers P.; O’Keefe H.; Oliff M. C.; Rise C. E.; Satz A. L.; Skinner S. R.; Svendsen J. L.; Tang L.; van Vloten K.; Wagner R. W.; Yao G.; Zhao B.; Morgan B. A. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol. 2009, 5, 647–654. [DOI] [PubMed] [Google Scholar]
- Olivos H. J.; Bachhawat-Sikder K.; Kodadek T. Quantum dots as a visual aid for screening bead-bound combinatorial libraries. ChemBioChem 2003, 4, 1242–1245. [DOI] [PubMed] [Google Scholar]
- Lim H. S.; Archer C. T.; Kodadek T. Identification of a peptoid inhibitor of the proteasome 19S regulatory particle. J. Am. Chem. Soc. 2007, 129, 7750–7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udugamasooriya D. G.; Dineen S. P.; Brekken R. A.; Kodadek T. A peptoid “antibody surrogate” that antagonizes VEGF receptor 2 activity. J. Am. Chem. Soc. 2008, 130, 5744–5752. [DOI] [PubMed] [Google Scholar]
- Lian W.; Upadhyaya P.; Rhodes C. A.; Liu Y.; Pei D. Screening bicyclic peptide libraries for protein–protein interaction inhibitors: Discovery of a tumor necrosis factor-α antagonist. J. Am. Chem. Soc. 2013, 135, 11990–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Tan P. H.; Zhang Y.; Pei D. On-bead screening of combinatorial libraries: Reduction of nonspecific binding by decreasing surface ligand density. J. Comb. Chem. 2009, 11, 604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintersteiner M.; Kimmerlin T.; Kalthoff F.; Stoeckli M.; Garavel G.; Seifert J. M.; Meisner N. C.; Uhl V.; Buehler C.; Weidemann T.; Auer M. Single bead labeling method for combining confocal fluorescence on-bead screening and solution validation of tagged one-bead one-compound libraries. Chem. Biol. 2009, 16, 724–735. [DOI] [PubMed] [Google Scholar]
- Raveendra B.; Hao W.; Baccala R.; Reddy M. M.; Schilke J.; Bennett J. L.; Theofiliopolous A. N.; Kodadek T. Discovery of peptoid ligands for anti-Aquaporin 4 antibodies. Chem. Biol. 2013, 20, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon V. A.; Wingerchuk D. M.; Kryzer T. J.; Pittock S. J.; Lucchinetti C. F.; Fujihara K.; Nakashima I.; Weinshenker B. G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [DOI] [PubMed] [Google Scholar]
- Astle J. M.; Simpson L. S.; Huang Y.; Reddy M. M.; Wilson R.; Connell S.; Wilson J.; Kodadek T. Seamless bead to microarray screening: Rapid identification of the highest affinity protein ligands from large combinatorial libraries. Chem. Biol. 2010, 17, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintersteiner M.; Buehler C.; Auer M. On-bead screens sample narrower affinity ranges of protein–ligand interactions compared to equivalent solution assays. ChemPhysChem 2012, 13, 3472–3480. [DOI] [PubMed] [Google Scholar]
- Reddy M. M.; Wilson R.; Wilson J.; Connell S.; Gocke A.; Hynan L.; German D.; Kodadek T. Identification of candidate IgG biomarkers for Alzheimer’s Disease via combinatorial library screening. Cell 2011, 144, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran T. M.; Kodadek T. A liquid array platform for the multiplexed analysis of synthetic molecule–protein interactions. ACS Chem. Biol. 2014, 92339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Kodadek T. Synthesis and screening of stereochemically diverse combinatorial libraries of peptide tertiary amides. Chem. Biol. 2013, 20, 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquino C.; Sarkar M.; Chalmers M. J.; Mendes K.; Kodadek T.; Micalizio G. A biomimetic polyketide-inspired approach to small molecule ligand discovery. Nat. Chem. 2011, 4, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figliozzi G. M.; Goldsmith R.; Ng S. C.; Banville S. C.; Zuckermann R. N. Synthesis of N-substituted glycine peptoid libraries. Methods Enzymol. 1996, 267, 437–447. [DOI] [PubMed] [Google Scholar]
- Sarma B. K.; Kodadek T. Acylhydrazides as peptoid sub-monomers. Chem. Commun. 2011, 47, 10590–10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma B. K.; Kodadek T. Sub-monomer synthesis of a hybrid peptoid–azapeptoid library. ACS Comb. Sci. 2012, 14, 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.