

Abstract
Introduction
Metastasis is the main cause of breast cancer morbidity and mortality. Processes that allow for tumor cell migration and invasion are important therapeutic targets. Here we demonstrate that receptor-interacting protein kinase 2 (RIP2), a kinase known to be involved in inflammatory processes, also has novel roles in cancer cell migration and invasion.
Methods
A total of six breast cancer expression databases, including The Cancer Genome Atlas, were assessed for RIP2 expression among various clinical subtypes and its role as a prognostic biomarker. mRNA fluorescence in situ hybridization (FISH) for RIP2 was performed on 17 stage III breast cancers to determine if there was a correlation between RIP2 expression and lymph node involvement. RNA-interference was used to knock-down RIP2 expression in MDA-MB-231, Htb126, SUM149PT, MCF7, T47D, and HCC1428 cells. Cell migration and invasion were measured in vitro by scratch/wound healing and transwell migration assays. A xenograft mouse model was used to assess tumor growth and chemosensitivity to docetaxel in vivo in MDA-MB-231 cells with and without RIP2 small hairpin RNA knockdown. Western blot and immunofluorescence imaging were used to evaluate protein expressions.
Results
Interrogation of expression databases showed that RIP2 expression is significantly over-expressed in triple-negative breast cancers (TNBC: estrogen-receptor (ER) negative, progesterone-receptor (PR) negative, Her2/neu- (Her2) negative), compared to other clinical subtypes. High RIP2 expression correlates with worse progression-free survival using a combined breast cancer expression array dataset consisting of 946 patients. Multivariate analysis shows RIP2 as an independent prognostic biomarker. Knock-down of RIP2 significantly decreases migration in both scratch/wound healing and transwell migration assays in MDA-MB-231, Htb126, SUM149PT, MCF7, and T47D cells and is correlated with decreased Nuclear Factor-kappaB and c-Jun N-terminal kinase (JNK) activation. Finally, RIP2 knock-down leads to increased sensitivity to docetaxel and decreased tumor mass and lung metastases in a xenograft mouse model.
Conclusion
These results highlight RIP2 as a pro-metastasis kinase in patients with advanced breast cancer. These results also illustrate a novel role for this kinase in addition to its known role in inflammation, and suggest that targeting RIP2 may improve outcomes in advanced breast cancer patients, in which it is overexpressed.
Introduction
Receptor-interacting protein kinase 2 (RIP2, also known as RIPK2, RICK and CARDIAK) is a serine/threonine/tyrosine kinase with a carboxy-terminal caspase activation and recruitment domain (CARD) known for its role in inflammation and immunity [1-3]. RIP2 association with the TNF receptor (TNFR) causes direct activation of NF-kappa B and induction of apoptosis [4-6]. We had previously demonstrated an unrecognized role of RIP2 in breast cancer and as a potential chemosensitizer [7]. Here we investigate the functional significance of RIP2 expression in breast cancer.
RIP2 has been associated with activation of the NF-kappa B, c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and mitogen-activated protein kinase (p-38) pathways [3,6,8,9]. Involvement in metastasis has been implicated in all of these pathways. NF-kappa B has been shown to be important for promoting migration and metastasis [10] and upregulating the expression of matrix metalloproteinases, urokinase-type plasminogen activator, and cytokines in highly metastatic cancer cell lines [11-13]. In contrast, although JNK is important in inflammation, proliferation, and apoptosis, it also regulates cell migration by maintaining the labile adhesions required for rapid cell migration [14,15]. In addition, both ERK [16] and p-38 [17] have been shown to be involved in tumor cell migration.
In this study, we found that RIP2 overexpression is most significant in triple-negative breast cancer (TNBC) and that RIP2 expression correlates with worse progression-free survival (PFS). RIP2 is an independent prognostic biomarker in multivariate analysis. mRNA fluorescence in situ hybridization (FISH) analysis of patients with locally advanced breast cancer and extensive lymph node metastases demonstrates increased RIP2 expression compared to patients with limited lymph node metastasis. When RIP2 expression is knocked down in MDA-MB-231, Htb126, SUM149PT, MCF7, and T47D breast cancer cells, there is significantly decreased migration as demonstrated by functional assays in vitro. Furthermore, RIP2 knockdown decreases tumor cell growth during chemotherapy in vivo and reduces lung metastases from MDA-MB-231 xenografts. We found that RIP2 expression regulates NF-kappa B and JNK activation in breast cancer cell lines. In addition, PP2, an RIP2 small molecule chemical inhibitor, decreases JNK activation and leads to decreased migration in vitro. Together, our findings demonstrate a novel role for RIP2 as a kinase involved in the migration of TBNC cells and in selected estrogen receptor (ER)-positive breast cancer cells.
Methods
Cells and chemicals
MDA-MB-231 and Htb126 cells were cultured in basal medium supplemented with 10% fetal calf serum and were originally purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and kindly provided by M White, Department of Cell Biology, University of Texas Southwestern Medical School, Dallas, TX. SUM149PT was purchased from Asterand (Detroit, MI, USA) and grown as per supplier’s instructions. MCF7, Htb126, and HCC1428 were purchased from ATCC and grown per supplier’s instructions. PP2 was obtained from Tocris Bioscience (Bristol, UK) and used at the indicated concentrations.
Expression array analysis and statistics
The Cancer Genome Atlas (TCGA) breast cancer expression data and clinical annotations were downloaded from the TCGA website [18]. Five other publically available breast cancer expression datasets [19-21] were separately normalized then pooled for analysis (n = 946) as previously described [7]. Data were downloaded from GEO [22] [GSE3494 [19]; GSE6532 [20]; GSE1456 [23]; GSE7390 [24]] and from NKI [25]. These datasets were chosen for having clinical annotations that include ER status, axillary lymph node involvement, PFS and/or overall survival information. Briefly, probes were mapped using Entrez identifiers and Human Genome Organization gene symbols and then averaged for gene-level analysis. Missing values were imputed using nearest neighbor averaging. All data analysis was conducted using tools in R/Bioconductor. Datasets were merged and standardized by scaling the columns, then the rows, and finally checked using principal component analysis (PCA) as previously reported [7]. Triple-negative samples were predicted from the annotated ER-negative samples using the subtyping tool, TNBCtype [26]. Univariate and multivariate Cox regression with Pearson correlation coefficient was performed using the R survival package version 2.36-14. Wald test P-values are reported for univariate Cox proportional hazards model; local Wald and overall log-rank P-values are reported for the multivariate model using Cox proportional hazards regression model. Survival curves were evaluated by Kaplan-Meier estimators with log-rank P-values reported. The median expression of RIP2 was used to split each dataset into two cohorts to examine expression patterns in samples that belong to the four clinical subtypes of breast cancer. P-values within boxplots were determined using the Welch two-sample t-test.
