

Abstract
HER2/Neu/ERBB2 is a receptor tyrosine kinase overexpressed in approximately 20% of human breast tumors. Truncated or mutant isoforms which show increased oncogenicity compared to the wild-type receptor are found in many breast tumors. Here we report that constitutively active ERBB2 sensitizes human breast epithelial cells to agents that induce endoplasmic reticulum (ER) stress, altering the unfolded protein response (UPR) of these cells. Deregulation of the ERK, AKT and mTOR activities elicited by mutant ERBB2 were involved in mediating this differential UPR response, elevating the response to ER stress and apoptotic cell death. Mechanistic investigations revealed that the increased sensitivity of mutant ERBB2-expressing cells to ER stress relied upon a UPR effector signaling involving the PERK-ATF4-CHOP pathway, upregulation of the proapoptotic cell surface receptor TRAIL-R2 and activation of proapoptotic caspase-8. Collectively, our results offer a rationale for the therapeutic exploration of treatments inducing ER stress against mutant ERBB2-expressing breast tumor cells.
Keywords: ER stress, ERBB2, ERK, Akt, mTOR, TRAIL-R2
Introduction
In response to different environmental and physiological stress conditions that increase the load of unfolded proteins in the ER, protein sensors located in the luminal face of the ER membrane activate the unfolded protein response (UPR) (1). Activation of this signaling pathway leads to a reduction in the influx of proteins into the ER, activates protein degradation pathways and increases the folding capacity of the ER (2). In vertebrates, three different types of ER stress transducers have been identified. Each type defines a distinct branch of the UPR that is mediated by protein kinase RNA (PKR)-like ER kinase (PERK) (3), inositol-requiring protein-1 (Ire1) (4) or activating transcription factor-6 (ATF6) (5). In each case, an integral membrane protein senses the protein-folding status in the ER lumen and transmits this information across the ER membrane to the cytosol and nucleus (2). These mechanisms allow adaptive and repair mechanisms that re-establish homeostasis. However, above a certain threshold, unresolved ER stress results in apoptosis (6).
As a major regulator of cell growth, metabolism and survival, the mammalian target of rapamycin (mTOR) pathway is commonly activated during oncogenesis (7). Thus, inactivating mutations or deletions of PTEN lead to Akt and mTOR activation and occur frequently in human cancers (8). Furthermore, loss of the upstream regulators of the mTOR pathway TSC1 and TSC2 leads to constitutive activation of mTORC1 and results in the development of tumors (9). Interestingly, tumor cells harbouring an activated mTOR pathway are more sensitive to ER stress-induced cell death (10-12), although the mechanism underlying this cell death process remains to be elucidated.
ERBB2 is a member of the ERBB receptor family, which also includes the epidermal growth factor receptor (EGFR, ERBB1), ERBB3, and ERBB4. Ligand binding to the extracellular domains of EGFR, ERBB3, and ERBB4 leads to the formation of catalytically active homo- and heterodimers to which ERBB2 is recruited as a preferred partner (13). Activation of the ERBB receptors leads to receptor autophosphorylation of C-terminal tyrosines and recruitment to these sites of cytoplasmic signal transducers that activate several downstream signaling pathways, such as the extracellular signal-regulated kinase and the phosphoinositide-3-kinase/AKT/mTOR pathways (14). Amplification of a genomic region containing the ERBB2 gene on chromosome 17q12 has been observed in approximately 25% of invasive breast tumors (15). ERBB2 overexpression is frequently accompanied by the occurrence of truncated forms of the receptor which are characterized by enhanced oncogenic potential (16, 17). In addition, somatic mutations in the ERBB2 gene have been reported in a number of tumors, including breast carcinomas, some of which results in a gain-of-function compared with wild-type ERBB2 (18, 19).
Herein, we have addressed the issue of the sensitivity to ER stress of human breast epithelial cells expressing an activated form of the ERBB2 oncogene. We show that mutant ERBB2 expression in breast epithelial cells leads to an overactivation of the UPR and markedly sensitizes these cells to ER stress-induced apoptosis. Hyperactivation of the PERK/ATF4/CHOP pathway in mutant ERBB2 cells following ER stress up-regulates TRAIL-R2 expression resulting in the induction of a caspase-8-mediated and mitochondria-operated apoptotic pathway. Importantly, deregulation of the PERK/ATF4/CHOP/TRAIL-R2 pathway and sensitivity of mutant ERBB2 to ER stress is abrogated by inhibition of the MAPK/ERK, Akt or mTOR activities. These results point at ER stress as a biochemical target by which tumor cells expressing gain-of-function ERBB2 mutants may be approached for therapeutic intervention.
Materials and Methods
Cell culture
MCF10A cells were maintained in DMEM/F12 supplemented with 5% donor horse serum (Gibco), 2 mM L-glutamine, 20 ng of epidermal growth factor (EGF)/ml, 10 μg of insulin/ml, 100 ng of cholera toxin/ml, 0.5 μg of hydrocortisone/ml, 50 U of penicillin/ml and 50 μg of streptomycin/ml at 37°C in a 5% CO2-humidified, 95% air incubator. The human tumor cell line MDA-MB231 was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 50 U of penicillin/ml, and 50 μg of streptomycin/ml. SKBr3 and BT-474 cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 50 U of penicillin/ml and 50 μg of streptomycin/ml.
Retroviral vectors and virus production
pbabe-NeuN vector (wild type ERBB2) for stable gene expression has been described previously (20). Constitutively active ERBB2 mutant (pbabe-NeuT) was kindly provided by Danielle Carroll (Harvard Medical School, Boston, MA). pbabe-BCL-xL was a gift from Dr. Cristina Muñoz (IDIBELL, Barcelona, Spain). Retroviruses for protein overexpression were produced by transfection of HEK293-T cells by the calcium phosphate method with the corresponding retroviral vectors. Retrovirus-containing supernatants were collected 48 hours after transfection and concentrated by ultracentrifugation at 22.000 rpm for 90 minutes at 4°C.
Generation of MCF10A Cell Lines
Cells were infected with the retroviruses mentioned above. Stable populations were obtained by selection with 1.5 μg/ml puromycin during 48 hours.
Determination of apoptosis
Cells (3×105/well) were treated in 6-well plates as indicated in the figure legends. After treatment, hypodiploid apoptotic cells were detected by flow cytometry according to published procedures (21). Briefly, cells were washed with phosphate buffered saline (PBS), fixed in cold 70% ethanol and then stained with propidium iodide while treating with RNAse. Quantitative analysis of subG1 cells was carried out in a FACSCalibur cytometer using the Cell Quest software (Becton Dickinson, Mountain View, CA). In BT-474 cells, phosphatidylserine exposure on the surface of apoptotic cells was detected by flow cytometry after staining with Anexin-V-FLUOS (Boehringer Mannheim, Germany).
