

Abstract
Introduction
Segmental duplications (low-copy repeats) are the recently duplicated genomic segments in the human genome that display nearly identical (> 90%) sequences and account for about 5% of euchromatic regions. In germline, duplicated segments mediate nonallelic homologous recombination and thus cause both non-disease-causing copy-number variants and genomic disorders. To what extent duplicated segments play a role in somatic DNA rearrangements in cancer remains elusive. Duplicated segments often cluster and form genomic blocks enriched with both direct and inverted repeats (complex genomic regions). Such complex regions could be fragile and play a mechanistic role in the amplification of the ERBB2 gene in breast tumors, because repeated sequences are known to initiate gene amplification in model systems.
Methods
We conducted polymerase chain reaction (PCR)-based assays for primary breast tumors and analyzed publically available array-comparative genomic hybridization data to map a common copy-number breakpoint in ERBB2-amplified primary breast tumors. We further used molecular, bioinformatics, and population-genetics approaches to define duplication contents, structural variants, and haplotypes within the common breakpoint.
Results
We found a large (> 300-kb) block of duplicated segments that was colocalized with a common-copy number breakpoint for ERBB2 amplification. The breakpoint that potentially initiated ERBB2 amplification localized in a region 1.5 megabases (Mb) on the telomeric side of ERBB2. The region is very complex, with extensive duplications of KRTAP genes, structural variants, and, as a result, a paucity of single-nucleotide polymorphism (SNP) markers. Duplicated segments are varied in size and degree of sequence homology, indicating that duplications have occurred recurrently during genome evolution.
Conclusions
Amplification of the ERBB2 gene in breast tumors is potentially initiated by a complex region that has unusual genomic features and thus requires rigorous, labor-intensive investigation. The haplotypes we provide could be useful to identify the potential association between the complex region and ERBB2 amplification.
Introduction
Gene amplification is a cellular process characterized by a selective increase of a particular genomic region without a proportional increase of the entire genome [1-4]. The selective increase accompanies the overexpression of a particular gene within the genomic region that confers a growth advantage to the cell. The growth advantage derived from gene amplification has long been recognized as an important problem for cancer patients. Increased copy numbers of proto-oncogenes, such as MYC, MYCN, and ERBB2, leads to the overexpression of oncogene products that drives abnormal cell proliferation [5-9]. Abnormal cell proliferation results in cancer progression and poor patient survival [10,11]. In addition, gene amplification is an underlying mechanism for acquired therapy resistance, as cancer cells counteract therapeutic agents by overactivating either therapy-target genes (for example, BCR-ABL amplification) or alternative survival pathways (for example, MET amplification) [12-17]. Despite these adverse effects on survival of cancer patients, little is known about amplification mechanisms, and in particular, about the initiating processes of gene amplification.
During the processes of gene amplification, extra copies of large genomic segments accumulate in a cell. The accumulation could be initiated either (a) by aberrant recombination that results in the unequal distribution of chromosomal materials between daughter cells [18-22] or (b) by the loss of DNA-replication control that leads to the extra round of segmental DNA replication within a single cell cycle [23-25]. In normal cells, these processes are tightly regulated and are less likely to initiate gene amplification [26,27]. In contrast, cancer cells often lack these controls and could initiate the processes. Furthermore, cellular surveillance systems (checkpoints) that ensure genome integrity at several stages of the cell cycle are impaired in cancer cells [28,29] and could fail to eliminate cells with extra copies. Once the accumulation is initiated, it could lead to further accumulation by the growth advantage conferred by the amplified gene(s). Therefore, defining initiating processes is the key for the better understanding of the amplification mechanism. However, defining initiation processes in tumors in vivo is not an easy task, as current methods for evaluating gene amplification may not be feasible for capturing the amplification mechanism. Gene amplification has been measured as the increase of copy-numbers of particular genomic regions by array-comparative genomic hybridization (array-CGH) [30,31]. Although array-CGH covers the entire genome and identifies amplified regions that are important for tumor phenotypes with high confidence, such highly amplified regions may not be the initiating regions but rather the end products of adaptive evolution of cancer genomes. Next-generation sequencing could provide both copy-number profiles and somatic breakpoint sequences in cancer genomes [32,33]. Because of the copy-number increases, breakpoint sequences tend to be biased toward amplified regions and may represent late events during amplicon formation.
The difficulty in identifying initiation processes in tumors in vivo is typified by the ERBB2 amplification in breast cancer [34,35]. ERBB2 (v-erb-b2 erythroblastic leukemia viral oncogene homolog 2) encodes an epidermal growth-factor receptor HER2 (human epidermal growth factor receptor 2) and is amplified in 10% to 20% of invasive breast tumors [5,11]. As increased HER2 protein stimulates growth-factor signaling pathway and drives cell proliferation, ERBB2-amplified (HER2-positive) tumors are associated with advanced stages, recurrence, and poor patient survival [36,37]. Although the clinically significant phenotype has been known for more than two decades, the amplification mechanism remains elusive. Such information could be important for the better understanding of the etiology of ERBB2-amplified tumors and may have implications in future clinical practice. ERBB2-amplified tumors have been treated with the monoclonal antibody trastuzumab [38]. Trastuzumab binds to HER2 and downregulates growth signaling and thus has significantly improved treatment outcomes for patients with HER2-positive tumors [39-41]. An accurate diagnosis of ERBB2 amplification is critical, because trastuzumab is solely designed (and effective) only for tumors with ERBB2 amplification. Not only the mechanism of action, but also fatal cardiac side effects [42,43] and high costs (more than $100,000/year per patient) [44-46] indicate the necessity of accurate diagnosis. Currently, fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC) are two major diagnostic tests for identifying responders and nonresponders to trastuzumab [47]. However, these current diagnostic tests have some issues, including variable results between institutions and ambiguous diagnoses, such as "equivocal" in IHC [48,49]. Preanalytic factors, such as the processing of specimens, the fixation method, and the choice of antibodies also introduce variability [50].
Amplification mechanisms could provide new information that may be useful to clarify issues associated with current tests. ERBB2 amplification occurs as the amplification of a genomic region surrounding ERBB2. A particular haplotype within the region may be more susceptible to ERBB2 amplification than other haplotypes. In this scenario, defining haplotypes by using patients' normal DNA could help to clarify ambiguous cases. From the tumor-biology point of view, it is not known why a subset of tumors develops ERBB2 amplification. For example, according to the cell-of-origin model [51], only a subset of breast tumors derived from luminal progenitor cells is HER2 positive. A better understanding of the amplification mechanism could tell us whether the lineage determination is random or has any genetic basis.
To understand the initiating mechanisms of ERBB2 amplification, we took integrated genomic, molecular, and bioinformatic approaches. Array-CGH data indicated that ERBB2-amplified tumors showed a unique pattern of copy-number transitions [52] that could result from a specific amplification mechanism (breakage-fusion- bridge (BFB) cycles). By using the BFB cycles as a guide, we identified a genomic region that could initiate ERBB2 amplification. The region displays a large (300-kb), complex block of duplicated segments (sequence similarity ≥ 90%) and several deletion polymorphisms. Such repeated sequences could be important in the initiation of ERBB2 amplification, as it has been observed in model systems that the frequency of gene amplification is shown to be determined by the presence of repeated sequences at the recombination sites [53-55]. Deletion polymorphisms of such repeated sequences may affect the initiation, and thus the frequency of ERBB2 amplification. Our results indicate an important role of a complex genomic region in the etiology of primary breast tumors.
Materials and methods
Ethics statement
This study was approved under the Cleveland Clinic internal Institutional Review Board (IRB07-136: EXEMPT: Chromosome Breakage and DNA Palindrome Formation). Specimens were obtained under the auspices of IRB 7881 (Evaluation of Genetic and Molecular Markers in Patients with Breast Cancer). All patients consented to allow their cancer specimens to be used by researchers in an anonymized fashion. The consent form indicates that publication will take place without identifiers to protect the identity of any specific individual.
Samples and DNA extraction
Breast cancer tissues were obtained from the tissue archives in the Pathology Department, specifically from consenting patients (IRB 7881). HER2 status of these tumors was determined with FISH. We first examined hematoxylin/eosin (HE)-stained sections and confirmed that at least 80% of cellularities were derived from tumors. Five 10-mm sections were subject to DNA extraction.
Noncancerous normal DNA (HapMap DNA samples) was obtained from the Coriell Institute. The sample ID is listed in Additional file 1, Table S4.
To extract DNA, tissue sections were incubated in the lysis buffer (100 mM NaCl/10 mM Tris⋅HCl, pH 8.0/25 mM EDTA/0.5% SDS/proteinase K) for 24 hours at 37°C, followed by phenol/chloroform extraction and ethanol precipitation, as described previously [54].
Array-CGH data analysis
Array-CGH datasets for 200 Her2-positive breast tumors and control normal samples (GSE21259) [52] were obtained from Gene Expression Omnibus (GEO) repository in the National Center for Biotechnology Information (NCBI) website. Partek Genomics Suite (Partek) was used to analyze the data. Raw data were normalized by using the Robust Multi-Array Average (RMA) method. RMA consists of three steps: a background adjustment, quantile normalization, and final summary. Normalized data were used to calculate the copy number of chromosome 17 in breast tumors.
Real-time polymerase chain reaction
We used real-time PCR for measuring copy numbers in genomic DNA. Primers were designed for repeat-masked sequences of the human genome (hg18) by using MacVector (see Additional file 1, Table S5). We designed primers that amplify 100- to 200-bp genomic regions. Light Cycler 480 (Roche, Indianapolis, IN, USA) was used for real-time PCR.
For primer sets of ERBB2, 1, 2, 4, 5, 7, and H19, PCR reactions were carried out in a three-step 40-cycle reaction of 95°C for 30 seconds, 60°C for 3 seconds, and 72°C for 30 seconds by using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). For primer sets of 3, 6, and 8, reactions were carried out in a two-step 40-cycle reaction of 95°C for 15 seconds and 60°C for 60 seconds. We used 5 ng/μl of genomic DNA for each reaction. Each sample was run in triplicate and was normalized to the internal control of H19 on chromosome 11. The primers used for this analysis are described in Additional file 1, Table S5.
In silico analysis of duplication contents within the complex genomic region
A 400-kb region of chromosome 17 (36,350,000 to 36,750,000 in the human genome (hg18)) was divided into 500-bp segments (see Additional file 1, Table S1). Each segment was scanned for similar regions throughout the human genome with BLAT at the UCSC Genome Browser. To exclude the possibility of missing some of the duplicated segments that are located at the boundary of the 500-bp window, we rescanned the region by using a 2,000-bp window. We used similar criteria of sequence homology > 90%, size > 100 bp, to define (a) intraregional duplications (duplications within the 400-kb region), (b) intrachromosomal duplications (duplications between the 400-kb region and somewhere in chromosome 17 other than the 4,000-kb region), and (c) interchromosomal duplications (duplications between the 400-kb region and somewhere in the human genome other than chromosome 17).