Xenografts and human tissues
We injected 2,000,000 MDA-MB-231 cells into each dorsal flank subcutaneously in 6-week old NOD/SCID mice. For primary tumor growth experiments, sh-control and shRIP2 cells were injected into contralateral flanks. For lung metastasis studies, either sh-control or shRIP2 cells were injected into each mouse. Four weekly IP injections of docetaxel (5 mg/kg) were administered starting at 7 days after xenograft placement. Tumor volumes were measured weekly and were calculated as:
At 10 weeks after xenograft placement, primary tumors and lungs were harvested and fixed in formalin for paraffin embedding. A mid-sagittal cut was made to each lung. Two sections per lung (200 microns apart) were mounted to glass slides and stained with hematoxylin and eosin (H&E) (Thermo Scientific, Kalamazoo, MI, USA), and imaged with Zeiss Axioskop2Plus (Thornwood, NY, USA). All animal work was approved by University of Texas Southwestern (UTSW) Institutional Animal Care and Use Committee (IACUC) and performed per institutional guidelines. De-identified primary human breast tumors were obtained from the UTSW Tissue Repository where tissues were originally obtained with patient consent and used with approval from UTSW Institutional Review Board.
Plasmids and transfections
pGIPZ RIP2 lentiviral shRNA was obtained from Open Biosystems (clone V2LHS_17021, Thermo Scientific, Inc. Pittsburgh, PA, USA). siRNAs designated as #D and #J were obtained from Dharmacon (clones D-003602-01-0005 and J-003602-09-0005, respectively from Thermo Scientific) and used with Lipofectamine RNAi Max reagent (Invitrogen, Grand Island, NY, USA) as per manufacturer’s protocol. pcDNA4 Omni-tagged RIP2 was kindly provided by D Abbott [27] (Department of Pathology, Case Western Reserve University, Cleveland, OH, USA) [28]. cDNA transfections were performed with Lipofectamine LTX reagent (Invitrogen) as per manufacturer’s protocol.
Viral transductions and stable selections
For lentivirus production, 1 μg of pGIPZ-shRNA plasmid together with 1 μg of helper plasmids (0.4 μg pMD2G and 0.6 μg psPAX2) were transfected into 293FT cells with Effectene reagent (Qiagen, Valencia, CA, USA). Viral supernatants were collected 48 hours after transfections and cleared through a 0.45-μm filter. Cells were infected with viral supernatants containing 4 μg/mL polybrene (Sigma, St. Louis, MO) and selected with puromycin for 7 days.
Western blot analysis and immunofluorescence
Total cell lysates were prepared by harvesting cells in Laemmli SDS reducing buffer. Protein concentrations were measured using a Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL, USA), resolved on an 8% to 10% polyacrylamide gel, and transferred to a polyvinylidine fluoride membrane. The following antibodies are from Cell Signaling (Beverly, MA, USA): glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (2118), p-JNK (4668), JNK (9258), p-p38 (4511), p38 (8690), p-NFkappa (3031), NFkappa (8242), p-ERK1/2 (4377), ERK 1/2(4695), p-RIP2 (4364). RICK (RIP2) antibody (sc-136059) was from Santa Cruz Biotech (Dallas, TX, USA). Focal adhesion kinase (FAK) (ab40794) and p-FAK (ab4803) antibodies were from Abcam (Cambridge, MA). Detection of peroxidase activity from HRP-conjugated antibodies was done with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Rockford, IL, USA). Images were captured with the G:BOX F3 with GeneSys software (SynGene, Frederick, MD). Alexa Fluor 568 anti-rabbit secondary antibody (Invitrogen) was used for immunofluorescence and images were taken at 100× with Zeiss Axiovert 200M (Thornwood, NY, USA).
Scratch/wound healing assay and Matrigel™ invasion assays
Cells were grown in normal growth media to monolayer confluence in 6-well tissue culture plates. They were then treated with mitomycin C (10 μg/mL) (Sigma, St. Louis, MO, USA) for 2 hours prior to scratching with a 1-mL pipette tip. A perpendicular pen mark on the tissue culture plate was used to mark the place for repeated imaging with an EVOS microscope (AMG/Life Technologies, Grand Prairie, TX, USA). For Matrigel™ invasion assays, cells were serum-starved for 16 hours, then 100,000 cells were seeded in the top chamber, in serum-free media, of 24-well invasion chambers, 8-μm pores (BD Biosciences, San Jose, CA, USA) with growth media in bottom chamber. Cells are allowed to migrate for 16 hours with processing procedure per manufacturer’s protocol. Hoechst 33342 (Sigma, St. Louis, MO, USA) was used to stain nuclei for imaging at 20× with the Zeiss Axiovert 200M and quantification with ImageJ (NIH, Bethesda, MD, USA).
Fluorescence in situ hybridization
Stellaris™ mRNA FISH probes against RIP2, CAL Fluor Red 610, was obtained from Biosearch Technologies (Novato, CA, USA) and hybridization performed as per manufacturer’s protocol. Images were obtained with Deltavision (Applied Precision, Issaquah, WA, USA) and quantification was with ImageJ.
Results
RIP2 overexpression correlates with triple-negative status and poor progression-free survival in breast cancer
We had previously shown that KIF14, a protein-protein interactor of RIP2 [29], is significantly over-expressed in triple-negative breast cancer. We examined TCGA [30] to see if RIP2 overexpression is also more common among triple-negative primary breast cancers. TBNCs, even though representing only 15% (71/459) of total cases, have a higher proportion of high RIP2 expression (Figure 1A, 87.3%, left side, ER-/PR-/Her2-) relative to the other clinical subtypes (Figure 1A, middle and right side). We next tested if high-RIP2 expression is also over-represented in other datasets. We chose five large, publicly available breast cancer expression array datasets that have adequate clinical annotations including ER status, axillary lymph node involvement, progression-free and/or overall survival data (n = 946 for PFS and n = 652 for overall survival, see Methods and as previously described [7]). As Her2 status is not annotated in these other datasets, we used the annotated ER-negative samples and obtained predicted triple-negative status from a validated public software platform TNBCtype [27]. Here, we also found that high-RIP2 expression is over-represented in the (predicted) triple-negative subgroup compared to others (see Figure 1B). RIP2 expression correlated to PFS but not overall survival outcomes analyzed by univariate Cox regression (P = 1.247e-4 and P = 0.281, respectively). Kaplan-Meier analysis of PFS when expression values were dichotomized at the median demonstrated the significant prognostic value of RIP2 expression (Figure 1C, red line, high levels of RIP2 versus black, low level of RIP2). The significance increased when patients in the top versus the bottom 30% of RIP2 expression were compared (Figure 1D). As RIP2 was highly correlated with triple-negative status, we examined this subgroup of patients (predicted from TNBCtype, n = 119; see Methods) but did not find that RIP2 expression within the TNBC cohort correlated with PFS when expression values were dichotomized at the median (P = 0.35, hazard ratio (HR) 0.8, 95% CI 0.4, 1.3) or when only the top versus bottom 30% of RIP2 expression were used (P = 0.68, HR 1.3, 95% CI 0.4, 4.1). On the other hand, multivariate analysis using Cox proportional hazards regression analysis using RIP2 expression and clinical features (triple-negative status, axillary lymph node involvement, ER status, size, and age) indicated that RIP2 is an independent prognostic biomarker (P = 0.0033, HR 1.43, 95% CI 1.10, 1.77) in addition to tumor size greater than 2cm (P <0.0001, HR 1.97, 95% CI 1.65, 2.29) (multivariate analysis with and without RIP2 expression in Figure 2A and B, respectively). Other clinical factors such as axillary lymph node involvement, ER status, and age were not independent prognostic factors for PFS on multivariate analysis although triple-negative status and younger age (younger than median age) trended towards worse survival (Figure 2A and B). To further validate RIP2 expression as an independent prognostic biomarker, we evaluated the prognostic value of RIP2 expression for only ER-positive patients (n = 465). High and low RIP2 expression (top versus bottom 30% of the overall median expression value of the combined dataset) segregated patients into a high- and low-risk group, respectively (Figure 2C). There was no direct correlation between RIP2 expression and tumor size (r = 0.173, Pearson’s correlation, data not shown). However, within the larger tumor cohort (size >2 cm), higher RIP2 expression is also prognostic of worse PFS (Figure 2D).