Immunoblot analysis of proteins
Cells (3 × 105) were washed with phosphate-buffered saline (PBS) and lysed in RIPA buffer. Protein content was measured with the Bradford reagent (Bio-Rad Laboratories, USA), before adding Laemmli sample buffer. Proteins were resolved on SDS-polyacrylamide minigels and detected as described previously (22). Tubulin and GAPDH were used as protein loading controls.
Reverse transcriptase (RT) and PCR assays
Total RNA was isolated from MCF10A cells with the Trizol reagent (Life Technologies) as recommended by the supplier. Total RNA was used as a template for cDNA synthesis using a RT-PCR kit (Perkin-Elmer). PCRs were carried out using specific primers (Supplementary materials). RT-PCR product of β-actin was used as a control for mRNA input.
Real-time –PCR
mRNA expression was analyzed in triplicate by RT-qPCR on the ABI Prism7500 sequence detection system using predesigned Assay-on-demand primers and probes (Applied Biosystems). Hypoxanthine-guanine phosphoribosyltransferase (HPRT1 Hs01003267_m1) was used as an internal control and mRNA expression levels of CHOP, TRAIL and TRAIL-R2 were given as fraction of mRNA levels in control cells. Primers and probes used were: ATF4 (Hs00909568_g1), CHOP (Hs01090850_m1), TRAIL (Hs00921974_m1) and TRAIL-R2 (Hs00366278_m1).
RNA interference
siRNAs against TRAIL-R2, Bid, Ire1, ATF4, ATF6, Caspase-8, Raptor, Rictor, Noxa, Bim and non-targeting scrambled control (Supplementary materials) were synthesized by Sigma (St. Louis, MO). Flexitube siRNAs against PERK, CHOP and TRAIL were purchased from Qiagen. Cells were transfected with siRNAs using DharmaFECT-1 (Dharmacon) as described by the manufacturer. After 6 h, transfection medium was replaced with regular medium and cells were further incubated for 42 h before further analysis.
Statistical analysis
All data are presented as the mean ± SE of at least three independent experiments. The differences among different groups were determined by the Student’s t test. P<0.05 was considered significant. *P<0.05; **P<0.01; ***P<0.001. n.s. not statistically significant.
Results
Enhanced sensitivity of mutant ERBB2-expressing cells to ER stress
Recent findings have demonstrated that the presence of ERBB2 somatic mutations is an alternative mechanism to activate ERBB2 in breast cancer (19). Different somatic mutations of ERBB2 enhance the tyrosine kinase activity and the oncogenic potential of this protein (18, 19, 23). One of the consequences of ERBB2 activation is the deregulated activation of the MAPK/ERK and PI3K/Akt/mTOR pathways, which promotes cell growth, proliferation, increased metabolism and motility (13). Given the reported crosstalk between the UPR and mTOR signaling pathways (10-12, 24) we investigated the impact of ERBB2 activation on the sensitivity of breast epithelial cells to ER stress. Results depicted in figure 1A indicate that human breast epithelial cells MCF10A expressing a constitutively active ERBB2 (NeuT) (20) showed constitutive phosphorylation of ERBB2 at Tyr1248 and were markedly more sensitive to the ER stress inducer thapsigargin than control cells (pbabe) or cells expressing wild type ERBB2 (NeuN). Time course experiments further confirmed the increased sensitivity of NeuT cells to ER stress (Fig. S1A). Tunicamycin, another ER stress inducer, also activated apoptosis differentially in NeuT cells (Fig. S1B). Furthermore, results shown in figure 1B demonstrate that the ERBB2 tyrosine kinase inhibitor lapatinib markedly reduced thapsigargin-induced apoptosis in NeuT cells strongly suggesting that sensitivity to ER stress is a result of the constitutive activation of ERBB2. In agreement with the data of the lower sensitivity of NeuN cells, breast tumor cell lines overexpressing either naturally (BT-474, SkBr3) or ectopically (MDA-MB231) the wild type form of ERBB2 showed a reduced sensitivity to ER stress (Fig. 1C). Evaluation of sensitivity to ER stress in breast tumor cells ectopically overexpressing the NeuT oncogene was not possible because these cells underwent premature senescence (Fig. S1C), as it has been previously reported (25).
Figure 1. Apoptotic response of ERBB2-expressing cells to ER stress.
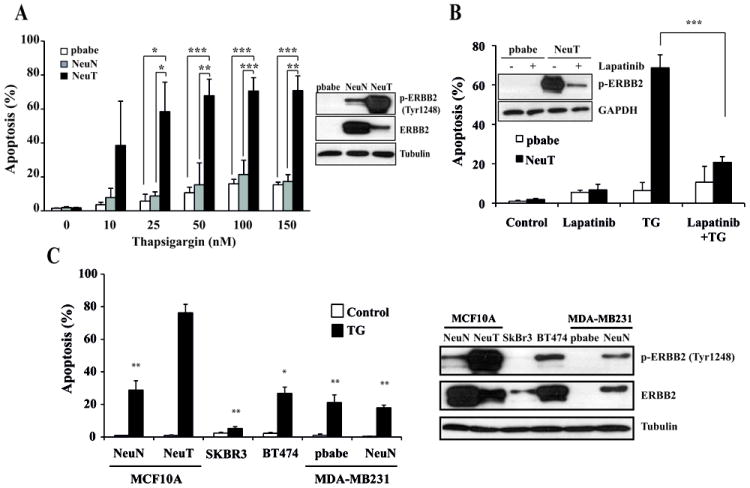
(A) Cells were treated with the indicated doses of thapsigargin for 30 hours and apoptosis was determined as described under Materials and Methods. Insert shows the expression of total ERBB2 and p-ERBB2 (Tyr1248). (B) pbabe and NeuT cells were incubated with or without Lapatinib (5 μM) for 48 hours and then treated with thapsigargin (100 nM) for 30 hours in the presence or the absence of Lapatinib. Apoptosis was determined as described under Materials and Methods. Insert shows the expression of p-ERBB2 (Tyr1248) after treatment with or without Lapatinib (5μM) for 48 hours. Error bars represent S.D. from three independent experiments. *P<0.05, **P<0.01, ***P<0.001. (C) Indicated cell lines were treated with 100 nM thapsigargin during 30 h and apoptosis (left panel) was measured as described under Materials and Methods. Right panel shows the expression of p-ERBB2 (Tyr1248) and total ERBB2 in the different cell lines tested. Error bars represent S.D. from three independent experiments. *P<0.05, **P<0.01, comparing the various cell lines with NeuT cells.