For segments that showed similarity within the 400-kb region, a line was drawn for connecting the two fragments. Each line corresponded to a 500-bp region that includes the segment (sequence homology > 90%, size > 100 bp) mapping to another 500-bp region within the region.
Segments with > 90% homology and > 100 bp were further separated into subclassifications based on the sequence similarities and sizes. We binned the size of segments into seven groups (100 to 500 bp, 501 to 1,000 bp, 1,001 to 1,500 bp, 1,501 to 2,000 bp, 2,001 to 2,500 bp, 2,501 to 3,000 bp, and > 3,000 bp). After separating fragments into different-size bins, we defined the degree of sequence homology for each segment.
Deletion polymorphism and PCR genotyping assay
In total, 83 structural variants were found in the Database of Genomic Variants (DGV) [56] over a 350-kb region (36.35 to 36.7 Mb). These variants were characterized by a number of different studies by using a variety of different assays (microarrays and deep sequencing) and different numbers of samples (from one individual to HapMap population). Two studies determined genotypes of structural variants for three major HapMap populations [57,58]. Only one deletion polymorphism had a minor allele frequency > 5%.
To obtain genotypes for the deletion polymorphism, two independent primer sets were designed for amplifying either the deletion allele or the nondeletion allele (see Additional file 1, Table S5). PCR was carried out in a final reaction volume of 50 μl with 1.0 U Taq polymerase (GoTaq, Promega, Madison, WI, USA), 1.5 mM MgCl2, 200 μM dNTPs, 10 pmol of each primer, and 100 ng of genomic DNA. The thermal-cycling conditions used for amplification consisted of an initial denaturation step at 95°C for 2 minutes, followed by 30 cycles of denaturation at 95°C for 30 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 30 seconds.
Repeated masked sequences
To determine whether "high-copy" repetitive elements are enriched within the complex genomic region, we scanned a 3-Mb region of chromosome 17: 35,000,000 to 38,000,000 (hg18) by Repeat Masker. Repeat Masker identifies interspersed repeats and low-complexity DNA and annotates these repeats into classes: SINE, LINE, LTR DNA elements, low complexity, small RNA, simple repeats, and unclassified. We binned the 3-Mb sequence into sixty 50-kb regions and made a summary of the total bp composition of each element (see Additional file 1, Figure S1).
Linkage disequilibrium analysis
We used the HapMap SNP genotyping data (from Release 28 of International HapMap project) for three population sets: CEU, YRI, and CHB plus JPT. We took all SNP genotypes from chromosome 17: 36,350,000 to 36,800,000. To determine linkage disequilibrium between SNPs and the deletion polymorphism, we incorporated the genotype of deletion polymorphism (CNVR7096.1) from the study by Conrad et al. [58]. For convenience, we converted the genotypes of 0 (homozygous deletion), 1 (heterozygous), and 2 (homozygous nondeletion) to a format that could be incorporated into our existing snp data by assigning 0 to AA, 1 to AG, and 2 to GG. We incorporated the converted Conrad genotype data into the HapMap release 28 data and excluded (a) individuals from Release 28 that had not been genotyped for the CNVR7096.1 and (b) individuals for whom more than 50% of SNP genotypes were not determined. That left us with 178 YRI, 174 CEU, and 86 CHB+JPT individuals. D', LOD, and r2 values were calculated by using Haploview 4.2 [59].
Triangular plots were generated by using Haploview 4.2. Currently Haploview 4.2 does not support the most recent release (number 28). The previous rerelease (number 27) does have a paucity of SNPs for the 110-kb region within the complex genomic region, and we cannot generate a triangular blot for the entire region. Therefore, to include the SNPs from the release 28 into Haploview, we used SNP tools of Microsoft Excel [60] to convert the genotypes into a .ped file and .map file that are recognized by Haploview.
Results
A series of recombination events from a single break could establish the gradient of copy-number increase toward ERBB2
The ERBB2 gene is located at chromosome 17q11.2-12. Previous studies have shown that the amplified regions (ERBB2 amplicon) reside within chromosomes as homogeneously staining regions (HSRs) [61-65], but not in extrachromosomal, double-minute chromosomes (DMs). Deletions of the telomeric side of ERBB2 are common [66,67], indicating the involvement of DNA breaks in the ERBB2 amplification. A large genomic region surrounding the ERBB2 gene is amplified, and within the amplified region, ERBB2 is located in the most highly amplified segment [52,68,69]. Copy number decreases gradually as it goes farther from ERBB2, and ends as copy-number loss (a gradient of copy-number increase). Therefore, elucidating underlying mechanisms (a) for the intrachromosomal amplification and (b) for the gradient of copy-number increase could lead to the better understanding of the mechanism of ERBB2 amplification.
One mechanism underlying intrachromosomal amplification is a well-established amplification mechanism called the breakage-fusion-bridge (BFB) cycle. The BFB cycle consists of a series of recombination events and is initiated by a chromosome break (Figure 1A) [18,19,22,70]. The replication of a broken chromosome would lead to a chromosome structure called sister chromatid fusion, in which sister chromatids are fused at a broken end. The resulting chromosome with two centromeres will have another chromosome break when two centromeres segregate into different daughter nuclei. Such a break could be resolved into sister-chromatid fusion and would initiate another round of a break and fusion. Therefore, the BFB cycles could result in the accumulation of genomic segments within the chromosome.
Figure 1.
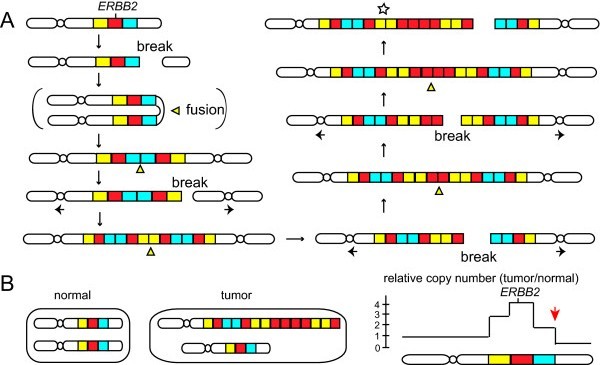
The breakage-fusion-bridge (BFB) cycles in chromosome 17 create the gradient of copy-number increases for ERBB2 amplification (model). (A) A break at the telomeric side of the ERBB2 gene can initiate the BFB cycles and can result in ERBB2 amplification. Genomic segments harboring the ERBB2 gene are shown in red; the flanking centromeric segment is shown in yellow; and a telomeric fragment is shown in blue. In this figure, the initiating break between the blue and the white segments leads to a series of chromatid fusions and inverted duplications (centers shown in yellow triangles) that results in a chromosome with the amplified ERBB2 gene (star). (B) The BFB cycles can result in the gradient of copy-number increases on an array-CGH platform. Illustrated are a normal cell with two normal chromosomes and a tumor cell with a chromosome generated by the BFB cycles (star in A) and a normal chromosome. An array-CGH experiment for measuring relative copy number (tumor/normal) shows the gradient of copy-number increase toward the ERBB2 gene in the tumor cell (right). Red arrow, the copy-number transition that marks the initiating region of ERBB2 amplification.
The accumulation of genomic segments by the BFB cycles could result in the gradient of copy-number increase (Figure 1B). An initial break could occur at the telomeric side (a blue segment) and lead to the formation of a dicentric chromosome. In the following cycle, a chromosome break at the centromeric side (a yellow segment) would be resolved into another dicentric chromosome. Further duplications and breaks would create a chromosome that accumulates segments within the chromosome. A chromosome having a segment harboring ERBB2 (a red segment) at very high copy number (Figure 1A, marked by a star) could be favored because of the growth advantage from ERBB2 overexpression. In such a chromosome, genomic segments flanking the ERBB2-harboring segment would also accumulate; however, because the flanking segments do not confer a growth advantage, their copy number would not be as high as that of the ERBB2-harboring segment. As a result, copy-number analysis for such a chromosome would show the different degree of copy-number increases between segments, and the highest increase would be seen for the segment harboring ERBB2 (Figure 1B). Importantly, such a scenario could predict a copy number transition for the initiating region of the BFB cycles. The initiating region (next to the blue segment) is marked by the transition from the copy-number loss to the low-level copy-number gain (red arrow) and is situated on the telomeric side of the ERBB2 gene.
A common copy-number breakpoint of ERBB2 amplicon
Where is the copy-number transition from a loss to a low-level gain for the ERBB2 amplicon? Although capturing low-level amplification is not as easy as detecting highly amplified regions with array-CGH, several studies have described such regions as the boundaries of the ERBB2 amplicon. For example, Sircoulomb et al. [71] analyzed 54 ERBB2-amplified breast tumors by using high-density array-CGH microarray and showed that a common telomeric boundary was predicted to be near the KRT40 (keratin 40) gene. The region was also described in another study as the boundary among ERBB2/TOP2A co-amplified tumors [72]. To determine whether the KRT40 region exhibits a common copy-number breakpoint, we analyzed a publically available array-CGH dataset that was obtained from 200 ERBB2-amplified tumors by using tiling-path BAC arrays (Figure 2) [52]. In the dataset, most of the tumors undergo copy-number transition from a high-level copy-number gain (ERBB2 region, showing in red) to a loss (regions in blue) within a 3-Mb region (chr17:35-Mb to 38-Mb in hg18) (Figure 2, top). Some tumors clearly show the copy-number transition from a gain to a loss near the KRT40 gene (Figure 2, bottom).
Figure 2.
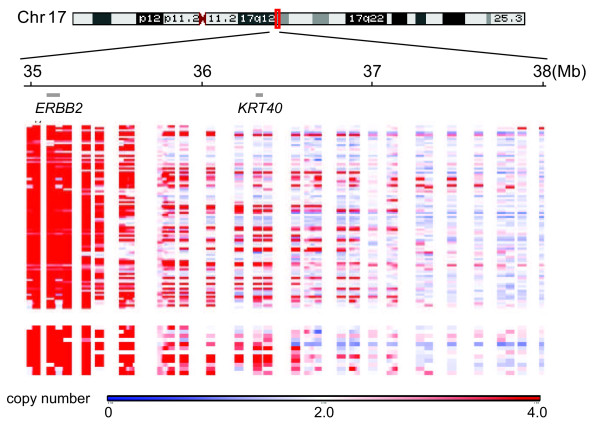
A common copy-number breakpoint near the KRT40 gene. Heat maps were created from the tiling-path BAC array data by Staaf et al. [52] and are shown for a 3-Mb region (35 to 38 Mb in hg18) of chromosome 17. The locations of ERBB2 and KRT40 are shown. Top, heat maps of 200 ERBB2-amplified breast tumors, and bottom, heat maps of a subset (11) of tumors with copy-number breakpoints near KRT40. Red, copy-number increase; blue, copy-number loss.