Figure 1.
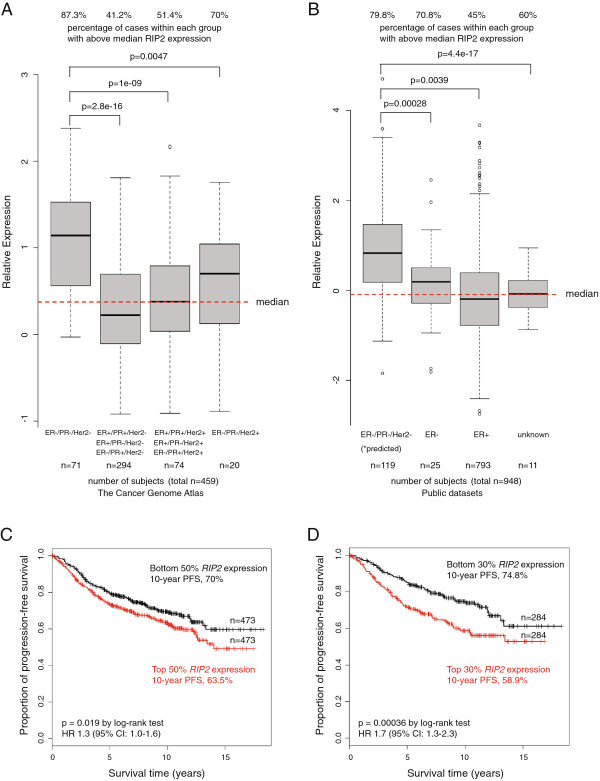
Distribution of high- and low- RIP2 expressions (dichotomized at the median) of the Cancer Genome Atlas (A) and public breast cancer expression array datasets (B) (see Methods) within each designated clinical subtype of breast cancer. Survival curves using Kaplan-Meier estimators using RIP2 as a biomarker (dichotomized about the median in C and using only top and bottom 30% of all patients in D for progression-free survival (PFS) with log-rank P-values reported. Red denotes higher expression and black denotes lower expression. HR, hazard ratio.
Figure 2.
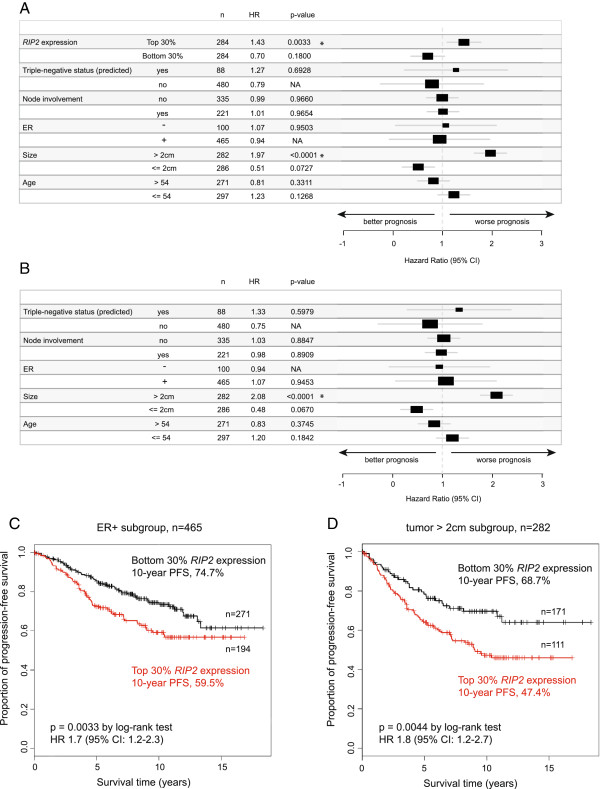
Multivariate Cox regression analysis using various clinical features with (A) and without (B) RIP2 expression for progression-free survival (PFS). *P <0.05. Kaplan-Meier estimator using top and bottom 30% RIP2 expression to segregate ER-positive (C) and tumor >2 cm patient cohorts (D). HR, hazard ratio; n, number of patients.
RIP2 expression affects migration and invasion
As tumor metastasis is the main cause of morbidity and mortality, we investigated the effect of RIP2 expression on cell migration. Metastasis of breast cancer primarily occurs through the regional lymphatic system with the extent of axillary lymph node involvement as a key prognostic factor for the disease. We obtained 17 cases of stage-III breast cancers with either little or extensive axillary lymph node involvement, from the University of Texas Southwestern (UTSW) Tissue Repository. Specifically, we evaluated RIP2 expression by mRNA FISH in stage-III breast cancer cases with either fewer than four axillary lymph nodes (N0, N1) or with significant axillary lymph node involvement (N3 or tumor metastasis in more than 10 axillary lymph nodes, infraclavicular lymph nodes, supraclavicular lymph nodes, or clinically detected ipsilateral internal mammary lymph nodes along with other axillary lymph nodes). We found that RIP2 expression appears to be more prominent in breast cancers that have significant axillary lymph node spread (Figure 3A,B). The differential RIP2 expression is not due to triple-negative status alone (percentage of triple-negative patients in N0/N1 groups was 33% and in N3 group 25%) and suggests a specific role for RIP2 in migration.