Role of the UPR branches in ER stress-induced apoptosis in mutant ERBB2-transformed cells
To investigate the mechanism of the enhanced sensitivity of NeuT cells to ER stress we examined the role of the UPR branches which are known to regulate cell fate upon ER stress (2). Thus, we determined the processing by Ire1α of the mRNA encoding the transcription factor X box-binding protein 1 (XBP1) to generate the active transcription factor XBP1s. In control cells, initial Ire1α signaling was substantially reduced upon prolonged ER stress (Fig. 2A), as previously reported in other cell systems (26). In contrast, in NeuT cells Ire1α activity remained elevated as indicated by the sustained expression of XBP1s upon ER stress. However, Ire1α knockdown did not result in the abrogation of ER stress-induced apoptosis (Fig. S2A). Likewise, ATF6 silencing did not reduce significantly apoptosis induced by thapsigargin (Fig. S2B).
Figure 2. Role of the PERK/ATF4/CHOP pathway in ER stress-induced apoptosis.
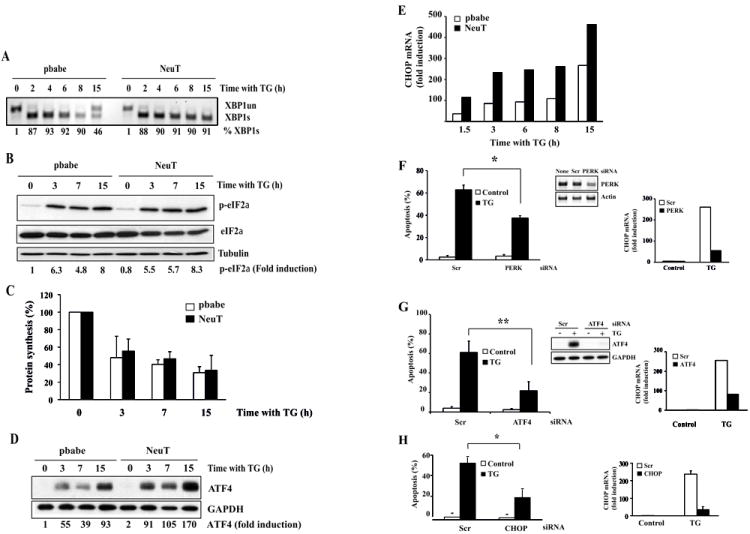
pbabe and NeuT cells were treated with thapsigargin (100 nM) for the indicated times. Following these treatments, XBP1 splicing was analyzed by RT-PCR (A). phosphorylation of eIF2alpha (B) and ATF4 induction (D) were assessed by western blotting, protein synthesis was measured by the incorporation of [3H]leucine (10 μCi/ml) into acid-precipitable material (C) and CHOP mRNA levels were determined by RT-qPCR (E). Quantification of RT-PCR and western blot signals was performed by densitometry with ImageQuant TL software after scanning the films on ImageScanner II (GE Healthcare). The relative expression of proteins was normalized to that of loading controls. Results are representative of 3 independent experiments. NeuT cells were transfected either with a scrambled oligonucleotide (Scr) or with siRNAs targeting PERK (F), ATF4 (G) or CHOP (H) for 48 hours. Cells were then treated with o without thapsigargin (100 nM) for 30 hours and apoptosis was determined. Error bars represent S.D. from three independent experiments. *P<0.05, **P<0.01. ATF4 knockdown was determined by western blotting. Silencing of PERK was assessed by RT-PCR. CHOP levels were determined by RT-qPCR.
We next examined the activity of the PERK/ATF4/CHOP pathway in both control and NeuT cells upon thapsigargin treatment. We found no differences in the activation of PERK upon thapsigargin treatment, as determined by the phosphorylation of the eukaryotic initiation factor 2 α (eIF2α) (Fig. 2B). Likewise, inhibition of general protein synthesis following ER stress occurred to the same extent and with similar kinetics in both control and NeuT cells (Fig. 2C). Interestingly, although we found no differences in ATF4 mRNA levels between control and NeuT cells following thapsigargin treatment (Fig. S2C), expression of ATF4 was significantly enhanced at the protein level in NeuT cells as compared to control cells upon ER stress (Fig. 2D). In addition, expression of the ATF4 target gene CHOP (27) was also markedly up-regulated in NeuT cells as compared to control cells, upon ER stress (Fig. 2E). Regarding sensitivity to ER stress, a significant inhibition of ER stress-induced CHOP expression and apoptosis was observed in NeuT cells following PERK knockdown (Fig. 2F). Further evidences for the role of the PERK pathway in ER stress-induced apoptosis in mutant ERBB2-transformed cells were obtained in experiments silencing the expression of downstream effectors of this pathway. As shown in figure 2G, ATF4 knockdown caused a marked inhibition of CHOP expression and apoptosis upon thapsigargin treatment. Similarly, silencing CHOP expression resulted in an important inhibition of thapsigargin-induced apoptosis in NeuT cells (Fig. 2H).
TRAIL-R2-mediated apoptosis in NeuT cells upon ER stress
We next investigated the role of caspases and the mitochondria in this cell death process. The pan-caspase inhibitor Z-VAD-fmk completely abrogated cell death induced by thapsigargin (Fig. 3A). Furthermore, caspase-9 and caspase-3 processing were also clearly enhanced in NeuT cells as compared to control cells (Fig. 3B). Strikingly, in Bcl-xL-overexpressing NeuT cells thapsigargin-induced apoptosis was clearly inhibited (Fig. 3C), which demonstrated that the increased sensitivity of NeuT cells to ER stress was due to the enhanced activation of a mitochondria-operated apoptotic pathway.
Figure 3. ER stress activates caspase-dependent cell death through a mitochondria-operated apoptotic pathway.
