

Abstract
Ubiquinone (UQ), also known as coenzyme Q (CoQ), is a redox-active lipid present in all cellular membranes where it functions in a variety of cellular processes. The best known functions of UQ are to act as a mobile electron carrier in the mitochondrial respiratory chain and to serve as a lipid soluble antioxidant in cellular membranes. All eukaryotic cells synthesize their own UQ. Most of the current knowledge on the UQ biosynthetic pathway was obtained by studying Escherichia coli and S. cerevisiae UQ-deficient mutants. The orthologues of all the genes known from yeast studies to be involved in UQ biosynthesis have subsequently been found in higher organisms. Animal mutants with different genetic defects in UQ biosynthesis display very different phenotypes, despite the fact that in all these mutants the same biosynthetic pathway is affected. This review summarizes the present knowledge of the eukaryotic biosynthesis of UQ, with focus on the biosynthetic genes identified in animals, including C. elegans, rodents and humans. Moreover, we review the phenotypes of mutants in these genes and discuss the functional consequences of UQ deficiency in general.
Keywords: ubiquinone biosynthesis, coenzyme Q, ubiquinone deficiency, COQ genes, unicellular model systems, metazoan model systems, human patients
Introduction
Ubiquinone (UQ), also known as coenzyme Q (CoQ), is a redox-active lipid present in all cellular membranes where it acts in a variety of cellular processes (Crane, 2007). The best-understood physiological function of UQ is as a mobile electron carrier in the mitochondrial respiratory chain, transporting electrons from complex I and II to complex III. Its antioxidant function in cellular membranes and lipoproteins is also well recognized (Ernster and Forsmark-Andree, 1993, Bentinger et al., 2007). Moreover, UQ has been described to be an obligatory cofactor for dihydroorotate dehydrogenase, a key enzyme for de novo pyrimidine biosynthesis (Evans and Guy, 2004). It has also been implicated in modulating the mitochondrial Permeability Transition Pores (mtPTP) (Fontaine et al., 1998). Furthermore, UQ is an active part of the electron transport systems of the plasma membrane and lysosomes (Turunen et al., 2004, Nohl and Gille, 2005). While a more complete picture of the functions of UQ is emerging, many fundamental questions still remain regarding details of its biosynthesis, its intracellular mobilization and its turnover, as well as about the pathogenesis of UQ deficiency. In this review, we summarize the research on UQ biosynthesis in animals, with emphasis on the functional consequences of various lesions in the UQ biosynthetic pathway. For a review on other aspects of the biology of UQ, the interested readers should consult the comprehensive review by Turunen and references therein (Turunen et al., 2004).
The biosynthesis of UQ has been studied extensively in E. coli and the yeast Saccharomyces cerevisiae (S. cerevisiae) by characterization of the biosynthetic intermediates that accumulate in UQ-biosynthesis defective strains (Meganathan, 2001, Tran and Clarke, 2007). The knowledge gathered in these two single-celled organisms, especially budding yeast, has been successfully used to identify the gene products required for the biosynthesis of UQ in higher organisms. Below, we first provide a brief overview of the proposed pathway for UQ biosynthesis in eukaryotic cells in general, followed by a section reviewing specific biosynthetic genes identified in C. elegans, rodents and humans. In the next section we summarize the phenotypes of mutants in these genes, whether or not it is known that they are actually caused by UQ deficiency. Finally, we elaborate on cellular phenotypes associated with UQ deficiency.
Overview of the UQ Biosynthetic Pathway
UQ (Fig. 1) is a benzoquinone linked to a hydrophobic poly-isoprenoid tail that varies in length in different species (Ernster and Dallner, 1995). For example, the major isoforms in budding yeast and E. coli are UQ6 and UQ8 (the subscript denotes the number of isoprene subunits) respectively, whereas UQ9 is found in rodents and worms, and UQ10 in humans. In addition, UQ9 is a minor, rare form in humans while UQ10 is the minor form in mice. The quinone head is the functional group, which undergoes reversible redox cycling, whereas the isoprenoid tail serves primarily to anchor ubiquinone in the membrane (Crane, 2001). It is not yet well understood why different species make ubiquinone molecules with different tail lengths. Studies have shown that UQ species with different tail lengths can functionally substitute each other, at least partially, highlighting that the side chain length of UQ is not essential for its functions (Okada et al., 1998, Hihi et al., 2003).
Figure 1. The chemical structure of ubiquinone (UQ).
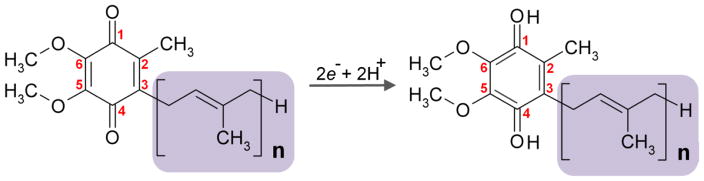
UQ, also called coenzyme Q (CoQ), is composed of a redox-active benzoquinone ring, conjugated to a polyisoprenoid tail that varies in length in different species. The numerical subscript n denotes the number of isoprene subunits in the tail. Yeast and E. coli utilize UQ6 and UQ8 respectively, whereas UQ9 is found in rodents and worm and UQ10 is the major UQ species in humans. Numbers in red refer to position of carbon atoms in the quinone ring. Addition of two electrons to oxidized UQ results in the full reduction of UQ.