We confirmed the copy-number transition in the subset of ERBB2-amplified tumors independently by using real-time (quantitative) PCR. We designed an eight-PCR primer set for copy-number measurements within the 1.5-Mb region of the telomeric side of the ERBB2 gene (Figure 3A). In particular, we measured copy numbers by using four primer sets for the 370-kb region surrounding KRT40. To develop a sensitive and specific assay, PCR conditions and primers were optimized to provide copy numbers that were nearly equal to 1 in seven normal HapMap DNA samples (Figure 3B). Fifteen breast-tumor tissues in which ERBB2 amplification was determined either as ERBB2-positive or -negative with FISH were subject to the copy-number measurements. Consistent with the diagnoses with FISH, ERBB2 copy number remained low in 10 ERBB2-negative (by FISH) breast tumors (Figure 3C). In contrast, all five ERBB2-positive tumors showed copy-number increases for the ERBB2 gene (2.3- to 14-fold). Copy number decreased dramatically within the 500-kb region between ERBB2 and the primer set 1; however, two tumors (red and blue) had a low-level copy-number gain up to the region surrounding KRT40. In both cases, copy number decreased to one or less within the 370-kb region.
Figure 3.
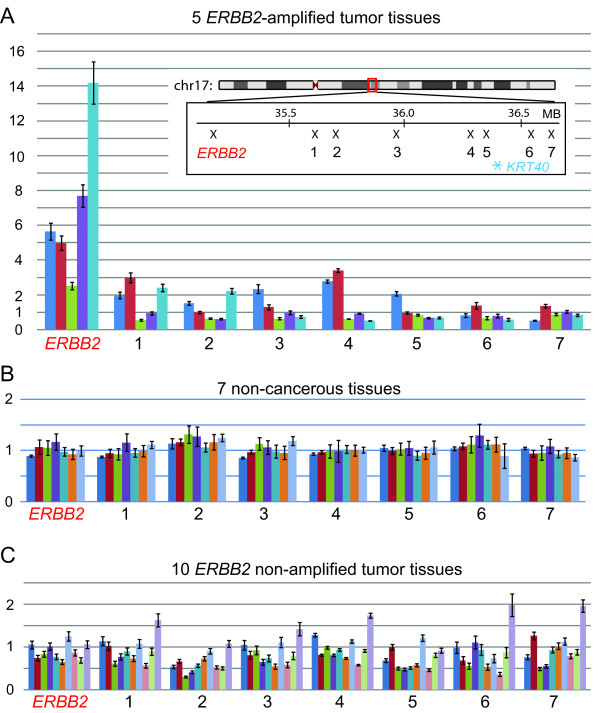
Real-time polymerase chain reaction (PCR)-based copy-number measurements for the telomeric half of the ERBB2 amplicon. Copy-number transitions for five ERBB2-amplified tumors (A), seven normal DNA samples from HapMap individuals (B), and 10 ERBB2-nonamplified tumors (C) are shown. Each color represents a copy-number transition of an individual tumor (or HapMap DNA in B). Note that two ERBB2-amplified tumors (blue and red in A) have a copy-number breakpoint near the KRT40 gene.
These results imply that a common copy-number breakpoint for ERBB2 amplification resides in the region near the KRT40 gene. Such a breakpoint between the copy-number gain and loss could possibly be an initiating region for ERBB2 amplification.
A large block of duplicated segments at the common copy-number breakpoint
What is a unique property of the genomic region surrounding the KRT40 gene? Is the region fragile and prone to DNA rearrangements? To address these questions, we conducted an extensive characterization of the region. The region consists of a gene family of keratin-associated protein (KRTAP) genes; 21 KRTAP genes are within the region (Figure 4-1). The KRTAP genes encode a major component of hair in mammals and play an essential role in the formation of rigid and resistant hair shafts [73,74]. Such a large number of genes for a single gene family could be derived from gene duplications during genome evolution and would create complex genomic regions harboring segments of high sequence identities.
Figure 4.
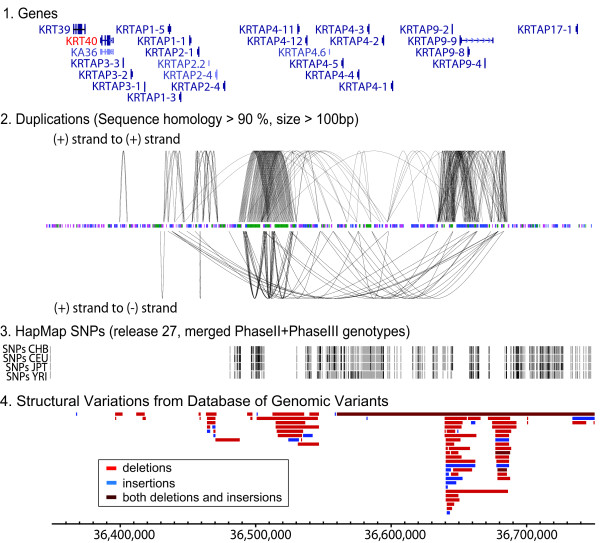
A complex genomic region at a common copy-number breakpoint of the ERBB2 amplicon. For the 400-kb region (from 36.350 to 36.750 Mb in hg18), duplicated segments (1), genes (2), the locations of HapMap SNPs in four major populations (Release 27) (3), and copy-number variants (from the Database of Genomic Variants) (4) are shown. In (1), duplicated segments are shown for either direct repeats (top) or inverted repeats (bottom). The distribution of repetitive sequences is also shown between the direct and inverted repeats. The (2), (3), and (4) were obtained from UCSC genome browser.
To determine the duplication contents, we scanned every 500-bp window in the region by Blat (UCSC genome Browser) and plotted segments that have more than 90% sequence homology (> 100-bp) with other windows (Figure 4-2 and Additional file, Table S1). We used a 100-bp cutoff rather than the conventional 1-kb cutoff, as such a short stretch of homology could still facilitate gene amplification [54,55]. A number of duplicated segments were identified within the region, both in the same strands (direct repeats, top) and between the complement strands (inverted repeats, bottom). Two large clusters of direct duplications are found (at around the coordinate 36.5-Mb and 36.65-Mb), and one of the duplications is 18-kb in size. These duplicated segments are not due to the extremely high content of repetitive elements, such as SINE elements, because the proportion of repetitive elements is very similar throughout the 3-Mb region surrounding the complex region (see Additional file, Figure S1).
Such extensive duplications create regions that are complex and difficult to investigate with current genomic approaches [75,76]. Failure to recognize duplications can lead to misinterpretation of marker genotypes [77,78]. For example, duplicated segments make it difficult to distinguish whether single-nucleotide changes are either the difference between duplicated segments (paralogous sequence variants) or allelic sequence variants (single-nucleotide polymorphisms, SNPs) [79,80]. Indeed, a set of SNPs that tag haploblocks in the human genome (HapMap SNPs, Release 27), an essential component of disease-association studies, is less well defined. An 110-kb region on the centromeric side does not have HapMap SNPs. Structural variants are common, and four deletion polymorphisms are within the region listed in the Database of Genomic Variants [81].
Sequence divergence between duplicated segments
Previous studies showed the association between somatic breakpoints in cancer genomes and evolutionary breakpoints [82,83]. Because segmental duplications colocalize with evolutionary breakpoints in primate genomes [84,85], duplication activities during primate evolution could illustrate the unstable nature of a complex genomic region.
First, we determined the frequency of duplicated segments for (a) duplications within the complex region, (b) duplications between the complex region and other regions in the same chromosome, and (c) duplications between the complex region and other regions in different chromosomes (Figure 5A). Duplications occurred predominantly (73.6%) within the complex region, suggesting that the recombination between duplicated segments within the region may also be frequent in somatic cells.
Figure 5.
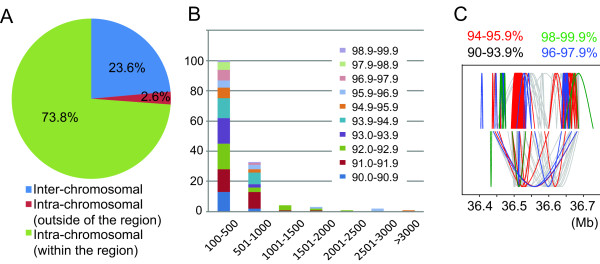
Recurrent duplications of genomic segments within the complex region during primate evolution. (A) A pie chart showing the proportion of duplications within the complex region, duplications between the complex region and outside of the region in the same chromosome, and duplications between the complex region and different chromosomes (interchromosomal duplications). Intrachromosomal duplications within the complex region account for three fourths of all the duplications. (B) Duplications within the complex region are binned based on size (x-axis), and the number of duplications for each bin is shown in the bar graph. A unique color is given based on the sequence identity between duplicated segments. (C) Inferred duplication activities within the complex region. Duplications are binned into four groups based on the sequence identity between duplicated segments, and older duplications (duplications with lower sequence identities) are overlaid by the newer duplications (duplications with higher sequence identities).
The frequency of duplication events during evolution could in part be addressed by sequence divergence between duplications. When a segment was duplicated, the resulting two segments were 100% identical in their DNA sequences. Mutations could have accumulated on each segment, which results in sequence divergence between two segments (the proportion of sequences that differs between duplicated segments). Assuming that mutations accumulate in a neutral fashion, whether duplications are newer or older could be in part inferred by using sequence divergence [86].
When we group the duplicated segments based on the sequence identities, sequence identities vary for each duplicated pair (Figure 5B). A large number of small duplicated segments (less than 1 kb) exist in which sequence identities differ between segments, ranging from 90% to nearly 100%. This is also the case for larger duplicated segments; the largest segment (18 kb) has sequence identity of 95.6%, whereas most of the 1- to 2-kb segments have 92% to 93% sequence identity.
Although gene conversion homogenizes duplicated segments and limits our ability to date duplications precisely by using sequence divergence [87,88], these results indicate that duplications have possibly occurred many times within the complex region during primate genome evolution.
Deletion polymorphisms within the complex region
Recombination between closely located repeats plays a critical role in the initiation of gene amplification in both mammalian cells and unicellular organisms [53-55,89]. We previously showed that as small as 79-bp DNA inverted repeats significantly increased the occurrence of gene amplification in mammalian cells [54]. Given the presence of duplicated segments and their structural variants within the region, a particular segment could promotes ERBB2 amplification, structural variants of which could be linked to the occurrence of ERBB2 amplification. Identifying such a segment directly might be difficult, however, because of the complexity of the region.