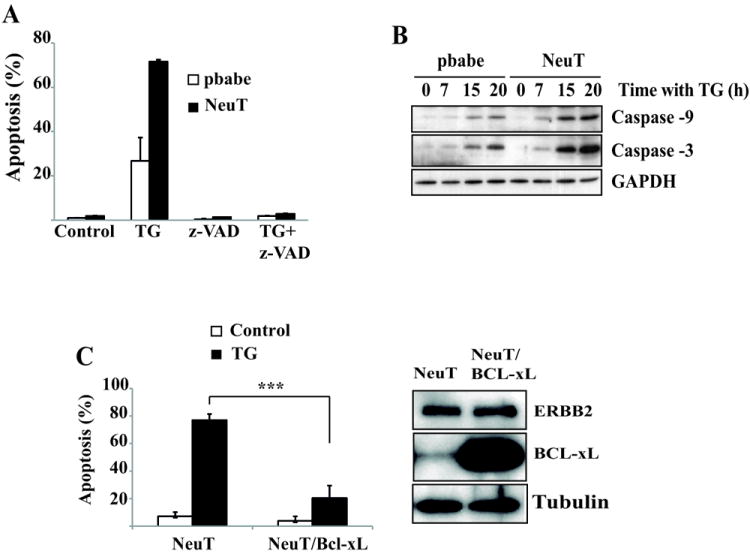
(A) Apoptosis was determined in pbabe and NeuT cells treated for 30 hours with thapsigargin (100 nM) in the presence or absence of z-VAD-fmk (10 μM). (B) pbabe and NeuT cells were treated with 100 nM thapsigargin for the indicated times. Activation of caspase-9 and caspase-3 were assessed by western blotting. Results are representative of 2 independent experiments. (C) Apoptosis was assessed in Bcl-xL – overexpressing NeuT cells treated with thapsigargin (100 nM) for 30 hours. Error bars represent S.D. from three independent experiments. *P<0.001. Bcl-xL overexpression was measured by western-blotting.
Transcriptional induction of BH3-only proteins by CHOP plays a role in ER stress-induced apoptosis in different systems (28, 29). We determined by reverse transcriptase-multiplex ligation-dependent probe amplification (RT-MLPA) the mRNA levels of twenty members of the Bcl-2 family, including ten BH3-only members. Although small differences were found between control and NeuT cells in the mRNA expression levels of Noxa and Bim upon ER stress (Fig. S3A), knockdown of these BH3-only proteins did not inhibit thapsigargin-induced apoptosis in NeuT cells (Fig. S3B). Collectively, these results suggested that activation of the mitochondrial pathway of apoptosis in NeuT cells by ER stress was independent of the up-regulation of BH3-only or down-regulation of anti-apoptotic Bcl-2 proteins expression.
CHOP involvement in ER stress-induced apoptosis has been previously linked to up-regulation of TRAIL-R2 expression and a potential CHOP-binding site has been identified in the 5’-flanking region of the TRAIL-R2 gene (30). Time course analysis of TRAIL-R2 levels indicated that CHOP-dependent TRAIL-R2 up-regulation following ER stress treatment was enhanced in NeuT cells compared to pbabe cells (Fig. 4A, S4, S5A and S5B). Moreover, TRAIL-R2 up-regulation induced by ER stress was accompanied by a marked decrease in FLIPL expression (Fig. 4B). Notably, caspase-8 activation was only observed in NeuT cells treated with thapsigargin (Fig. 4C). As FLIPL down-regulation was observed in both cell lines, these results suggested that the enhanced up-regulation of TRAIL-R2 in NeuT cells may be an important event in ER stress-induced apoptosis in these cells. In this respect, TRAIL-R2 knockdown markedly reduced ER stress-induced apoptosis (Fig. 4D and S5C). Furthermore, silencing caspase-8 expression markedly inhibited ER stress-induced apoptosis in NeuT cells (Fig. 4E and S5C), revealing a pivotal role of caspase-8 activation in ER stress-induced apoptosis in these cells. Activation of caspase-8 leads to the processing of the BH3-only substrate Bid generating a 15 kDa fragment which translocates to mitochondria to promote the release of apoptogenic factors (31). In NeuT cells, silencing Bid expression significantly reduced apoptosis induced by ER stress (Fig. 4F). Collectively, these results and data shown in figure 3C suggest a differential activation of a TRAIL-R2-mediated, mitochondria-operated apoptotic pathway in NeuT cells upon ER stress.
Figure 4. Involvement of TRAIL-R2 and caspase-8 in ER stress-induced apoptosis in NeuT cells.
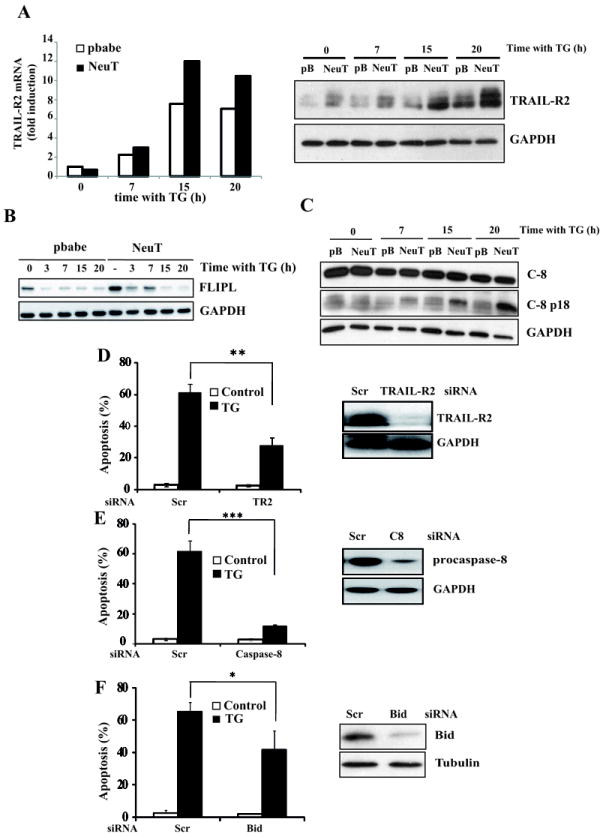
Cells were treated with 100 nM thapsigargin for the indicated times. Following these treatments, TRAIL-R2 induction (A) was determined by RT-qPCR and western blotting. FLIPL levels (B) and caspase-8 activation (C) were examined by western blotting. Results are representative of two independent experiments. In (D), (E) and (F) NeuT cells were transfected either with a scrambled oligonucleotide or siRNA oligonucleotides targeting TRAIL-R2 (D), caspase-8 (E) or Bid (F) for 48 hours as described in Methods. Cells were then incubated in the presence or absence of thapsigargin (100 nM) for 30 h. Protein knockdown and apoptosis were determined as previously described. Error bars represent S.D. from three independent experiments. *P<0.05, **P<0.01, ***P<0.001.
To determine the role of potentially secreted TRAIL in the apoptosis induced by ER stress we used a recombinant TRAIL-R2/Fc chimeric protein that has been shown to potently neutralize the apoptotic activity of exogenous TRAIL (32). However, at doses five times higher than those required to inhibit apoptosis by exogenously added TRAIL (Fig. S6A, right panel), TRAIL-R2/Fc did not inhibit apoptosis induced by thapsigargin (Fig. S6A, left panel). Furthermore, silencing TRAIL expression prior to ER stress treatment did not inhibit apoptosis by thapsigargin (Fig. S6B), which suggested the lack of involvement of endogenous TRAIL in ER stress-induced apoptosis.