Figure 3.
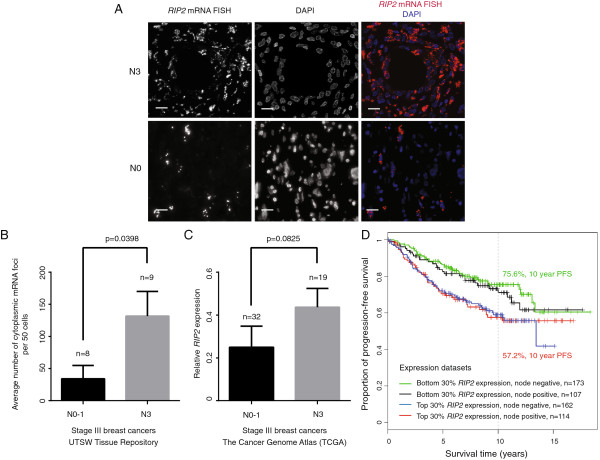
RIP2 mRNA expression in primary tumors suggests correlation with extensive axillary lymph node invasion in stage III breast cancer. (A)RIP2 mRNA fluorescence in situ hybridization (FISH) in primary breast tissue of a representative patient with N3 (top panels) and N0 (bottom panels) breast cancer. Scale bar = 20 μM. (B) Average number of cytoplasmic mRNA foci per 50 cells was used to compare the group of stage-III patients from the University of Texas Southwestern Medical Center (UTSW) Tissue Repository with extensive (N3) versus limited (N0, N1) lymph node involvement. Average of three imaging fields per case with error bars representing standard error of the mean (SEM). (C) Relative RIP2 expression of stage-III patients from The Cancer Genome Atlas (TCGA) with N3 versus N0/N1 disease. Statistics per two-sided Student t-test for B, C. (D) Kaplan-Meier estimator to evaluate RIP2 expression and lymph node status as prognostic indicator in the combined public expression array datasets. PFS, progression-free survival.
We interrogated TCGA for stage-III patients with N0 to N1 versus N3 disease and found a similar trend of higher expression in N3 disease, although the P-value for the two-sided t-test was not significant (P = 0.825, see Figure 3C). This needs to be examined further, especially in a larger dataset of stage-III patients.
Although the public datasets we examined lack adequate annotation for stage determination (only binary information given for lymph node status), we evaluated how lymph node status may affect RIP2 expression as a prognostic indicator (evaluating patients in the top versus bottom 30% of RIP2 expression) and found that high RIP2 expression with node positivity correlated with significantly worse PFS compared to low RIP2 expression with node negativity (log-rank P = 0.0042, HR 1.9, 95% CI 1.2, 2.8; Figure 3D).
To further evaluate the correlation of RIP2 expression with migration or invasion in vitro, we generated RIP2 knockdown using two short-interfering RNAs (siRNAs), designated si-RIP2 #J and si-RIP2 #D, in the TBNC cell lines MDA-MB-231, Htb126, and SUM149PT (Figure 4A, see Methods). Among available TBNC cell lines, we chose these triple-negative cell lines that have basal or mesenchymal/stem-like characteristics, as they represent subtypes of breast cancer with poor prognosis and no available targeted therapies [31]. Both the scratch/wound healing and Matrigel™ transwell invasion assays showed that decreased RIP2 expression led to decreased migration. Importantly, reconstituted RIP2 expression (pcDNA4 Omni-RIP2) largely restored migratory ability in all three triple-negative breast cancer cell lines (Figure 4B-E), indicating that the migration deficiency is unlikely to be an off-target effect of our siRNAs and that RIP2 has a clear role in migration.
Figure 4.
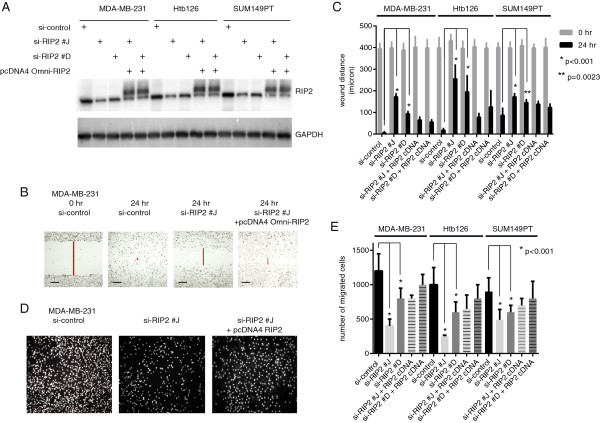
RIP2 knockdown decreases migration and invasion in triple-negative breast cancer (TNBC) cell lines. (A) Western blot showing relative protein expressions of RIP2 knockdown (si-RIP2 #J and si-RIP2 #D) and pcDNA4 Omni-tagged RIP2 in three TBNC cell lines. (B) Representative scratch/wound healing assay images for MDA-MB-231 cells. (C) Quantification of scratch/wound healing assay results for three triple-negative cell lines and error bars represent standard error of the mean (SEM). Data represent results from three separate experiments. Statistics per Student t-test as indicated. Scale bar = 100 μM. (D) Representative Matrigel™ invasion assays from MDA-MB-231 cells at 16 hours. (E) Quantification of Matrigel™ invasion assay results for three triple-negative cell lines with error bars representing SEM. Columns represent results from triplicates repeated in three separate experiments. Statistics per Student t-test.
As our in silico survival studies indicated a potential role for RIP2 in ER-positive breast cancer, we then tested if RIP2 expression affects migratory ability of ER-positive breast cancer cell lines MCF7, T47D, and HCC1428 (Figure 5A,B,C). ER-positive cell lines migrate less and slower in both the scratch/wound healing (Figure 5B) and the Matrigel™ invasion (Figure 5C) assays compared to the TNBC cell lines (Figure 4C,E). Knockdown of RIP2 decreases the migratory ability of MCF7 and T47D cells but not HCC1428 cells (Figure 5B,C).
Figure 5.