As an initial step, we defined haplotypes within the region. Different haplotypes could carry different genomic segments, and one haplotype could be associated with ERBB2 amplification. Because ERBB2 amplification occurs in 10% to 20% of breast tumors in all three major populations [90,91], the haplotype should likely be a common one in all populations. To define common haplotypes, we first searched for common deletion polymorphisms within the region from the Database of Genomic Variants (DGV) and the dbSNP database. Because of the paucity and the confounding effect from paralogous variants, SNP genotypes may not be as reliable as those of a deletion polymorphism. Furthermore, we could design a PCR-based genotyping assay for a deletion polymorphism to confirm that the variants are allelic, but not paralogous [92].
Although a number of studies reported deletion polymorphisms within the region, only two studies conducted genotyping on a population scale: copy-number variants studies from McCarroll et al. [57] for 270 HapMap samples and Conrad et al. [58] for 450 individuals of European, African, and East Asian ancestry: YRI (Yoruba in Ibadan, Nigeria), CEU (Utah residents with Northern and Western European ancestry from the CEPH collection), and CHB+JPT (Han Chinese in Beijing, China, and Japanese in Tokyo, Japan). Among the four (in MacCarroll et al.) and five (in Conrad et al.) deletion polymorphisms described in these studies within the region, only one is a common polymorphism (minor allele frequency > 5%). The polymorphism is located at the telomeric end of the complex region and overlaps with a 5.9-kb deletion polymorphism (rs72137527 from dbSNP database).
To confirm that rs72137527 is the deletion polymorphism, we developed a genotyping PCR assay and genotyped several HapMap individuals (Figure 6). First, the genotypes from 10 HapMap trios (father, mother, and offspring) were consistent with the pattern of mendelian inheritance. Thus, the deletion was confirmed as an allelic polymorphism, not as paralogous variants. Second, the genotyping results by PCR assay were highly consistent (35 of 38 individuals) with the previous study [58], indicating that rs72137527 is the deletion polymorphism genotyped by two studies. The deletion was likely to have occurred by nonallelic homologous recombination (NAHR [93-96]), as it is flanked by 679-bp duplicated segments that are 92.1% similar to each other.
Figure 6.
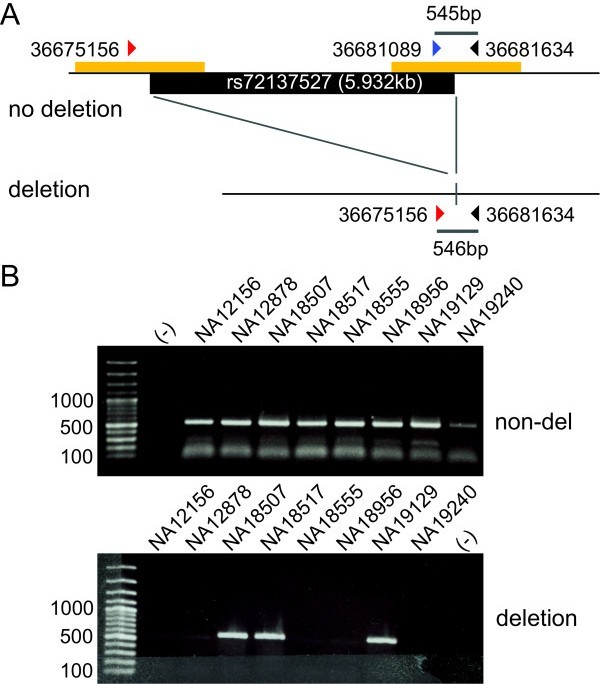
Genotyping polymerase chain reaction (PCR) for the deletion polymorphism rs72137527. (A) A PCR strategy for the deletion polymorphism located at the telomeric end of the complex region. Two independent forward primers (36675156 and 36681089) were paired with a common reverse primer (36681634) for amplifying either the nondeletion allele or the deletion allele. (B) Ethidium bromide staining gels are shown for either the PCR amplification of the deletion allele (deletion) or that of the nondeletion allele (non-del). DNA from 8 HapMap individuals was used.
Haploblocks within the complex genomic region
Deletion polymorphisms and SNPs are very often in linkage disequilibrium (LD) [97,98]. The extent of a haplotype (haploblock) harboring the deletion polymorphism can be determined by the LD analysis between the deletion polymorphism and HapMap SNPs. To define the LD, we calculated the squared correlation coefficient r2 between the deletion polymorphism and SNPs for three major populations (Figure 7 and see Additional file, Table S2). We found that several SNPs are in strong LD with the deletion polymorphism in all three populations. The LD blocks (r2 > 0.9) extend a longer distance for CEU (27 SNPs, 114.48-kb) and CHB+JPT (31 SNPs, 137.72-kb) than YRI (17 SNPs, 65.17 kb) (see Additional file, Table S2). We also noticed that LD decreases gradually with distance for YRI. In contrast, LD is discontinuous for both CEU and CHB+JPT. The smaller LD block for African populations is consistent with the previous observations and may reflect a population bottleneck when modern humans first left Africa [99].
Figure 7.
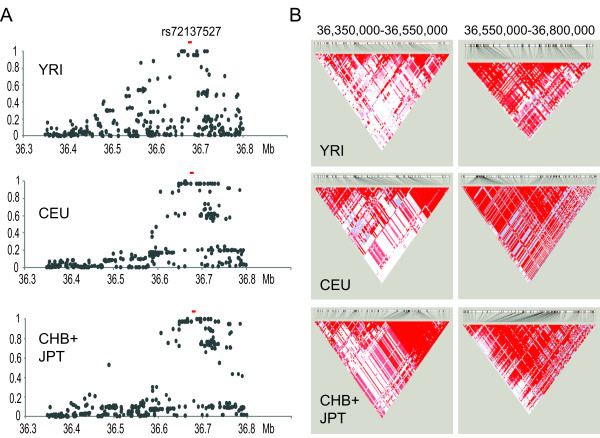
Haploblocks within the complex genomic region (chr17: 36,350,000-36,750,000 in hg18). (A) Linkage disequilibrium between the deletion polymorphism (rs72137527) and HapMap single-nucleotide polymorphisms (SNPs). The r2 values between the rs72137527 and each HapMap SNP are plotted against the physical locations of each SNP. (B) Haploblocks for the entire complex region. Triangular plots were generated by using Haploview for the HapMap SNP genotypes from three major populations (release 28) and are shown each for a centromeric half and a telomeric half. Red, strong linkage; white, no linkage.
We then used Haploview to illustrate haploblocks for the entire region by using the SNP genotypes from the HapMap Release 28 (Figure 7B), the newer release that fills the 110-kb SNP gap (Figure 4) in Release 27. Consistent with the LD analysis between the deletion polymorphism and SNPs, a large haploblock is found for the telomeric side of the complex genomic region. However, a haploblock is less clear and smaller for the centromeric side of the complex region. Given the fact that the centromeric regions do not have as many duplicated segments as the telomeric region (Figure 4), having a large gap in the HapMap Release 27 seems unexplainable. The centromeric side may have unusual features and will require further characterization for identifying better genotyping markers.
Discussion
In this study, we described a common copy-number breakpoint that potentially initiates ERBB2 amplification in primary breast tumors. The region is complex and consists of a large number of duplicated segments that form direct and inverted repeats. The sequence identities between duplicates are very high (> 90%), and some of them are more than 99% identical to each other. These duplicated segments are associated with the KRTAP gene family members, but not with high-copy repeats, such as SINE elements. Duplications appear to have occurred recurrently and predominantly within the region during primate evolution. These results suggest that the complex region could be more fragile than other unique loci and could play a mechanistic role in ERBB2 amplification.
Several lines of evidence support the unstable nature of complex genomic regions in the human genome. First, genomic regions with duplicated segments are preferred sites of non-disease-causing structural (copy number) variants [56,100]. The increased frequency of structural variants is due to the recombination between duplicated segments (non-allelic homologous recombination, NAHR [93-96]) in the germline. NAHR between segmental duplications leads to deletions, duplications, and inversions. Second, NAHR between duplicated segments also causes clinical phenotypes called genomic disorders [93-96,101]. NAHR between duplicated segments occurs recurrently and generates either duplications or deletions that determine the phenotypes of diseases. Recurrent NAHR for genomic disorders further supports the unstable nature of complex regions. Furthermore, the blocks of duplicated segments have been shown to be the most dynamic regions of the genome during primate evolution [102-104].
These facts would strongly argue for the unstable nature of complex genomic regions. Indeed, the important role of segmental duplications in creating somatic mutations in cancers is emerging. The breakpoints of isochromosome 17q, the most common isochromosome in human malignancy, was located within a large (> 30) inverted segmental duplication on 17p (isochromosome 17q, i17q) [105-107]. Translocation between chromosome 9 and 22, t(9;22)(q34;q11) causes the BCR/ABL gene fusion that is the underlying etiology of chronic myeloid leukemia (CML) [108]. From 10% to 20% of the translocation occurred between the 76-kb interchromosomal segmental duplications that are located either at the centromere proximal to ABL on chr 9 or the centromere distal to BCR on chr 22. The involvement of segmental duplications was also described for the microdeletion of PTEN tumor-suppressor gene in aggressive prostate cancers [109].
At the chromosome level, breakage-fusion-bridge (BFB) cycles are likely an underlying mechanism of ERBB2 amplification for at least a subset of breast tumors, as (a) the ERBB2 amplicons predominantly reside within a chromosome [61-65], and (b) copy-number loss at the telomeric side of the complex genomic regions (Figure 2) indicates chromosome breaks resulting in the loss of genetic materials. The BFB cycles have been shown to establish intrachromosomal amplicons for other oncogenes, such as CCND1 [110,111]. CCDN1 resides at chromosome 11q13 and is frequently amplified in head and neck tumors. CCND1 is surrounded by three clusters of segmental duplications. These clusters have been shown to colocalize with the boundaries of amplified regions [112], suggesting that a series of rearrangements could occur within these clusters during BFB cycles. In this regard, it is noteworthy that, in addition to the complex region described in this study, additional complex regions exist within ERBB2 amplicons. At the centromeric side, two large (a few hundred kb) euchromatic gaps of human genome assembly (hg18) are noted: one at 1.5 mega-base (Mb) and another at 3.3 Mb centromeric side of ERBB2 (see Additional file 1, Figure S2) [113]. Assembly gaps represent regions with full duplicated DNAs and/or complex, unclonable regions. Similar to CCND1 amplicon, these duplicated DNAs within gaps may serve as substrates for DNA rearrangements during BFB cycles.