Signaling pathways regulation upon ER stress in control and NeuT cells
To further investigate the mechanism of ER stress-induced apoptosis in NeuT cells, we first examined the activation of Akt and ERK in both pbabe and NeuT cells treated with thapsigargin. Basal levels of phosphorylated Akt were markedly different in pbabe and NeuT cells (Fig. 5A), as expected from the constitutive activation of the PI3K/Akt pathway by the mutant ERBB2 protein. As previously reported in other cell types (33), there was an acute activation of Akt in both pbabe and NeuT cells upon thapsigargin treatment, which was followed by a decrease in phosphorylated Akt to levels below the basal level in pbabe cells. In contrast, levels of phosphorylated Akt protein remained elevated after the initial peak of activation in NeuT cells treated with thapsigargin (Fig. 5A). Similarly to what was observed for Akt, basal levels of activated ERK were highly elevated in NeuT cells as compared to pbabe cells (Fig. 5B). Furthermore, in pbabe cells ER stress caused a marked inhibition of ERK phosphorylation starting at 7h after the addition of thapsigargin (Fig. 5B). Remarkably, in NeuT cells we observed a sustained activation of ERK upon ER stress which remained phosphorylated for up to 20h of thapsigargin treatment (Fig. 5B). The observed correlation between the sustained activation of the Akt and ERK pathways and the increased apoptosis of NeuT cells upon ER stress prompted us to assess the effect of specific inhibitors of these pathways on the apoptosis induced by thapsigargin. Incubation of NeuT cells either with an Akt (GSK690693) or a MEK1 (U0126) inhibitor partially inhibited apoptosis (Fig. 5C). Strikingly, in the presence of both inhibitors apoptosis was markedly inhibited, suggesting that the sustained activation of the Akt and ERK pathways observed upon ER stress played a key role in the induction of apoptosis.
Figure 5. Signaling pathways activated in cells undergoing ER stress.
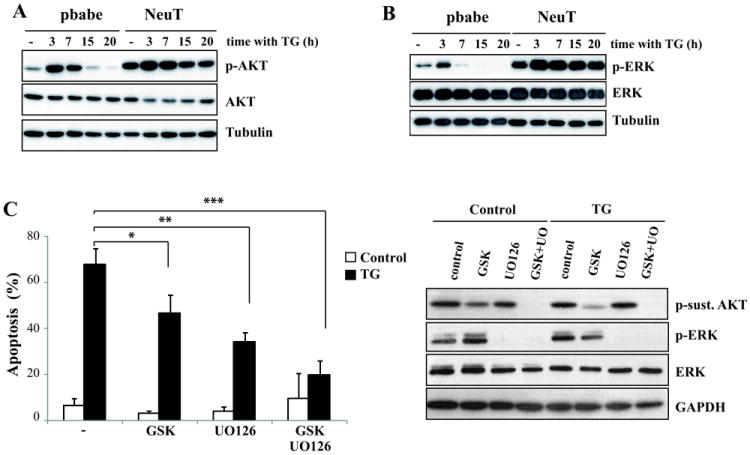
(A,B) Cells were treated with 100 nM thapsigargin for the indicated times. (A) Akt activation (p-AktSer473) or (B) Erk activation (p-ERK) were assessed by western blotting. Results are representative of 3 independent experiments. (C) Cells were treated with thapsigargin (100 nM) for 30 hours in the presence or absence of GSK690963 (10 μM), U0126 (10 μM) or both inhibitors and apoptosis was determined. Error bars represent S.D. from three independent experiments. *P<0.05, **P<0.01, ***P<0.001. The efficacy of the inhibitors was assessed by western-blotting.
TSC2 phosphorylation by either Akt or ERK/RSK leads to disruption of the TSC1/TSC2 complex, a negative regulator of mTOR activity (34, 35). Moreover, a number of evidences have revealed the existence of a bidirectional cross-talk between ER stress and mTOR signaling (24). In addition, a link between ERBB2 overexpression and activation of the Akt/mTOR/4E-BP1 pathway has been reported in breast cancer progression (36). As expected, basal levels of phosphorylated p70(S6K), a major mTORC1 substrate, was higher in NeuT than in control cells (Fig. 6A). Upon ER stress there was an acute activation of the mTORC1 pathway in both cell lines, with maximal activity at 3h post-thapsigargin addition. Thereafter, phosphorylated p70(S6K) returned to levels below the basal level in control cells. On the contrary, in NeuT cells the initial peak of p70(S6K) phosphorylation was followed by a sustained mTORC1 activity which remained elevated for up to 20h (Fig. 6A). We next examined the impact of sustained mTORC1 activity in the sensitivity of NeuT cells to ER stress. Intriguingly, at a dose that strongly inhibits p70(S6K) phosphorylation (Fig. 6B, right panel) the mTORC1 inhibitor rapamycin did not significantly reduce ER stress-induced apoptosis (Fig. 6B, left panel). We also tested the effect of torin1, a highly potent and selective ATP-competitive mTOR inhibitor which, unlike rapamycin, fully inhibits mTORC1 and mTORC2 complexes (37). Torin1 completely inhibited both rapamycin-sensitive and –insensitive mTORC1 activities in NeuT cells (Fig. 6B, right panel). Furthermore, torin1 also efficiently inhibited mTORC2 activity as indicated by the complete inhibition of AktSer473 phosphorylation (Fig. 6B, right panel). Strikingly, we observed an almost complete inhibition of apoptosis by torin1 (Fig. 6B, left panel), suggesting an important role of mTOR in this cell death process. The role of mTORC1 and mTORC2 activities in the sensitivity of NeuT cells to ER stress was further studied in experiments silencing Raptor or Rictor expression with siRNA. Interestingly, Raptor knockdown significantly reduced thapsigargin-induced apoptosis in NeuT cells (Fig. 6C, left panel). In contrast, silencing Rictor expression did not result in a significant inhibition of ER stress-induced apoptosis (Fig. 6C). Furthermore, a double Raptor/Rictor knockdown did not further reduce ER stress-induced apoptosis over the effect of Raptor siRNA (Fig. 6C). In view of these results, although inhibition of apoptosis by Raptor knockdown was not as strong as that observed with torin1, it is possible that the residual Raptor protein may be sufficient to sensitize the cells to thapsigargin (Fig. 6C, right panel). They also suggest that inhibition of rapamycin-insensitive mTORC1 activities may be responsible for the observed abrogation of ER stress-induced apoptosis by Torin1.