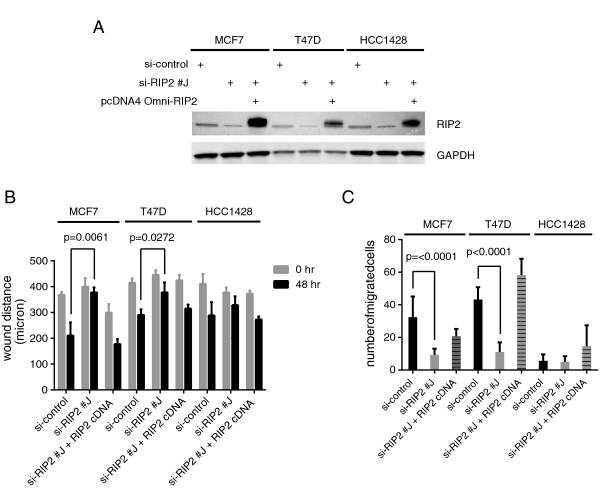
RIP2 knockdown decreases migration and invasion in certain estrogen receptor (ER)-positive cell lines. (A) Western blot showing relative protein expressions of RIP2 knockdown (siRIP2 #J) and pcDNA4 Omni-tagged RIP2 in 3 ER-positive breast cancer cell lines. (B) Quantification of scratch/wound healing assay results for three triple negative cell lines and error bars represent standard error of the mean (SEM). Data represent results from two separate experiments. Statistics per Student t-test as indicated. (C) Quantification of Matrigel™ invasion assay results for three ER-positive cell lines with error bars representing SEM. Columns represent results from triplicates repeated in two separate experiments. Statistics per Student t-test.
RIP2 expression affects chemosensitivity in vivo and tumor cell migration
We next investigated the in vivo effect of RIP2 knockdown using mouse xenograft studies. We injected equal numbers of short-hairpin (sh) RNA, sh-control and sh-RIP2 stably expressed in MDA-MB-231 cells (Figure 6A) into contralateral dorsal flanks of immunodeficient NOD-SCID mice. As RIP2 knockdown by itself has little effect on cell growth in vitro (Figure 6B), we used docetaxel (one of the most common chemotherapies given for breast cancer) to determine if RIP2 knockdown would chemosensitize tumor cells to docetaxel. RIP2 knockdown cells show a significant decrease in tumor mass after docetaxel treatments compared to non-silencing sh-control MDA-MB-231 cells injected into contralateral dorsal flanks (Figure 6C).
Figure 6.
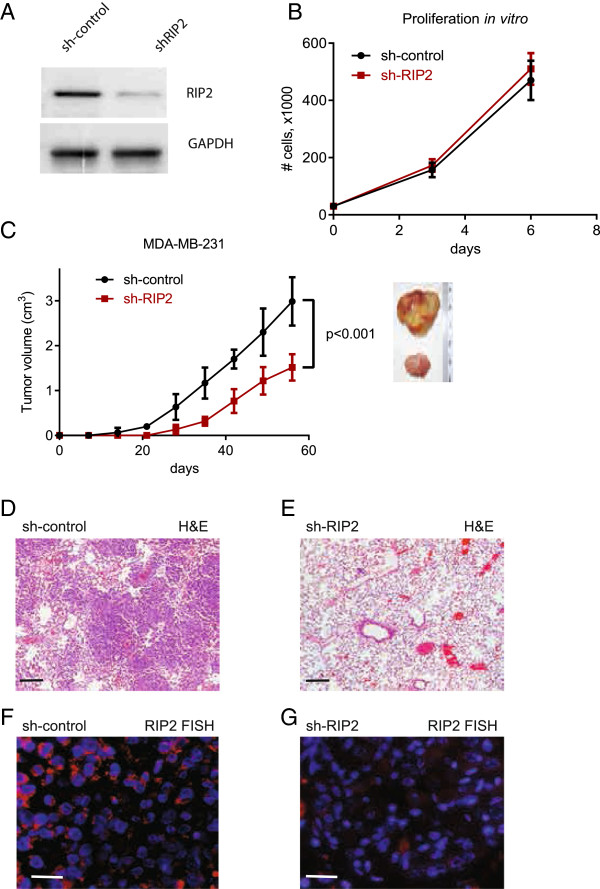
RIP2 knockdown does not cause decrease in proliferation but does cause decreased tumor growth and lung metastasis in vivo with docetaxel treatments. (A) Western blot showing relative RIP2 expression in sh-control and sh-RIP2 MDA-MB-231 cells. (B) Relative proliferation rate of sh-control and sh-RIP2 MDA-MB-231 cells in vitro. (C) Tumor volume at various time points after xenograft placements. Images are representative of final tumors at 10 weeks for sh-control (top) and sh-RIP2 (bottom). Statistics per Student t-test at final time point recorded. Representative H&E stains of lung sections at 10 weeks are shown (D and E). Scale bar represents 100 μm. Representative RIP2 fluorescence in situ hybridization (FISH) images of lung sections are shown in (F) and (G). Red, RIP2 mRNA signal. Blue, 4',6-diamidino-2-phenylindole (DAPI) nuclear stain. Scale bar represents 25 μm.
We also examined the metastatic potential of sh-control and shRIP2 MDA-MB-231 cells (these were originally derived from a malignant pleural effusion) to the lungs. Strikingly, after four weekly doses of docetaxel, lung metastases were noticeably present in all animals that had only sh-control xenografts (n = 3/3 animals) (Figure 6D) with large areas where the tumor cells replaced the lung parenchyma at the time of sacrifice (2.5 months after xenograft placement). In contrast, no obvious lung metastases were present in the animals with sh-RIP2 only xenografts (n = 6/6 animals) (Figure 6E). RIP2 FISH of the lungs showed that sh-control cells to the lung retain high RIP2 expression whereas the small amount of sh-RIP2 cells that metastasized to the lungs continued to have low RIP2 expression (Figure 6F and G, respectively). The significant difference in the presence of lung metastasis may in part be related to the differences seen in the primary tumor size in response to chemotherapy. Additional studies in vivo will be needed to more fully ascertain the role of RIP2 in metastasis.
RIP2 is an activator of the NF-kappa B and JNK pathways
To understand specific mechanisms underlying the migration defect seen with RIP2 knockdown in TBNC cells, we evaluated known pathways downstream of RIP2, including the NF-kappa B, ERK, JNK, and p-38 pathways [32]. In MDA-MB-231 and Htb126 cells, two of the cell lines with the greatest migration defects in our in vitro assays, RIP2 knockdown is associated with decreased phosphorylation of NF-kappa B and JNK but not p38 or ERK (Figure 7A). A slight increase in the phosphorylation of p38 can be observed (Figure 7A). Both JNK and NF-kappa B are associated with defects in adhesion [14,15,33]. Focal adhesion kinase (FAK) has been well-established as a gene significant for cancer cell migration and metastasis (reviewed in [34]) and is currently being used either as a drug target or as a biomarker in various solid tumors (see Clinicaltrials.gov). We therefore evaluated the expression of p-FAK to see if RIP2 expression correlates with this well-known biomarker for migration and metastasis. We found that although sh-control MDA-MB-231 cells have ample focal adhesions around the cells, RIP2 knockdown cells have fewer focal adhesions as reflected by decreased p-FAK protein (Figure 7A) and less continuous p-FAK staining around the cells (Figure 7B).
Figure 7.