We further found that other commonly amplified genes are also in close proximity to complex genomic regions. Among the 13 cancer genes that are most commonly amplified and overexpressed [114], five genes (ERBB2, CCND1, MYCL1, MDM4, and MYCN) are located within 1.5 Mb from either assembly gaps or blocks of duplicated segments (see Additional file, Table S3). Additionally, chromosome 1q21, a commonly amplified region in many tumor types, has 18 gaps within 6 Mb. In contrast, neither complex genomic regions nor assembly gaps are seen within the 6-Mb region surrounding MYC oncogene, which could explain a different mechanism for MYC amplification [115].
At the DNA level, sequence homology between duplicated segments could play an initiating role in BFB cycles and gene amplification. By using model systems, we and others showed that inverted repeats preexisting in the genome can nucleate the duplication of large genomic segments [22,53-55,89]. Duplicated segments could facilitate the initiation of BFB cycles in two ways.
First, inverted repeats can adopt Holliday junction-like structure by forming a cruciform. The resolution of a cruciform results in two chromosomal parts with hairpin-capped ends. The replication of a centromere-harboring part with a hairpin-capped end results in the formation of a dicentric chromosome and the initiation of BFB cycles.
Second, duplicated segments could adopt a complex secondary structure that can impose an obstacle to the progression of replication forks (Figure 8) [116,117]. As replication fork stalling and collapse could be processed into one-ended DNA breaks [118], the complex regions may have increased DNA breaks. The 5'- to 3'-end resection of one-ended DNA breaks exposes single-stranded DNA [119]. When the end of single-stranded DNA folds back and anneals to an inverted repeat sequence (intrastrand annealing [55]), it would prime DNA synthesis (break-induced replication, BIR [120]) and fill in the single-stranded gap to create a chromosome with a hairpin-capped end. Thus, the sequence homology between duplicated segments could be mutagenic and initiate BFB cycles.
Figure 8.
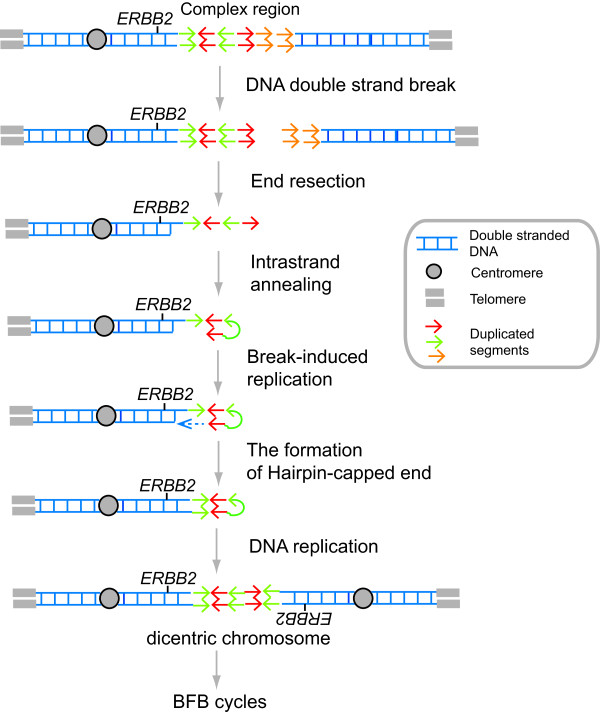
Nonallelic homologous recombination (NAHR) between duplicated segments initiates the BFB cycles (model). Duplicated segments within a complex region could adopt a complex secondary structure that can impose an obstacle for the progression of replication forks and would generate a DSB. The 5'- to 3'-end resection of a DSB exposes single-stranded DNA that would fold back and anneal between inverted repeat sequences (intrastrand annealing). BIR (break-induced replication) would prime DNA synthesis and fill the single-stranded gap to create a chromosome with a hairpin-capped end. The replication of the chromosome would generate a dicentric chromosome that initiates the BFB cycles.
In this regard, it is noteworthy that ERBB2 amplification is absent in breast tumors from BRCA1 mutation carriers [121]. BRCA1 binds to many proteins of DNA damage response and repair and thus plays a critical role in maintaining genome integrity [122]. BRCA1 is recruited to the chromatin with damaged DNA very early [123,124] and stimulates DNA end resection for homology-directed repair [125,126]. As BRCA1 mutant cells could lack efficient end resection, both mutation-free (conservative) and mutagenic homology-directed repair pathways could be impaired [127]. The conservative pathway is RAD51 dependent and repairs DSBs by using sister chromatids as a template, whereas the mutagenic pathway can be RAD51 independent [128] and could use repeated segments as a template. Therefore, the fact that ERBB2 amplification is rare in tumors with BRCA1 mutation may indicate that ERBB2 amplification is dependent on mutagenic homology-directed repair. In contrast, 15% of tumors derived from BRCA2 mutation carriers have ERBB2 amplification [121]. BRCA2 also functions for homology-directed repair; however, it has a more-specific role. BRCA2 has a RAD51-binding domain and plays an important role in conservative repair [129,130]. Indeed, in BRCA2 mutant cells, conservative repair was impaired, but mutagenic repair was not affected [127]. Therefore, the distinct ERBB2 amplification tendency between BRCA1 and BRCA2 mutant careers further suggests the involvement of recombination between repeated segments in ERBB2 amplification.
Alternatively, BIR initiated from one-ended DNA breaks at the sites of collapsed replication forks could be more processive, and repeated template switching (fork stalling and template switching, FoSTeS [131]) could result in complex genomic rearrangements and copy-number transition [132,133]. Newly established forks from one-ended DNA breaks could invade into either sister chromatid or homologues at nonallelic loci by using duplicated sequences or microhomology [134,135]. Invading strands can be unstable and often dissociate from template strands. The resulting free ends would repeat invasion several times at nonallelic loci to create complex genomic rearrangements. Copy-number increases from such complex rearrangements is relatively low, from twofold to threefold [132]. However, duplication and triplication of the segments could facilitate further rearrangements (for example, unequal sister chromatid exchange) and high-level amplification.
ERBB2 amplicons have been classified into two groups: a large amplicon including the TOP2A gene and a smaller, more-restricted amplicon (without TOP2A) surrounding the ERBB2 gene [66,67]. TOP2A encodes a DNA topoisomerase II (topoII) that controls and alters the topologic state of DNA in several aspects of DNA metabolism, such as chromosome segregation, transcription, and chromatin organization [136-138]. Because the complex region is located at the telomeric side of TOP2A gene, tumors having a breakpoint at the region belong to the TOP2A-coamplified tumors. Whether tumors without TOP2A amplification have independent common copy-number breakpoints is an important issue for future studies. It is also possible that an initiating break/recombination occurs at the complex region (or on a further telomeric side [139]) and, during the evolution of the amplicon, secondary rearrangements could delete both the region including TOP2A and the complex region from the amplicon. TOP2A deletion in ERBB2-amplified tumors is common [66,140]. Even in coamplified tumors, TOP2A and ERBB2 resided in different chromosomal domains [64,141], suggesting that secondary rearrangements separated the two genes from primary amplicons.
Given the established role of repeated segments in gene amplification in experimental systems, structural variants of such segments could have a significant effect in the occurrence of ERBB2 amplification [22]. Several structural variants are reported within the region, and some of them could be good candidates for the variants. However, defining the DNA sequences at breakpoints and identifying the segments responsible for ERBB2 amplification can be hampered by the complexity of the region. Therefore, as an initial step, we made an effort to define the haploblocks within the region. By combining the genotyping data from the deletion polymorphism and SNP genotypes, we were able to define two blocks, one of which showed a strong LD within the block. Our ongoing effort for further defining haploblocks and identifying genetic markers will provide a better understanding of the complex region. Such genetic markers could be useful, especially for the genomic regions where SNP markers are less well defined and genome-wide association studies [142,143] may have a limited power.
Conclusions
We show here a potential initiating role of a complex genomic region in ERBB2 amplification in breast cancer. The genomic sequence of the region is still ambiguous, as Genome Reference Consortium is providing an alternative sequence assembly for the region. Furthermore, two large sequence gaps (in hg18) exist on the centromeric side of ERBB2 (see Additional file, Figure S2). These sequence gaps likely contain many repeated sequences and structural variants and could also be fragile. Therefore, ERBB2 is flanked by many complex genomic regions that may not be sufficiently investigated by current genomic technologies. Investigating such regions in detail, including the patterns of DNA rearrangements at the nucleotide level, structural variants, and haplotypes within the regions, is important for the mechanistic study of ERBB2 amplification.
Abbreviations
Array-CGH: array-comparative genomic hybridization; BFB cycles: breakage-fusion-bridge cycles; CCND1: cyclin D1 gene; ERBB2: v-erb-b2 erythroblastic leukemia viral oncogene homolog 2; FISH: fluorescence in situ hybridization; HER2: human epidermal growth factor receptor 2; KRT40: keratin 40 gene; KRTAP: keratin-associated protein gene; IHC: immunohistochemistry; NAHR: nonallelic homologous recombination; TOP2A: topoisomerase 2A gene.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
MM and HT wrote the manuscript. MM and AI carried out the molecular genetic experiments. MM conducted SNP analyses. AK and RT conceived of the study, participated in its design and coordination, and helped to draft the manuscript. XC and RS conducted the microarray data analyses. GTB, JC, JL, and RT provided breast tumor tissues. All authors read and approved the final manuscript.
Supplementary Material
Figure S1. Repeat Masker Chr17 35-38 Mb (p.2). Figure S2. Sequence gaps near ERBB2 (in hg18) (p. 3). Table S1. Chromosome 17 BLAT results (p. 4 to 24). Table S2. LD map boundaries (p. 25). Table S3. Cancer gene amplification and complex genomic regions (p. 26). Table S4. HapMap sample ID list (p. 27). Table S5. PCR and qPCR primer list (p. 28).
Contributor Information
Michael Marotta, Email: marottm@ccf.org.
Xiongfong Chen, Email: chenx3@mail.nih.gov.
Ayako Inoshita, Email: ayaent@juntendo.ac.jp.
Robert Stephens, Email: bobs@ncifcrf.gov.
G Thomas Budd, Email: buddg@ccf.org.
Joseph P Crowe, Email: crowej@ccf.org.
Joanne Lyons, Email: lyonsj4@ccf.org.
Anna Kondratova, Email: kondraa@ccf.org.
Raymond Tubbs, Email: tubbsr@ccf.org.
Hisashi Tanaka, Email: tanakah@ccf.org.
Acknowledgements
We thank Drs George Stark, Charis Eng, and Scott Diede for comments on the manuscript. This work is supported by the funding from Cleveland Clinic, American Cancer Society, and National Cancer Institute (R01CA149385).