Figure 6. Role of mTOR in ER stress-induced apoptosis.
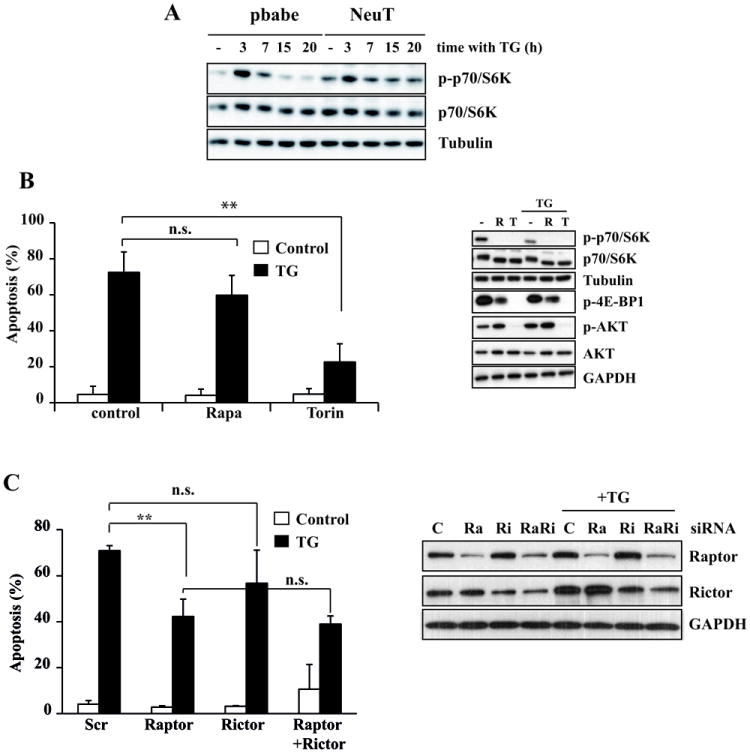
(A) Cells were treated with 100 nM thapsigargin for the indicated times. P-p70S6K and p70S6K levels were assessed by western blotting. (B) NeuT cells were treated with 100 nM thapsigargin for 30 h in the presence or absence of Rapamycin (500 nM) or torin1 (250 nM). Apoptosis (left panel) was measured as previously described. Error bars represent S.D. from three independent experiments. **P<0.01. P-p70S6K, p70S6K, p-4E-BP1, Akt Ser473 phosphorylation and Akt levels were assessed by western blotting (right panel). (C) NeuT cells were transfected either with a scrambled oligonucleotide or siRNA oligonucleotides targeting Raptor, Rictor or both for 72 h, and then treated with 100 nM thapsigargin during 30 h. Apoptosis was measured as previously described. Error bars represent S.D. from three independent experiments. **P<0.01. n.s. not statistically significant. Raptor and Rictor knockdown were assessed by western blotting.
Crosstalk between ERBB2-regulated signaling and the PERK/ATF4/CHOP/TRAIL-R2 pathway upon ER stress
To elucidate the step(s) in the PERK/ATF4/CHOP/TRAIL-R2 pathway that was affected by ERBB2-activated signaling in NeuT cells we first studied the up-regulation of ATF4 upon thapsigargin treatment. Of note, ATF4 induction by thapsigargin was reduced in the presence of GSK690693 and U0126 (Fig. 7A, left panel) or Torin1 (Fig. 7A, right panel). Likewise, results shown in figure 7B (left panel) demonstrated that the combination of GSK690693 and U0126 significantly inhibited CHOP expression upon ER stress. In addition, mTOR inhibition by Torin1 reduced CHOP mRNA induction by thapsigargin (Fig. 7B). Finally, we determined the effect of the various inhibitors on TRAIL-R2 expression in ER-stressed NeuT cells. Upregulation of TRAIL-R2 mRNA (Fig. 7B, right panel) and protein levels (Fig. 7C) by thapsigargin were considerably abrogated by the inhibitors. Interestingly, Raptor knockdown by siRNA markedly abrogated TRAIL-R2 up-regulation upon thapsigargin treatment (Fig. 7D), further confirming the role of mTORC1 activity in ER stress-induced apoptosis (Fig. 6C). Overall, our results support the model that sustained activation by mutant ERBB2 of signaling pathways that converge in mTORC1 favours the transition from an adaptive response to an ATF4/CHOP/TRAIL-R2-mediated apoptotic process in human breast epithelial cells undergoing ER stress.
Figure 7. Crosstalk between mutant ERBB2-regulated signaling and the ATF4/CHOP/TRAIL-R2 pathway upon ER stress.
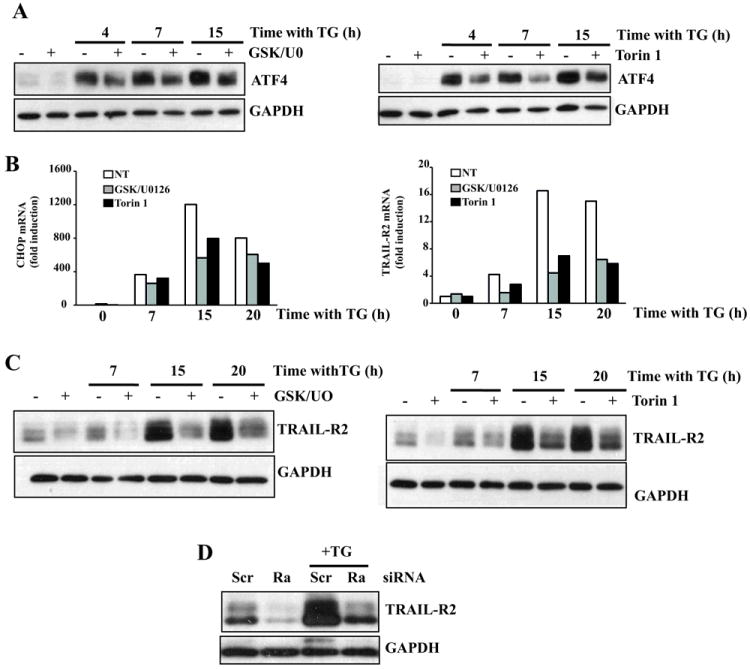
NeuT cells were treated with 100 nM thapsigargin for the indicated times in the presence or absence of GSK690963 (10 μM), U0126 (10 μM) or Torin1 (250 nM)). ATF4 (A) and TRAIL-R2 protein levels (C) were assessed by western blotting. Results are representative of 3 independent experiments. (B) NeuT cells were treated as in (A) and CHOP and TRAIL-R2 mRNA levels were assessed by RT-qPCR as described in Methods. (D) NeuT cells were transfected with either a siRNA oligonucleotide targeting Raptor or a scrambled oligonucleotide for 72 h, and then treated with 100 nM thapsigargin for 15 h. TRAIL-R2 levels were assessed by westerm blotting. Results are representative of 3 independent experiments.