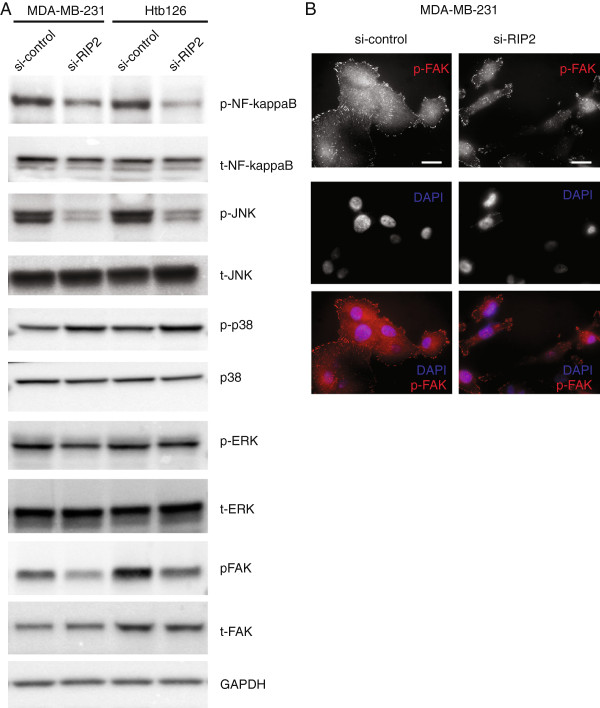
RIP2 affects activation of NF-kappaB and JNK pathways. (A) Western blots showing relative expressions of various proteins in si-control and si-RIP2 cells in 2 triple negative breast cancer cell lines. (B) pFAK immunofluorescence of MDA-MB-231 cells show less abundant pFAK staining around si-RIP2 cells relative to si-control cells. Scale bar represents 10 μm.
RIP2 kinase activity is necessary for JNK activation but not NF-kappa B
PP2 is a known small-molecule chemical inhibitor of RIP2 [35] and the Src family of tyrosine kinases [36]. We found that at 1 μM PP2 has a noticeable effect on the amount of RIP2 phosphorylation in MDA-MB-231 cells. At the same concentration of PP2, there was a correlated decrease in the amount of phosphorylation of JNK and FAK but not NF-kappa B (Figure 8A), implicating JNK activation as a key downstream effector for RIP2-mediated migration in MDA-MB-231 cells. As expected, there was a significant decrease in the number of cells that migrated in the Matrigel™ transwell invasion assay (Figure 8B,C) when cells were treated with PP2.
Figure 8.
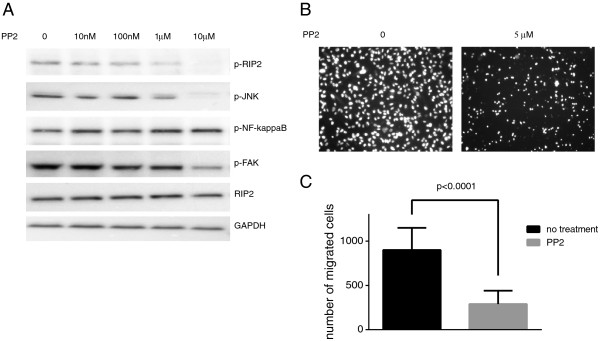
Chemical inhibition of RIP2 leads to decreased JNK and FAK activation. (A) Western blots showing relative expressions of various proteins in MDA-MB-231 cells treated with indicated doses of PP2. (B) Representative Matrigel™ invasion assays from MDA-MB-231 cells at 16 hours when treated with indicated doses of PP2. (C) Quantification of Matrigel™ invasion assay results for MDA-MB-231 cells treated with PP2 with error bars representing standard error of the mean (SEM). Columns represent results from two separate experiments done in triplicates. Statistics per Student t-test.
Discussion
To determine immediate actionable targets from our previous chemosensitivity screen, we have identified RIP2 as a gene important for breast cancer cell metastasis. RIP2 is a kinase with known central roles in inflammation and immunity. We have demonstrated a novel role for RIP2 in the metastatic potential of multiple breast cancer cells with high RIP2 expression both in vitro and in vivo and shown that it is a target for drug treatment in cancer therapy.
Although RIP2 is over-expressed more often in TNBC than other clinical subtypes, we demonstrated in the present study that RIP2 expression is an independent prognostic biomarker. Although high RIP2 expression correlated with triple-negative status, it was not a mere surrogate of triple-negative status, as high RIP2 expression also correlated with poor survival in ER-positive breast cancers and those with tumor size >2 cm. However, although RIP2 may also be a target for other types of breast cancer (that is, ER-positive), its high expression within the triple-negative subset identifies this cohort as the most likely to show immediate benefit from targeted treatment against RIP2.
Axillary lymph node metastasis in primary breast cancers has long been used clinically as a poor prognostic indicator [37] but there remains controversy about whether axillary lymph node metastasis reflects lead-time bias or true tumor biology (that is, metastatic potential or aggressiveness) [37-39]. Multivariate analysis of our combined expression array datasets did not reveal lymph node status to be prognostic for PFS although we found that RIP2 mRNA expression correlated with extensive axillary lymph node invasion in stage-III breast cancer. This suggests that a combination of lymph node status and RIP2 expression may prove to be of prognostic utility in the clinical setting. However, studies involving larger samples are needed to elucidate the role of RIP2 in axillary lymph node spread.
Here we provided in vitro evidence for the role of RIP2 in migration and invasion in TNBC and selected ER-positive breast cancers. The effect of RIP2 knockdown in ER-positive cell lines, although significant for MCF7 and T47D, was modest compared to the effect of RIP2 knockdown in the triple-negative cell lines. TNBC and ER-positive breast cancers have significantly different clinical courses, with TNBC generally being more likely to relapse, metastasize, and be resistant to treatment [40]. Expression array analysis of TBNC has revealed multiple subtypes, each with various potential targets [31]. Recent large clinical trials have failed to validate the clinical efficacy of various targets deemed to be important for subsets of TNBC (examples include iniparib against poly-ADP ribose polymerase [41] and bevacizumab against vascular endothelial growth factor receptor [42]). There is clearly an unmet need for additional therapeutic targets. As high expression of RIP2 is found in approximately 80 to 90% of TNBCs and approximately 50% of ER-positive breast cancers, RIP2 kinase is an attractive novel therapeutic target for the majority of TNBC and selected ER-positive breast cancers that have upregulated RIP2.
We have found that RIP2 knockdown correlates with decreased phosphorylation of JNK and NF-kappa B in TNBC. Both of these pathways have well-known roles in cancer progression. In our current studies, we observed a slight increase in the phosphorylation of p38, suggesting a potential compensatory effect. JNK and p38 integrate signals involved in proliferation, differentiation, survival and migration (reviewed in [43]). Conversely, NF-kappa B activation by RIP2 has been shown to be secondary to interactions from a multimeric complex of proteins and not directly from RIP2 kinase activity [44]. Our results suggest that although RIP2 may be a crucial member of the complex that controls NF-kappaB activation, its kinase activity is not necessary for NF-kappa B activation. However, the kinase activity of RIP2 appears to be important for the activation of JNK. Further experiments on the mechanism of RIP2 activation of JNK and NF-kappa B, along with potential compensatory alterations in other pathways are necessary.