Eukaryotic cells rely on endogenous synthesis for adequate UQ supply. To date, 11 nuclear genes have been identified in the budding yeast S. cerevisiae as being essential for endogenous production of UQ6. Nine genes are designated COQ1 through COQ9 to indicate a deficiency in Coenzyme Q (UQ) (Tran and Clarke, 2007, Ozeir et al., 2011). In addition, Pierrel and colleagues (Pierrel et al., 2010) recently discovered a new function of yeast ferredoxin and ferredoxin reductase (encoded by YAH1 and ARH1) in UQ6 biosynthesis. Mutants for all these genes are deficient in the production of UQ6 and incapable of growing on non-fermentable carbon sources, because UQ is necessary for mitochondrial respiration (Tran and Clarke, 2007). Fig. 2 illustrates the current model for the UQ6 biosynthesis pathway in budding yeast. The first committed step is assembly and elongation of the isoprenoid tail catalyzed by the Coq1p enzyme. Coq2p mediates the second step, which is the condensation of the isoprenoid tail with a quinone ring precursor, thus generating the first membrane-bound UQ precursor. 4-hydroxybenzoate (4-HB) is the long-known ring precursor of UQ (Tran and Clarke, 2007). Recently, two independent studies have identified para-aminobenzoic acid (pABA) as another precursor of the ubiquinone ring in budding yeast (Pierrel et al., 2010, Marbois et al., 2010). pABA differs from 4-HB only by one substituent of benzoic acid, an amine (NH2) rather than a hydroxyl group (OH). It is converted into UQ via the same UQ biosynthetic pathway, but needs an additional step(s) to replace NH2 by OH at C4. It has been speculated that pABA is likely a precursor of UQ only in yeast (Pierrel et al., 2010). E. coli mutants deficient in 4-HB lack UQ synthesis. Treatment of mammalian cells with pABA resulted in a decrease in UQ levels.
Figure 2. Schematic illustration of the UQ biosynthetic pathway in the yeast S. cerevisiae.

The pathway starts with assembly and elongation of the isoprenoid tail catalyzed by the enzyme Coq1p (not shown). Coq2p mediates the condensation of the isoprenoid tail with either one of two basic ring structures, para-hydroxybenzoate (4-HB) or para-aminobenzoate (pABA), producing 3-hexaprenyl-4-hydroxybenzoate (HHB) and 3-hexaprenyl-4-aminobenzoic acid (HAB) respectively. The basic ring moiety then undergoes a series of modifications to produce UQ. Enzymes required for 5 of the 7 modifications are shown in blue. A question mark (?) indicates that the protein catalyzing the reaction has yet to be identified. NH2 from pABA and C4-aminated intermediates are shown in red. NH2-to-OH conversion is thought to takes place prior to demethoxyubiquinone (DMQ6) formation. The intermediates that have been detected in yeast coq mutants are shown in brackets. They include HHB and HAB in “ coq3-9 mutants (Tran and Clarke, 2007, Pierrel et al., 2010, Marbois et al., 2010), 3-hexaprenyl-4-hydroxyphenol (4-HP) and 3-hexaprenyl-4-aminophenol (4-AP) in “ coq6 overexpressing Coq8p and in mutant cells deficient in Yalh1 or Ahr1 (Ozeir et al., 2011), 3-hexaprenyl-4-amino-5-hydroxybenzoic acid (HHAB) in “ coq4 cells overexpressing Coq8p and in the “ coq4-1 point mutant (Xie et al., 2012), Demethyl-DMQ6 (DDMQ6) in “ coq5 overexpressing Coq8p (Xie et al., 2012), DMQ6 in a coq7 point mutant (Padilla et al., 2004, Marbois et al., 2010), yeast cells with partial inactivation of Coq7p (Pierrel et al., 2010) and “ coq7 mutants overexpressing Coq8p (Xie et al., 2012); only minute amounts of 4-amino-DMQ6 (IDMQ6) were found in wild type yeast cells and in the coq7-1 point mutant as well as in “ coq6 and “ coq9 cells overexpressing Coq8p (Xie et al., 2012) Asterisks indicate compounds that are the main intermediates detected when either 4-HB or pABA are provided as ring precursors. Coq8p is a putative kinase, believed to have a regulatory role in UQ6 biosynthesis. Deletion of COQ8 affects the phosphorylation states of Coq3p, Coq5p and Coq7p (Xie et al., 2011, Tauche et al., 2008). In addition, Coq4p and Coq9p (not shown) are also required for UQ6 biosynthesis, although their precise roles remain to be elucidated (Tran and Clarke, 2007).