Discussion
ERBB2 activation leads to dysregulation of intracellular signaling pathways that control cell metabolism, growth and proliferation (13, 14). A number of ERBB2 somatic mutations found in ERBB2 gene amplification negative breast cancer patients are activating mutations that likely drive tumorigenesis and may confer resistance to ERBB2 tyrosine kinase inhibitors (19). The present study demonstrates that breast epithelial cells expressing a constitutively active form of ERBB2 are markedly sensitive to ER stress. Our work also identifies ERK, Akt and mTOR activities as responsible for the enhanced sensitivity of these cells to ER stress. Recent studies have reported that chronic activation of mTORC1 results in an increase in PERK activity and sensitivity to ER stress although the molecular mechanism leading to cell death was not elucidated (11, 12, 38, 39). Moreover, there are marked differences between our results in mutant ERBB2-expressing cells and those reported in TSC1/2 deficient cells. Thus, TSC1/2 deficient cells showed elevated eIF2α phosphorylation upon ER stress and a truncated UPR in which induction of other ER stress markers was severely compromised (11). In contrast, we did not observe an increased PERK activity upon ER stress in ERBB2 cells compared with control cells. Interestingly, we have found an enhanced induction of ATF4 and CHOP and sustained activation of the Ire1α branch in mutant ERBB2-expressing cells. Although certain links between Ire1α and ER stress-induced apoptosis have been suggested (40, 41), our results indicate that Ire1α silencing did not reduce apoptosis in ER-stressed ERBB2 cells, excluding a role of the Ire1α pathway in death induced by ER stress in these cells.
The PERK/ATF4/CHOP branch of the UPR has a dual role in cells undergoing ER stress. As part of the adaptive response to ER stress the PERK/ATF4/CHOP pathway has been related to the activation of cytoprotective autophagy upon ER stress in different cellular models (42, 43). Although at present we could not exclude a role of autophagy in the different sensitivity of mutant ERBB2-expressing cells to ER stress, our data demonstrate that knockdown of any of the PERK/ATF4/CHOP axis proteins resulted in a significant inhibition of ER stress-induced apoptosis in cells expressing constitutively active ERBB2. The proapoptotic role of the PERK/ATF4/CHOP pathway in ER-stress-induced apoptosis has been previously demonstrated. Thus, down-regulation of anti-apoptotic Bcl-2 family members and up-regulation of proapoptotic BH3-only proteins by CHOP have been frequently reported in the activation of apoptosis upon ER stress (6, 29). However, our data do not support a similar mechanism in mutant ERBB2-expressing cells. On the other hand, a number of evidences support the involvement of death receptors and in particular TRAIL-R2 in the death of cells undergoing ER stress (30, 44, 45). In addition, TRAIL-R2 up-regulation upon ER stress treatments has been suggested to play a prominent role in the sensitization of tumor cells to exogenous TRAIL by ER stress treatments (46). Furthermore, CHOP-dependent up-regulation of TRAIL-R2 and a potential CHOP binding site in the TRAIL-R2 promoter have been reported (30). However, the molecular mechanisms controlling TRAIL-R2 up-regulation by the PERK/ATF4/CHOP pathway have not been fully elucidated. We found that dysregulated activation of the ERK, Akt and mTORC1 in cells expressing a constitutively active form of ERBB2 critically enhances ATF4, CHOP and TRAIL-R2 levels upon ER stress which lead to the activation of a caspase-8-dependent, TRAIL-independent apoptotic process. Ligand-independent assembly of the DISC has been demonstrated in the TNF family of death receptors, most likely due to the homotypic association of receptors mediated by the pre-ligand-binding assembly domain (PLAD) (47). Furthermore, ectopic TRAIL-R2 expression has been previously demonstrated to be sufficient to induce apoptosis in the absence of ligand (48). As the DISC components may co-localize in an intracellular membrane fraction in breast epithelial cells in the absence of TRAIL (22), the increased expression of TRAIL-R2 and down-regulation of cFLIP induced by ER stress in ERBB2-expressing cells could result in the formation of a DISC containing TRAIL-R2, FADD and procaspase-8 in which caspase-8 is activated. Our findings underscore the complexity of the mechanisms involved in the apoptosis elicited by ER stress in mutant ERBB2-expressing cells and warrant further investigation to characterize the site of caspase-8 activation in these cells.
Although we do not know the mechanism underlying the increased expression of ATF4 protein in mutant ERBB2-expressing cells upon ER stress, available evidences suggest that a critical step in the regulation of ATF4 protein levels is the ATF4 translational control at the 5’-leader of the ATF4 mRNA, a region containing two upstream open reading frames (uORFs) that are well conserved among vertebrates (49). Furthermore, regulatory elements in the 5′-untranslated region (5′-UTR) located upstream of the translation start site (TSS) of certain mRNAs are also key components of the translational response to mTOR activation (50). Alternatively, ATF4 is a short-lived protein with a half-life of less than 30 min, whose degradation by the proteasome depends on the interaction with the SCF/βTrCP E3 ubiquitin ligase (51). In addition to an increase in ATF4 levels, other important mechanisms for posttranslational regulation of ATF4 activity are phosphorylation or interaction with other transcription factors thus increasing its transcriptional activity (52). Whether or not these mechanisms are responsible for the enhanced ATF4 expression and activity in mutant ERBB2 cells is an issue that requires further investigation. In addition, further studies to elucidate the role of mTORC1 activity in the control of ATF4 levels would provide new insights into the molecular basis of the the transition from an adaptive response to an apoptotic process in human breast epithelial cells undergoing ER stress.
In vivo, tumor microenvironment is characterized by severe hypoxia, glucose deprivation and acidosis. These combined factors lead to the accumulation of misfolded proteins in the ER which results in ER stress, triggering the UPR to facilitate tumor survival and growth. Chronic ER stress in tumor cells increases the expression of the ER chaperones which provides a survival advantage to tumor cells in an adverse microenvironment. Therefore, pharmacological interference or knockdown strategies to abrogate chaperone expression or function may represent a potentially relevant strategy to sensitize these cells to different chemotherapeutic agents. Alternatively, our results suggest that overactivating the proapoptotic branches of the UPR in tumor cells that are prone to ER stress due to environmental conditions or constitutive activation of signaling pathways may modulate the expression of proteins of the TRAIL pathway and activate a ligand-independent apoptotic program in these cells. This would be particularly relevant in tumor cells with activating mutations in the ERBB2 gene or expressing truncated p95ERBB2, which may be resistant to trastuzumab or tyrosine kinase inhibitors.