Conclusions
We previously found that RIP2 knockdown increases chemosensitivity [7]. Here, we demonstrated that RIP2 has a role in facilitating metastasis and is an independent prognostic marker for breast cancer. Its association with human breast tumors and patient outcome warrants further investigation. We have identified RIP2 as a gene that is upregulated in the majority of TBNCs and likely contributes to the metastatic process.
Abbreviations
ATCC: American Type Culture Collection; ER: estrogen receptor; ERK: extracellular signal-regulated kinase; FAK: Focal adhesion kinase; FISH: fluorescence in situ hybridization; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GEO: Gene Expression Omnibus; H&E: Hematoxylin and eosin; IP: intraperitoneal; JNK: c-Jun N-terminal kinase; p-38: mitogen-activated protein kinase; PFS: progression-free survival (the time elapsed between treatment initiation and tumor progression or death from any cause with censoring of patients who are lost to follow-up); PP2: 3-(4-chlorophenyl) 1-(1,1-dimethylethyl)-1H-pyrazolo(3,4-d)pyrimidin-4-amine; RIP: Receptor-interacting protein kinase 2; SEM: standard error of the mean; shRNA: short-hairpin RNA; siRNA: small-interfering RNA; TCGA: The Cancer Genome Atlas; TNBC: triple-negative breast cancer; TNF: tumor necrosis factor; UTSW: The University of Texas Southwestern Medical Center in Dallas.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SMS: conception and design, development of methodology, acquisition of data, analysis and interpretation of data, writing of the manuscript. CC: development of methodology, acquisition of data, manuscript revision. KB: development of methodology, acquisition of data, manuscript revision. GJ: acquisition of data, manuscript revision. SLB: acquisition of data, manuscript revision. GFL: development of methodology, acquisition of data, manuscript revision. WEW: analysis and interpretation of data, manuscript revision. JWS: analysis and interpretation of data, manuscript revision. All authors read and approved the final manuscript.
Contributor Information
Stina M Singel, Email: Stina.Singel@utsouthwestern.edu.
Kimberly Batten, Email: Kimberly.Batten@utsouthwestern.edu.
Crystal Cornelius, Email: Crystal.Cornelius@utsouthwestern.edu.
Gaoxiang Jia, Email: Gaoxiang.Jia@utsouthwestern.edu.
Gail Fasciani, Email: Gail.Fasciani@utsouthwestern.edu.
Summer L Barron, Email: Summer.Barron@utsouthwestern.edu.
Woodring E Wright, Email: Woody.Wright@utsouthwestern.edu.
Jerry W Shay, Email: Jerry.Shay@utsouthwestern.edu.
Acknowledgements
We would like to thank D Abbott (Department of Pathology, Case Western Reserve University, Cleveland, OH) for plasmids; M White (Department of Cell Biology, Department of Cell Biology, University of Texas Southwestern Medical School, Dallas, TX) for tissue culture cells, and breast cancer tissue from the University of Texas Southwestern Tissue Management Shared Resources. The project was supported by Simmons Cancer Center Support Grant (5P30 CA 142543-03), T32CA136515 from the National Cancer Institute and Susan G Komen Foundation Postdoctoral Fellowship for SMS, and The Southland Financial Corporation Distinguished Chair in Geriatric Research for JWS. This work was performed in laboratories constructed with support from National Institute of Health grant C06 RR30414.
References
- Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, Xavier RJ, Green DR, Lamkanfi M, Dinarello CA, Doherty PC, Kanneganti TD. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14:480–488. doi: 10.1038/ni.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes JG, Lee J, Geddes K, Rubino S, Philpott DJ, Girardin SE. Essential role of Rip2 in the modulation of innate and adaptive immunity triggered by Nod1 and Nod2 ligands. Eur J Immunol. 2011;41:1445–1455. doi: 10.1002/eji.201040827. [DOI] [PubMed] [Google Scholar]
- Tigno-Aranjuez JT, Asara JM, Abbott DW. Inhibition of RIP2’s tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. 2010;24:2666–2677. doi: 10.1101/gad.1964410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, del Peso L, Koseki T, Chen S, Nunez G. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J Biol Chem. 1998;273:12296–12300. doi: 10.1074/jbc.273.20.12296. [DOI] [PubMed] [Google Scholar]
- McCarthy JV, Ni J, Dixit VM. RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J Biol Chem. 1998;273:16968–16975. doi: 10.1074/jbc.273.27.16968. [DOI] [PubMed] [Google Scholar]
- Thome M, Hofmann K, Burns K, Martinon F, Bodmer JL, Mattmann C, Tschopp J. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr Biol. 1998;8:885–888. doi: 10.1016/S0960-9822(07)00352-1. [DOI] [PubMed] [Google Scholar]
- Singel SM, Cornelius C, Batten K, Fasciani G, Wright WE, Lum L, Shay JW. A Targeted RNAi screen of the breast cancer genome identifies KIF14 and TLN1 as genes that modulate docetaxel chemosensitivity in triple-negative breast cancer. Clin Cancer Res. 2013;19:2061–2070. doi: 10.1158/1078-0432.CCR-13-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas TA, Baldwin DT, Stewart TA. RIP2 is a Raf1-activated mitogen-activated protein kinase kinase. J Biol Chem. 1999;274:33684–33690. doi: 10.1074/jbc.274.47.33684. [DOI] [PubMed] [Google Scholar]
- Jacquet S, Nishino Y, Kumphune S, Sicard P, Clark JE, Kobayashi KS, Flavell RA, Eickhoff J, Cotten M, Marber MS. The role of RIP2 in p38 MAPK activation in the stressed heart. J Biol Chem. 2008;283:11964–11971. doi: 10.1074/jbc.M707750200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig G, Christopherson KW 2nd, Bhat-Nakshatri P, Kumar S, Kishimoto H, Miller KD, Broxmeyer HE, Nakshatri H. NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. 2003;278:21631–21638. doi: 10.1074/jbc.M300609200. [DOI] [PubMed] [Google Scholar]
- Kang H, Lee M, Choi KC, Shin DM, Ko J, Jang SW. N-(4-hydroxyphenyl)retinamide inhibits breast cancer cell invasion through suppressing NF-KB activation and inhibiting matrix metalloproteinase-9 expression. J Cell Biochem. 2012;113:2845–2855. doi: 10.1002/jcb.24159. [DOI] [PubMed] [Google Scholar]