References
- Schwab M. Oncogene amplification in solid tumors. Semin Cancer Biol. 1999;14:319–325. doi: 10.1006/scbi.1999.0126. [DOI] [PubMed] [Google Scholar]
- Albertson DG. Gene amplification in cancer. Trends Genet. 2006;14:447–455. doi: 10.1016/j.tig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;14:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, Debatisse M, Giulotto E, Wahl GM. Recent progress in understanding mechanisms of mammalian DNA amplification. Cell. 1989;14:901–908. doi: 10.1016/0092-8674(89)90328-0. [DOI] [PubMed] [Google Scholar]
- Di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, Aaronson SA. erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science. 1987;14:178–182. doi: 10.1126/science.2885917. [DOI] [PubMed] [Google Scholar]
- Little CD, Nau MM, Carney DN, Gazdar AF, Minna JD. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature. 1983;14:194–196. doi: 10.1038/306194a0. [DOI] [PubMed] [Google Scholar]
- Collins S, Groudine M. Amplification of endogenous myc-related DNA sequences in a human myeloid leukaemia cell line. Nature. 1982;14:679–681. doi: 10.1038/298679a0. [DOI] [PubMed] [Google Scholar]
- Schwab M, Varmus HE, Bishop JM, Grzeschik KH, Naylor SL, Sakaguchi AY, Brodeur G, Trent J. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature. 1984;14:288–291. doi: 10.1038/308288a0. [DOI] [PubMed] [Google Scholar]
- Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, Gilbert F, Alt FW. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell. 1983;14:359–367. doi: 10.1016/0092-8674(83)90169-1. [DOI] [PubMed] [Google Scholar]
- Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;14:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;14:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;14:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;14:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Goker E, Waltham M, Kheradpour A, Trippett T, Mazumdar M, Elisseyeff Y, Schnieders B, Steinherz P, Tan C, Berman E, Bertino JR. Amplification of the dihydrofolate reductase gene is a mechanism of acquired resistance to methotrexate in patients with acute lymphoblastic leukemia and is correlated with p53 gene mutations. Blood. 1995;14:677–684. [PubMed] [Google Scholar]
- Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–2899. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 2003;14:75–85. doi: 10.1016/S1470-2045(03)00979-3. [DOI] [PubMed] [Google Scholar]
- Shannon KM. Resistance in the land of molecular cancer therapeutics. Cancer Cell. 2002;14:99–102. doi: 10.1016/S1535-6108(02)00101-0. [DOI] [PubMed] [Google Scholar]
- Smith KA, Gorman PA, Stark MB, Groves RP, Stark GR. Distinctive chromosomal structures are formed very early in the amplification of CAD genes in Syrian hamster cells. Cell. 1990;14:1219–1227. doi: 10.1016/0092-8674(90)90417-D. [DOI] [PubMed] [Google Scholar]
- Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 1997;14:215–225. doi: 10.1016/S0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- Gisselsson D, Pettersson L, Hoglund M, Heidenblad M, Gorunova L, Wiegant J, Mertens F, Dal Cin P, Mitelman F, Mandahl N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci USA. 2000;14:5357–5362. doi: 10.1073/pnas.090013497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo AW, Sabatier L, Fouladi B, Pottier G, Ricoul M, Murnane JP. DNA amplification by breakage/fusion/bridge cycles initiated by spontaneous telomere loss in a human cancer cell line. Neoplasia. 2002;14:531–538. doi: 10.1038/sj.neo.7900267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Yao MC. Palindromic gene amplification: an evolutionarily conserved role for DNA inverted repeats in the genome. Nat Rev Cancer. 2009;14:216–224. doi: 10.1038/nrc2591. [DOI] [PubMed] [Google Scholar]
- Ganley AR, Ide S, Saka K, Kobayashi T. The effect of replication initiation on gene amplification in the rDNA and its relationship to aging. Mol Cell. 2009;14:683–693. doi: 10.1016/j.molcel.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Green BM, Finn KJ, Li JJ. Loss of DNA replication control is a potent inducer of gene amplification. Science. 2010;14:943–946. doi: 10.1126/science.1190966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ, Payen C, Raghuraman MK, Dunham MJ. Origin-dependent inverted-repeat amplification: a replication-based model for generating palindromic amplicons. PLoS Genet. 2011;14:e1002016. doi: 10.1371/journal.pgen.1002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty TD, White A, Sanchez J. Suppression of gene amplification in human cell hybrids. Science. 1992;14:1425–1427. doi: 10.1126/science.1542791. [DOI] [PubMed] [Google Scholar]
- Tlsty TD. Normal diploid human and rodent cells lack a detectable frequency of gene amplification. Proc Natl Acad Sci USA. 1990;14:3132–3136. doi: 10.1073/pnas.87.8.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;14:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;14:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- van Beers EH, Nederlof PM. Array-CGH and breast cancer. Breast Cancer Res. 2006;14:210. doi: 10.1186/bcr1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkel D, Albertson DG. Comparative genomic hybridization. Annu Rev Genomics Hum Genet. 2005;14:331–354. doi: 10.1146/annurev.genom.6.080604.162140. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, Greenman CD, Jia M, Latimer C, Teague JW, Lau KW, Burton J, Quail MA, Swerdlow H, Churcher C, Natrajan R, Sieuwerts AM, Martens JW, Silver DP, Langerød A, Russnes HE, Foekens JA, Reis-Filho JS, van't Veer L, Richardson AL, Børresen-Dale AL. et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;14:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, Teague JW, Menzies A, Goodhead I, Turner DJ, Clee CM, Quail MA, Cox A, Brown C, Durbin R, Hurles ME, Edwards PA, Bignell GR, Stratton MR, Futreal PA. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;14:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiu KK, Natrajan R, Geyer FC, Ashworth A, Reis-Filho JS. DNA amplifications in breast cancer: genotypic-phenotypic correlations. Future Oncol. 2010;14:967–984. doi: 10.2217/fon.10.56. [DOI] [PubMed] [Google Scholar]
- Reis-Filho JS. Next-generation sequencing. Breast Cancer Res. 2009;14(Suppl 3):S12. doi: 10.1186/bcr2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, Kitchen P. Clinical significance of HER-2/neu oncogene amplification in primary breast cancer: The South Australian Breast Cancer Study Group. J Clin Oncol. 1993;14:1936–1942. doi: 10.1200/JCO.1993.11.10.1936. [DOI] [PubMed] [Google Scholar]
- Andrulis IL, Bull SB, Blackstein ME, Sutherland D, Mak C, Sidlofsky S, Pritzker KP, Hartwick RW, Hanna W, Lickley L, Wilkinson R, Qizilbash A, Ambus U, Lipa M, Weizel H, Katz A, Baida M, Mariz S, Stoik G, Dacamara P, Strongitharm D, Geddie W, McCready D. Neu/erbB-2 amplification identifies a poor-prognosis group of women with node-negative breast cancer: Toronto Breast Cancer Study Group. J Clin Oncol. 1998;14:1340–1349. doi: 10.1200/JCO.1998.16.4.1340. [DOI] [PubMed] [Google Scholar]
- Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA. 1992;14:4285–4289. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;14:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, Gianni L, Baselga J, Bell R, Jackisch C, Cameron D, Dowsett M, Barrios CH, Steger G, Huang CS, Andersson M, Inbar M, Lichinitser M, Láng I, Nitz U, Iwata H, Thomssen C, Lohrisch C, Suter TM, Rüschoff J, Suto T, Greatorex V, Ward C, Straehle C, McFadden E. et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;14:1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM, Pisansky TM, Fehrenbacher L, Kutteh LA, Vogel VG, Visscher DW, Yothers G, Jenkins RB, Brown AM, Dakhil SR, Mamounas EP, Lingle WL, Klein PM, Ingle JN, Wolmark N. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;14:1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- Tan-Chiu E, Yothers G, Romond E, Geyer CE Jr, Ewer M, Keefe D, Shannon RP, Swain SM, Brown A, Fehrenbacher L, Vogel VG, Seay TE, Rastogi P, Mamounas EP, Wolmark N, Bryant J. Assessment of cardiac dysfunction in a randomized trial comparing doxorubicin and cyclophosphamide followed by paclitaxel, with or without trastuzumab as adjuvant therapy in node-positive, human epidermal growth factor receptor 2-overexpressing breast cancer: NSABP B-31. J Clin Oncol. 2005;14:7811–7819. doi: 10.1200/JCO.2005.02.4091. [DOI] [PubMed] [Google Scholar]
- Seidman A, Hudis C, Pierri MK, Shak S, Paton V, Ashby M, Murphy M, Stewart SJ, Keefe D. Cardiac dysfunction in the trastuzumab clinical trials experience. J Clin Oncol. 2002;14:1215–1221. doi: 10.1200/JCO.20.5.1215. [DOI] [PubMed] [Google Scholar]
- Ferrusi IL, Marshall DA, Kulin NA, Leighl NB, Phillips KA. Looking back at 10 years of trastuzumab therapy: what is the role of HER2 testing? A systematic review of health economic analyses. Per Med. 2009;14:193–215. doi: 10.2217/17410541.6.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MC, Figg WD, Thurman PW. No rational theory for drug pricing. J Clin Oncol. 2004;14:962–963. doi: 10.1200/JCO.2004.99.318. [DOI] [PubMed] [Google Scholar]
- Elkin EB, Weinstein MC, Winer EP, Kuntz KM, Schnitt SJ, Weeks JC. HER-2 testing and trastuzumab therapy for metastatic breast cancer: a cost-effectiveness analysis. J Clin Oncol. 2004;14:854–863. doi: 10.1200/JCO.2004.04.158. [DOI] [PubMed] [Google Scholar]
- Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, van de Vijver M, Wheeler TM, Hayes DF. American Society of Clinical Oncology; College of American Pathologists. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;14:118–145. doi: 10.1200/JCO.2006.09.2775. [DOI] [PubMed] [Google Scholar]
- Roche PC, Suman VJ, Jenkins RB, Davidson NE, Martino S, Kaufman PA, Addo FK, Murphy B, Ingle JN, Perez EA. Concordance between local and central laboratory HER2 testing in the breast intergroup trial N9831. J Natl Cancer Inst. 2002;14:855–857. doi: 10.1093/jnci/94.11.855. [DOI] [PubMed] [Google Scholar]
- Paik S, Bryant J, Tan-Chiu E, Romond E, Hiller W, Park K, Brown A, Yothers G, Anderson S, Smith R, Wickerham DL, Wolmark N. Real-world performance of HER2 testing: National Surgical Adjuvant Breast and Bowel Project experience. J Natl Cancer Inst. 2002;14:852–854. doi: 10.1093/jnci/94.11.852. [DOI] [PubMed] [Google Scholar]
- Gown AM. Current issues in ER and HER2 testing by IHC in breast cancer. Mod Pathol. 2008;14(Suppl 2):S8–S15. doi: 10.1038/modpathol.2008.34. [DOI] [PubMed] [Google Scholar]