Supplementary Material
Acknowledgments
Financial support: This work was supported by grants SAF2009-07163 and SAF2012-32824 (ALR) and SAF2010-20519 (JG) from Ministerio de Ciencia e Innovación, Red Temática de Investigación Cooperativa en Cáncer (RTICC: RD06/0020/0068 and RD12/0036/0026 to ALR and RD12/0036/0029 to JG), the European Community through the regional development funding program (FEDER) and Junta de Andalucía (P09-CVI-4497) to ALR. RMP, CP and RY were supported by contracts from Ministerio de Economía y Competitividad (MINECO) and Junta de Andalucía, respectively. ACG was supported by a FPI fellowship from MINECO. We thank FJ Fernandez-Farrán for excellent technical assistance. We also thank Tania Sánchez-Pérez for scientific discussions.
Abbreviations
- ER
endoplasmic reticulum
- UPR
unfolded protein response
- PERK
protein kinase RNA (PKR)-like ER kinase
- Ire1α
inositol-requiring protein-1
- ATF6
activating transcription factor-6
- mTOR
mammalian target of rapamycin
- CHOP
CAAT/enhancer binding protein homologous protein
- TRAIL-R2
tumor necrosis factor-related apoptosis-inducing ligand receptor 2
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- FLIP
FLICE-inhibitory protein
- DISC
death-inducing signaling complex
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–4. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- 2.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 3.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 4.Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. Embo J. 1998;17:5708–17. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–9. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 6.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–63. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 9.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 10.Fang M, Shen Z, Huang S, Zhao L, Chen S, Mak TW, et al. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell. 2010;143:711–24. doi: 10.1016/j.cell.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Kang YJ, Lu MK, Guan KL. The TSC1 and TSC2 tumor suppressors are required for proper ER stress response and protect cells from ER stress-induced apoptosis. Cell Death Differ. 2011;18:133–44. doi: 10.1038/cdd.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008;29:541–51. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 14.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–16. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 15.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 16.Anido J, Scaltriti M, Bech Serra JJ, Santiago Josefat B, Todo FR, Baselga J, et al. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. Embo J. 2006;25:3234–44. doi: 10.1038/sj.emboj.7601191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christianson TA, Doherty JK, Lin YJ, Ramsey EE, Holmes R, Keenan EJ, et al. NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer research. 1998;58:5123–9. [PubMed] [Google Scholar]
- 18.Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10:25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 19.Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–37. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haenssen KK, Caldwell SA, Shahriari KS, Jackson SR, Whelan KA, Klein-Szanto AJ, et al. ErbB2 requires integrin alpha5 for anoikis resistance via Src regulation of receptor activity in human mammary epithelial cells. J Cell Sci. 2010;123:1373–82. doi: 10.1242/jcs.050906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong J, Traganos F, Darzynkiewicz Z. A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Analytical biochemistry. 1994;218:314–9. doi: 10.1006/abio.1994.1184. [DOI] [PubMed] [Google Scholar]
- 22.Yerbes R, Palacios C, Reginato MJ, Lopez-Rivas A. Cellular FLIP(L) plays a survival role and regulates morphogenesis in breast epithelial cells. Biochim Biophys Acta. 2011;1813:168–78. doi: 10.1016/j.bbamcr.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Bargmann CI, Weinberg RA. Increased tyrosine kinase activity associated with the protein encoded by the activated neu oncogene. Proc Natl Acad Sci U S A. 1988;85:5394–8. doi: 10.1073/pnas.85.15.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012;22:274–82. doi: 10.1016/j.tcb.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Trost TM, Lausch EU, Fees SA, Schmitt S, Enklaar T, Reutzel D, et al. Premature senescence is a primary fail-safe mechanism of ERBB2-driven tumorigenesis in breast carcinoma cells. Cancer research. 2005;65:840–9. [PubMed] [Google Scholar]
- 26.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–9. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281:7260–70. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 29.Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–49. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279:45495–502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- 31.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–90. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 32.Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–97. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J Biol Chem. 2004;279:49420–9. doi: 10.1074/jbc.M407700200. [DOI] [PubMed] [Google Scholar]
- 34.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 35.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–93. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 36.Zhou X, Tan M, Stone Hawthorne V, Klos KS, Lan KH, Yang Y, et al. Activation of the Akt/mammalian target of rapamycin/4E-BP1 pathway by ErbB2 overexpression predicts tumor progression in breast cancers. Clin Cancer Res. 2004;10:6779–88. doi: 10.1158/1078-0432.CCR-04-0112. [DOI] [PubMed] [Google Scholar]
- 37.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121:2181–96. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012;19:310–20. doi: 10.1038/cdd.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–75. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–6. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 42.Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. 2011;31:3616–29. doi: 10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, et al. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29:4424–35. doi: 10.1038/onc.2010.191. [DOI] [PubMed] [Google Scholar]
- 44.Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009;138:377–88. doi: 10.1016/j.cell.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He Q, Lee DI, Rong R, Yu M, Luo X, Klein M, et al. Endoplasmic reticulum calcium pool depletion-induced apoptosis is coupled with activation of the death receptor 5 pathway. Oncogene. 2002;21:2623–33. doi: 10.1038/sj.onc.1205345. [DOI] [PubMed] [Google Scholar]
- 46.Shiraishi T, Yoshida T, Nakata S, Horinaka M, Wakada M, Mizutani Y, et al. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer research. 2005;65:6364–70. doi: 10.1158/0008-5472.CAN-05-0312. [DOI] [PubMed] [Google Scholar]
- 47.Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–4. doi: 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- 48.Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–21. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 49.Yusta B, Baggio LL, Estall JL, Koehler JA, Holland DP, Li H, et al. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006;4:391–406. doi: 10.1016/j.cmet.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Gentilella A, Thomas G. Cancer biology: The director’s cut. Nature. 2012;485:50–1. doi: 10.1038/485050a. [DOI] [PubMed] [Google Scholar]
- 51.Lassot I, Segeral E, Berlioz-Torrent C, Durand H, Groussin L, Hai T, et al. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol Cell Biol. 2001;21:2192–202. doi: 10.1128/MCB.21.6.2192-2202.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. 2007;9:2357–71. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- 53.MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, Alnemri ES. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem. 1997;272:25417–20. doi: 10.1074/jbc.272.41.25417. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.