- Moreau M, Mourah S, Dosquet C. beta-Catenin and NF-kappaB cooperate to regulate the uPA/uPAR system in cancer cells. Int J Cancer. 2011;128:1280–1292. doi: 10.1002/ijc.25455. [DOI] [PubMed] [Google Scholar]
- Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, Zhang Y, Mazumdar A, Hilsenbeck SG, Mills GB, Brown PH. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013;73:3470–3480. doi: 10.1158/0008-5472.CAN-12-4524-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. JNK phosphorylates paxillin and regulates cell migration. Nature. 2003;424:219–223. doi: 10.1038/nature01745. [DOI] [PubMed] [Google Scholar]
- Lee MH, Koria P, Qu J, Andreadis ST. JNK phosphorylates beta-catenin and regulates adherens junctions. FASEB J. 2009;23:3874–3883. doi: 10.1096/fj.08-117804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara E, Maenaka S, Shimada E, Nishimura Y, Sakurai H. Dynamic regulation of extracellular signal-regulated kinase (ERK) by protein phosphatase 2A regulatory subunit B56gamma1 in nuclei induces cell migration. PLoS One. 2013;8:e63729. doi: 10.1371/journal.pone.0063729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore HM, Gonzalez ME, Toy KA, Cimino-Mathews A, Argani P, Kleer CG. EZH2 inhibition decreases p38 signaling and suppresses breast cancer motility and metastasis. Breast Cancer Res Treat. 2013;138:741–752. doi: 10.1007/s10549-013-2498-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Data Portal. https://tcga-data.nci.nih.gov/tcga/
- Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, Klijn JG, Larsimont D, Buyse M, Bontempi G, Delorenzi M, Piccart MJ, Sotiriou C. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007;25:1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Gene Expression Omnibus (GEO) public functional genomics data repository. http://www.ncbi.nlm.nih.gov/geo/
- Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, Liu ET, Miller L, Nordgren H, Ploner A, Sandelin K, Shaw PM, Smeds J, Skoog L, Wedren S, Bergh J. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7:R953–R964. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, Viale G, Delorenzi M, Zhang Y, d’Assignies MS, Bergh J, Lidereau R, Ellis P, Harris AL, Klijn JG, Foekens JA, Cardoso F, Piccart MJ, Buyse M, Sotiriou C. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007;13:3207–3214. doi: 10.1158/1078-0432.CCR-06-2765. [DOI] [PubMed] [Google Scholar]
- Computational Cancer Biologty, Division of Molecular Carcinogenesis, Netherlands Cancer Institute Data portal. http://bioinformatics.nki.nl/data.php.
- A subtyping tool for triple-negative breast cancer. https://cbc.mc.vanderbilt.edu/tnbc/ [DOI] [PMC free article] [PubMed]
- Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol. 2004;14:2217–2227. doi: 10.1016/j.cub.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Gruneberg U, Neef R, Li X, Chan EH, Chalamalasetty RB, Nigg EA, Barr FA. KIF14 and citron kinase act together to promote efficient cytokinesis. J Cell Biol. 2006;172:363–372. doi: 10.1083/jcb.200511061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O’Connor L, Li M, Taylor R, Dharsee M, Ho Y, Heilbut A, Moore L, Zhang S, Ornatsky O, Bukhman YV, Ethier M, Sheng Y, Vasilescu J, Abu-Farha M, Lambert JP, Duewel HS, Stewart II, Kuehl B, Hogyue K, Colwill K, Gladwish K, Muskat B. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. 2010;7:243–249. doi: 10.1038/cmi.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko BS, Chang TC, Chen CH, Liu CC, Kuo CC, Hsu C, Shen YC, Shen TL, Golubovskaya VM, Chang CC, Shyue SK, Liou JY. Bortezomib suppresses focal adhesion kinase expression via interrupting nuclear factor-kappa B. Life Sci. 2010;86:199–206. doi: 10.1016/j.lfs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Golubovskaya VM, Cance W. Focal adhesion kinase and p53 signal transduction pathways in cancer. Front Biosci. 2010;15:901–912. doi: 10.2741/3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windheim M, Lang C, Peggie M, Plater LA, Cohen P. Molecular mechanisms involved in the regulation of cytokine production by muramyl dipeptide. Biochem J. 2007;404:179–190. doi: 10.1042/BJ20061704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Jatoi I, Hilsenbeck SG, Clark GM, Osborne CK. Significance of axillary lymph node metastasis in primary breast cancer. J Clin Oncol. 1999;17:2334–2340. doi: 10.1200/JCO.1999.17.8.2334. [DOI] [PubMed] [Google Scholar]
- van der Heiden-van der Loo M, Schaapveld M, Ho VK, Siesling S, Rutgers EJ, Peeters PH. Outcomes of a population-based series of early breast cancer patients with micrometastases and isolated tumour cells in axillary lymph nodes. Ann Oncol. 2013;24:2794–2801. doi: 10.1093/annonc/mdt243. [DOI] [PubMed] [Google Scholar]
- Chang MC, Escallon JM, Colgan TJ. Prognostic significance of a positive axillary lymph node fine-needle aspirate in patients with invasive breast carcinoma. Cancer cytopathology. 2013;122:138–144. doi: 10.1002/cncy.21354. [DOI] [PubMed] [Google Scholar]
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- O’Shaughnessy J, Schwartzberg L, Danso M, Rugo H, Miller K, Yardley D, Carlson R, Finn R, Charpentier E, Freese M, Gupta S, Blackwood-Chirchir A, Winer EP. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC) J Clin Oncol. 2011;29 doi: 10.1200/JCO.2014.55.2984. suppl; abstr 1007. [DOI] [PubMed] [Google Scholar]
- Cameron D, Brown J, Dent R, Jackisch C, Mackey J, Pivot X, Steger GG, Suter TM, Toi M, Parmar M, Laeufle R, Im YH, Romieu G, Harvey V, Lipatov O, Pienkowski T, Cottu P, Chan A, Im SA, Hall PS, Bubuteishvili-Pacaud L, Henschel V, Deurloo RJ, Pallaud C, Bell R. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): primary results of a randomised, phase 3 trial. Lancet Oncol. 2013;14:933–942. doi: 10.1016/S1470-2045(13)70335-8. [DOI] [PubMed] [Google Scholar]
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Conde C, Rambout X, Lebrun M, Lecat A, Di Valentin E, Dequiedt F, Piette J, Gloire G, Legrand S. The inositol phosphatase SHIP-1 inhibits NOD2-induced NF-kappaB activation by disturbing the interaction of XIAP with RIP2. PLoS One. 2012;7:e41005. doi: 10.1371/journal.pone.0041005. [DOI] [PMC free article] [PubMed] [Google Scholar]