- Polyak K. Breast cancer: origins and evolution. J Clin Invest. 2007;14:3155–3163. doi: 10.1172/JCI33295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staaf J, Jonsson G, Ringner M, Vallon-Christersson J, Grabau D, Arason A, Gunnarsson H, Agnarsson BA, Malmstrom PO, Johannsson OT, Loman N, Barkardottir RB, Borg A. High-resolution genomic and expression analyses of copy number alterations in HER2-amplified breast cancer. Breast Cancer Res. 2010;14:R25. doi: 10.1186/bcr2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;14:183–193. doi: 10.1016/S0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Tapscott SJ, Trask BJ, Yao MC. Short inverted repeats initiate gene amplification through the formation of a large DNA palindrome in mammalian cells. Proc Natl Acad Sci USA. 2002;14:8772–8777. doi: 10.1073/pnas.132275999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Cao Y, Bergstrom DA, Kooperberg C, Tapscott SJ, Yao MC. Intrastrand annealing leads to the formation of a large DNA palindrome and determines the boundaries of genomic amplification in human cancer. Mol Cell Biol. 2007;14:1993–2002. doi: 10.1128/MCB.01313-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;14:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, Elliott AL, Parkin M, Hubbell E, Webster T, Mei R, Veitch J, Collins PJ, Handsaker R, Lincoln S, Nizzari M, Blume J, Jones KW, Rava R, Daly MJ, Gabriel SB, Altshuler D. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;14:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AW, Robson S, Stirrups K, Valsesia A, Walter K, Wei J. Wellcome Trust Case Control Consortium; Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME. Origins and functional impact of copy number variation in the human genome. Nature. 2010;14:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;14:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Chen B, Wilkening S, Drechsel M, Hemminki K. SNP_tools: a compact tool package for analysis and conversion of genotype data for MS-Excel. BMC Res Notes. 2009;14:214. doi: 10.1186/1756-0500-2-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XY, Meltzer PS, Dalton WS, Trent JM. Identification of cryptic sites of DNA sequence amplification in human breast cancer by chromosome microdissection. Nat Genet. 1994;14:155–161. doi: 10.1038/ng1094-155. [DOI] [PubMed] [Google Scholar]
- Muleris M, Almeida A, Gerbault-Seureau M, Malfoy B, Dutrillaux B. Detection of DNA amplification in 17 primary breast carcinomas with homogeneously staining regions by a modified comparative genomic hybridization technique. Genes Chromosomes Cancer. 1994;14:160–170. doi: 10.1002/gcc.2870100303. [DOI] [PubMed] [Google Scholar]
- Muleris M, Almeida A, Gerbault-Seureau M, Malfoy B, Dutrillaux B. Identification of amplified DNA sequences in breast cancer and their organization within homogeneously staining regions. Genes Chromosomes Cancer. 1995;14:155–163. doi: 10.1002/gcc.2870140302. [DOI] [PubMed] [Google Scholar]
- Nielsen KV, Muller S, Moller S, Schonau A, Balslev E, Knoop AS, Ejlertsen B. Aberrations of ERBB2 and TOP2A genes in breast cancer. Mol Oncol. 2010;14:161–168. doi: 10.1016/j.molonc.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalla P, Helbig R, Drieschner N, Flohr AM, Krohn M, Bullerdiek J. Molecular-cytogenetic analysis of fragmentation of chromosome 17 in the breast cancer cell line EFM-19. Anticancer Res. 2002;14:1987–1992. [PubMed] [Google Scholar]
- Jarvinen TA, Tanner M, Rantanen V, Barlund M, Borg A, Grenman S, Isola J. Amplification and deletion of topoisomerase IIalpha associate with ErbB-2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer. Am J Pathol. 2000;14:839–847. doi: 10.1016/S0002-9440(10)64952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner M, Jarvinen P, Isola J. Amplification of HER-2/neu and topoisomerase IIalpha in primary and metastatic breast cancer. Cancer Res. 2001;14:5345–5348. [PubMed] [Google Scholar]
- Kauraniemi P, Barlund M, Monni O, Kallioniemi A. New amplified and highly expressed genes discovered in the ERBB2 amplicon in breast cancer by cDNA microarrays. Cancer Res. 2001;14:8235–8240. [PubMed] [Google Scholar]
- Kauraniemi P, Kuukasjarvi T, Sauter G, Kallioniemi A. Amplification of a 280-kilobase core region at the ERBB2 locus leads to activation of two hypothetical proteins in breast cancer. Am J Pathol. 2003;14:1979–1984. doi: 10.1016/S0002-9440(10)63556-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. The stability of broken ends of chromosomes in Zea mays. Genetics. 1941;14:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircoulomb F, Bekhouche I, Finetti P, Adelaide J, Ben Hamida A, Bonansea J, Raynaud S, Innocenti C, Charafe-Jauffret E, Tarpin C, Ben Ayed F, Viens P, Jacquemier J, Bertucci F, Birnbaum D, Chaffanet M. Genome profiling of ERBB2-amplified breast cancers. BMC Cancer. 2010;14:539. doi: 10.1186/1471-2407-10-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriola E, Marchio C, Tan DS, Drury SC, Lambros MB, Natrajan R, Rodriguez-Pinilla SM, Mackay A, Tamber N, Fenwick K, Jones C, Dowsett M, Ashworth A, Reis-Filho JS. Genomic analysis of the HER2/TOP2A amplicon in breast cancer and breast cancer cell lines. Lab Invest. 2008;14:491–503. doi: 10.1038/labinvest.2008.19. [DOI] [PubMed] [Google Scholar]
- Wu DD, Irwin DM, Zhang YP. Molecular evolution of the keratin associated protein gene family in mammals, role in the evolution of mammalian hair. BMC Evol Biol. 2008;14:241. doi: 10.1186/1471-2148-8-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers MA, Langbein L, Winter H, Ehmann C, Praetzel S, Korn B, Schweizer J. Characterization of a cluster of human high/ultrahigh sulfur keratin-associated protein genes embedded in the type I keratin gene domain on chromosome 17q12-21. J Biol Chem. 2001;14:19440–19451. doi: 10.1074/jbc.M100657200. [DOI] [PubMed] [Google Scholar]
- Human genome: What's been most surprising? Cell. 2011;14:9–10. doi: 10.1016/j.cell.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Lupski JR. 2002 Curt Stern Award Address: Genomic disorders recombination-based disease resulting from genomic architecture. Am J Hum Genet. 2003;14:246–252. doi: 10.1086/346217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, Chakravarti A, Patel PI. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;14:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- Matise TC, Chakravarti A, Patel PI, Lupski JR, Nelis E, Timmerman V, Van Broeckhoven C, Weeks DE. Detection of tandem duplications and implications for linkage analysis. Am J Hum Genet. 1994;14:1110–1121. [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE. Diversity of human copy number variation and multicopy genes. Science. 2010;14:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman D, White SJ, Potter S, Eichler EE, Den Dunnen JT, Brookes AJ. Complex SNP-related sequence variation in segmental genome duplications. Nat Genet. 2004;14:861–866. doi: 10.1038/ng1401. [DOI] [PubMed] [Google Scholar]
- Zhang J, Feuk L, Duggan GE, Khaja R, Scherer SW. Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet Genome Res. 2006;14:205–214. doi: 10.1159/000095916. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Larkin DM, Everts-van der Wind A, Bourque G, Tesler G, Auvil L, Beever JE, Chowdhary BP, Galibert F, Gatzke L, Hitte C, Meyers SN, Milan D, Ostrander EA, Pape G, Parker HG, Raudsepp T, Rogatcheva MB, Schook LB, Skow LC, Welge M, Womack JE, O'brien SJ, Pevzner PA, Lewin HA. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science. 2005;14:613–617. doi: 10.1126/science.1111387. [DOI] [PubMed] [Google Scholar]
- Darai-Ramqvist E, Sandlund A, Muller S, Klein G, Imreh S, Kost-Alimova M. Segmental duplications and evolutionary plasticity at tumor chromosome break-prone regions. Genome Res. 2008;14:370–379. doi: 10.1101/gr.7010208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samonte RV, Eichler EE. Segmental duplications and the evolution of the primate genome. Nat Rev Genet. 2002;14:65–72. doi: 10.1038/nrg705. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;14:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- She X, Liu G, Ventura M, Zhao S, Misceo D, Roberto R, Cardone MF, Rocchi M, Green ED, Archidiacano N, Eichler EE. A preliminary comparative analysis of primate segmental duplications shows elevated substitution rates and a great-ape expansion of intrachromosomal duplications. Genome Res. 2006;14:576–583. doi: 10.1101/gr.4949406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay SJ, Khajavi M, Lupski JR, Hurles ME. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am J Hum Genet. 2006;14:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta M, Piontkivska H, Tanaka H. Molecular trajectories leading to the alternative fates of duplicate genes. PLoS One. 2012;14:e38958. doi: 10.1371/journal.pone.0038958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan V, Mieczkowski PA, Kim HM, Petes TD, Lobachev KS. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell. 2006;14:1283–1296. doi: 10.1016/j.cell.2006.04.042. [DOI] [PubMed] [Google Scholar]
- Kwong A, Mang OW, Wong CH, Chau WW, Law SC. Breast cancer in Hong Kong, Southern China: the first population-based analysis of epidemiological characteristics, stage-specific, cancer-specific, and disease-free survival in breast cancer patients: 1997-2001. Ann Surg Oncol. 2011;14:3072–3078. doi: 10.1245/s10434-011-1960-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund MJ, Butler EN, Hair BY, Ward KC, Andrews JH, Oprea-Ilies G, Bayakly AR, O'Regan RM, Vertino PM, Eley JW. Age/race differences in HER2 testing and in incidence rates for breast cancer triple subtypes: a population-based study and first report. Cancer. 2010;14:2549–2559. doi: 10.1002/cncr.25016. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Marotta M, Eichler EE, Eng C, Tanaka H. Linkage disequilibrium between two high-frequency deletion polymorphisms: implications for association studies involving the glutathione-S transferase (GST) genes. PLoS Genet. 2009;14:e1000472. doi: 10.1371/journal.pgen.1000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders ten years on. Genome Med. 2009;14:42. doi: 10.1186/gm42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;14:74–82. doi: 10.1016/S0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/S0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;14:211–220. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, Dallaire S, Gabriel SB, Lee C, Daly MJ, Altshuler DM. International HapMap Consortium. Common deletion polymorphisms in the human genome. Nat Genet. 2006;14:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet. 2006;14:82–85. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]
- Wall JD, Pritchard JK. Haplotype blocks and linkage disequilibrium in the human genome. Nat Rev Genet. 2003;14:587–597. doi: 10.1038/nrg1123. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, Oseroff VV, Albertson DG, Pinkel D, Eichler EE. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;14:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, Fitzpatrick CA, Segraves R, Richmond TA, Guiver C, Albertson DG, Pinkel D, Eis PS, Schwartz S, Knight SJ, Eichler EE. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;14:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Baertsch R, Kent WJ, Haussler D, Eichler EE. Hotspots of mammalian chromosomal evolution. Genome Biol. 2004;14:R23. doi: 10.1186/gb-2004-5-4-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armengol L, Pujana MA, Cheung J, Scherer SW, Estivill X. Enrichment of segmental duplications in regions of breaks of synteny between the human and mouse genomes suggest their involvement in evolutionary rearrangements. Hum Mol Genet. 2003;14:2201–2208. doi: 10.1093/hmg/ddg223. [DOI] [PubMed] [Google Scholar]
- Johnson ME, Cheng Z, Morrison VA, Scherer S, Ventura M, Gibbs RA, Green ED, Eichler EE. Recurrent duplication-driven transposition of DNA during hominoid evolution. Proc Natl Acad Sci USA. 2006;14:17626–17631. doi: 10.1073/pnas.0605426103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbouti A, Stankiewicz P, Nusbaum C, Cuomo C, Cook A, Hoglund M, Johansson B, Hagemeijer A, Park SS, Mitelman F, Lupski JR, Fioretos T. The breakpoint region of the most common isochromosome, i(17q), in human neoplasia is characterized by a complex genomic architecture with large, palindromic, low-copy repeats. Am J Hum Genet. 2004;14:1–10. doi: 10.1086/380648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Willner GA, Lopez-Terrada D, Bhattacharjee MB, Patel KU, Stankiewicz P, Lupski JR, Pfeifer JD, Perry A. Early recurrence in standard-risk medulloblastoma patients with the common idic(17)(p11.2) rearrangement. Neuro Oncol. 2012;14:831–840. doi: 10.1093/neuonc/nos086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. Copy number variation at the breakpoint region of isochromosome 17q. Genome Res. 2008;14:1724–1732. doi: 10.1101/gr.080697.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albano F, Anelli L, Zagaria A, Coccaro N, D'Addabbo P, Liso V, Rocchi M, Specchia G. Genomic segmental duplications on the basis of the t(9;22) rearrangement in chronic myeloid leukemia. Oncogene. 2010;14:2509–2516. doi: 10.1038/onc.2009.524. [DOI] [PubMed] [Google Scholar]
- Yoshimoto M, Ludkovski O, DeGrace D, Williams JL, Evans A, Sircar K, Bismar TA, Nuin P, Squire JA. PTEN genomic deletions that characterize aggressive prostate cancer originate close to segmental duplications. Genes Chromosomes Cancer. 2012;14:149–160. doi: 10.1002/gcc.20939. [DOI] [PubMed] [Google Scholar]
- Shuster MI, Han L, Le Beau MM, Davis E, Sawicki M, Lese CM, Park NH, Colicelli J, Gollin SM. A consistent pattern of RIN1 rearrangements in oral squamous cell carcinoma cell lines supports a breakage-fusion-bridge cycle model for 11q13 amplification. Genes Chromosomes Cancer. 2000;14:153–163. doi: 10.1002/(SICI)1098-2264(200006)28:2<153::AID-GCC4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Reshmi SC, Roychoudhury S, Yu Z, Feingold E, Potter D, Saunders WS, Gollin SM. Inverted duplication pattern in anaphase bridges confirms the breakage-fusion-bridge (BFB) cycle model for 11q13 amplification. Cytogenet Genome Res. 2007;14:46–52. doi: 10.1159/000097425. [DOI] [PubMed] [Google Scholar]
- Gibcus JH, Kok K, Menkema L, Hermsen MA, Mastik M, Kluin PM, van der Wal JE, Schuuring E. High-resolution mapping identifies a commonly amplified 11q13.3 region containing multiple genes flanked by segmental duplications. Hum Genet. 2007;14:187–201. doi: 10.1007/s00439-006-0299-6. [DOI] [PubMed] [Google Scholar]
- Zody MC, Garber M, Adams DJ, Sharpe T, Harrow J, Lupski JR, Nicholson C, Searle SM, Wilming L, Young SK, Abouelleil A, Allen NR, Bi W, Bloom T, Borowsky ML, Bugalter BE, Butler J, Chang JL, Chen CK, Cook A, Corum B, Cuomo CA, de Jong PJ, DeCaprio D, Dewar K, FitzGerald M, Gilbert J, Gibson R, Gnerre S, Goldstein S. et al. DNA sequence of human chromosome 17 and analysis of rearrangement in the human lineage. Nature. 2006;14:1045–1049. doi: 10.1038/nature04689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer. 2010;14:59–64. doi: 10.1038/nrc2771. [DOI] [PubMed] [Google Scholar]
- Storlazzi CT, Fioretos T, Surace C, Lonoce A, Mastrorilli A, Strombeck B, D'Addabbo P, Iacovelli F, Minervini C, Aventin A, Dastugue N, Fonatsch C, Hagemeijer A, Jotterand M, Mühlematter D, Lafage-Pochitaloff M, Nguyen-Khac F, Schoch C, Slovak ML, Smith A, Solè F, Van Roy N, Johansson B, Rocchi M. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet. 2006;14:933–942. doi: 10.1093/hmg/ddl010. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci USA. 2008;14:9936–9941. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD. Chromosomal translocations in yeast induced by low levels of DNA polymerase: a model for chromosome fragile sites. Cell. 2005;14:587–598. doi: 10.1016/j.cell.2004.12.039. [DOI] [PubMed] [Google Scholar]
- Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat Res. 2003;14:103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Haber JE. Partners and pathwaysrepairing a double-strand break. Trends Genet. 2000;14:259–264. doi: 10.1016/S0168-9525(00)02022-9. [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Haber JE. Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem. 2006;14:111–135. doi: 10.1146/annurev.biochem.74.082803.133234. [DOI] [PubMed] [Google Scholar]
- Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;14:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;14:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;14:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;14:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;14:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. 1998;14:25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;14:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1996;14:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esashi F, Galkin VE, Yu X, Egelman EH, West SC. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Struct Mol Biol. 2007;14:468–474. doi: 10.1038/nsmb1245. [DOI] [PubMed] [Google Scholar]
- Galkin VE, Esashi F, Yu X, Yang S, West SC, Egelman EH. BRCA2 BRC motifs bind RAD51-DNA filaments. Proc Natl Acad Sci USA. 2005;14:8537–8542. doi: 10.1073/pnas.0407266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;14:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Carvalho CM, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, McCall A, Pivnick EK, Hines-Dowell S, Seaver LH, Friehling L, Lee S, Smith R, Del Gaudio D, Withers M, Liu P, Cheung SW, Belmont JW, Zoghbi HY, Hastings PJ, Lupski JR. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet. 2011;14:1074–1081. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, Bacino CA, Campos-Acevedo LD, Delgado MR, Freedenberg D, Garnica A, Grebe TA, Hernández-Almaguer D, Immken L, Lalani SR, McLean SD, Northrup H, Scaglia F, Strathearn L, Trapane P, Kang SH, Patel A, Cheung SW, Hastings PJ, Stankiewicz P. et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;14:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;14:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;14:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal N, Parvin JD. DNA topoisomerase IIalpha is required for RNA polymerase II transcription on chromatin templates. Nature. 2001;14:435–438. doi: 10.1038/35096590. [DOI] [PubMed] [Google Scholar]
- Uemura T, Ohkura H, Adachi Y, Morino K, Shiozaki K, Yanagida M. DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell. 1987;14:917–925. doi: 10.1016/0092-8674(87)90518-6. [DOI] [PubMed] [Google Scholar]
- Holm C, Goto T, Wang JC, Botstein D. DNA topoisomerase II is required at the time of mitosis in yeast. Cell. 1985;14:553–563. doi: 10.1016/S0092-8674(85)80028-3. [DOI] [PubMed] [Google Scholar]
- Lamy PJ, Fina F, Bascoul-Mollevi C, Laberenne AC, Martin PM, Ouafik L, Jacot W. Quantification and clinical relevance of gene amplification at chromosome 17q12-q21 in human epidermal growth factor receptor 2-amplified breast cancers. Breast Cancer Res. 2011;14:R15. doi: 10.1186/bcr2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks DG, Yoder BJ, Pettay J, Swain E, Tarr S, Hartke M, Skacel M, Crowe JP, Budd GT, Tubbs RR. The incidence of topoisomerase II-alpha genomic alterations in adenocarcinoma of the breast and their relationship to human epidermal growth factor receptor-2 gene amplification: a fluorescence in situ hybridization study. Hum Pathol. 2005;14:348–356. doi: 10.1016/j.humpath.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Jarvinen TA, Tanner M, Barlund M, Borg A, Isola J. Characterization of topoisomerase II alpha gene amplification and deletion in breast cancer. Genes Chromosomes Cancer. 1999;14:142–150. doi: 10.1002/(SICI)1098-2264(199910)26:2<142::AID-GCC6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Broeks A, Schmidt MK, Sherman ME, Couch FJ, Hopper JL, Dite GS, Apicella C, Smith LD, Hammet F, Southey MC, Van 't Veer LJ, de Groot R, Smit VT, Fasching PA, Beckmann MW, Jud S, Ekici AB, Hartmann A, Hein A, Schulz-Wendtland R, Burwinkel B, Marme F, Schneeweiss A, Sinn HP, Sohn C, Tchatchou S, Bojesen SE, Nordestgaard BG, Flyger H, Ørsted DD. et al. Low penetrance breast cancer susceptibility loci are associated with specific breast tumor subtypes: findings from the Breast Cancer Association Consortium. Hum Mol Genet. 2011;14:3289–3303. doi: 10.1093/hmg/ddr228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou AC, Wang X, Fredericksen ZS, McGuffog L, Tarrell R, Sinilnikova OM, Healey S, Morrison J, Kartsonaki C, Lesnick T, Ghoussaini M, Barrowdale D. EMBRACE; Peock S, Cook M, Oliver C, Frost D, Eccles D, Evans DG, Eeles R, Izatt L, Chu C, Douglas F, Paterson J, Stoppa-Lyonnet D, Houdayer C, Mazoyer S, Giraud S, Lasset C. et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat Genet. 2010;14:885–892. doi: 10.1038/ng.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Repeat Masker Chr17 35-38 Mb (p.2). Figure S2. Sequence gaps near ERBB2 (in hg18) (p. 3). Table S1. Chromosome 17 BLAT results (p. 4 to 24). Table S2. LD map boundaries (p. 25). Table S3. Cancer gene amplification and complex genomic regions (p. 26). Table S4. HapMap sample ID list (p. 27). Table S5. PCR and qPCR primer list (p. 28).