After the attachment of the isoprenoid tail to one of the two basic ring structures (4-HB or pABA), the ring moiety is subjected to a series of modifications. Coq6p catalyzes the first ring modification, hydroxylation at the position C5 of the benzoquinone ring (Ozeir et al., 2011). Coq3p carries out two different O-methylation reactions. Coq5p is required for the only C-methylation step. Coq7p is responsible for the penultimate step introducing the last hydroxyl group (Miyadera et al., 2001, Jonassen et al., 1998). In addition, the C5-hydroxylation is deficient in yeast cells depleted for Yah1p or Arh1p (Pierrel et al., 2010); however, the exact mechanisms by which these two genes affect UQ6 biosynthesis are not understood.
Biosynthesis of UQ8 in E. coli relies on ten different ubi genes (ubiA-ubiH). Compared to the UQ6 biosynthetic pathway in budding yeast, the order of reactions of ring modification in E. coli is slightly different. In E. coli the steps following the fusing of the octaprenyl side chain onto the ring intermediate 4-HB are decarboxylation, hydroxylation and methylation rather than hydroxylation, methylation and then decarboxylation (Meganathan, 2001). E. coli ubi mutants are devoid of UQ8 and accumulate distinct intermediates depending on which reaction is affected (Gibson and Young, 1978, Meganathan, 2001). In contrast, full loss-of-function yeast coq3 to coq9 mutant strains accumulate the same earlier intermediate, 3-hexaprenyl-4-hydroxybenzoate (HHB) when grown in the presence of 4-HB, or 3-hexaprenyl-4-aminobenzoic acid (HAB) when making use of pABA as a ring precursor (Pierrel et al., 2010, Marbois et al., 2010, Tran and Clarke, 2007). On the basis of these and other findings, in particular the dependence of several Coq proteins on each other for theirs stability, and the co-migration of several of these as a high molecular mass complex, a model of a large UQ biosynthetic complex which involve most Coq proteins has been proposed for yeast (Tran and Clarke, 2007). The model suggests that no Coq enzyme that modifies the UQ ring can function outside the complex and all Coq polypeptides are necessary for the stability of the complex. The details of the organization of the complex, however, have not been resolved.
In addition to the Coq proteins that have been assigned a specific catalytic function, the exact molecular functions of Coq4p, Coq8p and Coq9p, have not yet been fully elucidated. Coq8p has been postulated to function as a kinase and to play a regulatory role in UQ biosynthesis (Leonard et al., 1998). Over-expression of Coq8p in yeast “ coq5, “ coq6, or “ coq7 strains allows for accumulation of the substrate of the missing enzyme, suggesting that overexpression of Coq8p prevents the complex to fall apart (Xie et al., 2012, Ozeir et al., 2011, Padilla et al., 2009). Moreover, it has been shown that the low steady-state levels of several other Coq proteins that are observed in “ coq2-7 and “ coq9-10 strains can be restored by Coq8p overexpression (Zampol et al., 2010, Xie et al., 2012). These findings indicate that Coq polypeptides are stabilized and the Coq polypeptide complex is restored in the presence of more Coq8p. However, the molecular mechanisms that underpin these observations remain to be clarified. No enzymatic function is known for Coq4p and it has been proposed to function in the assembly and/or maintenance of the UQ biosynthetic complex (Marbois et al., 2005, Marbois et al., 2009). “ coq4 yeast cells in which Coq8p is overexpressed were shown to have a deficiency in the O5-methylation catalyzed by Coq3p, as well as an impairment in at least one other step downstream of the earliest Coq3p-dependent reaction (Xie et al., 2012). In the case of mutants lacking Coq9p, overexpression of Coq8p leads to the accumulation of UQ biosynthetic intermediates diagnostic of insufficient activity of both Coq6p and Coq7p enzymes (Xie et al., 2012). How Coq4p and Coq9p influence the activities of other COQ proteins remains to be determined (Johnson et al., 2005, Xie et al., 2012). Unlike other coq mutants, a yeast coq10 null mutant produces near normal amounts of UQ6 but like the others, it is also respiration-defective. Thus it is held that Coq10p is not involved in UQ biosynthesis but instead required for the proper function of UQ6 in the respiratory chain (Barros et al., 2005). Over-expression of Coq8p increases UQ6 synthesis 2-fold and partially rescues the respiratory defect of coq10 null mutations (Barros et al., 2005). However, an excess of Coq10p itself has an adverse effect on respiration (Zampol et al., 2010).
Functional conservation of the UQ Biosynthetic Pathway
Homologues of yeast COQ genes have been identified in a range of higher organisms including worms, rodents and humans (Table 1). In C. elegans, eight genes have thus far been identified to be involved in UQ biosynthesis (Hihi et al., 2002, Asencio et al., 2003, Ewbank et al., 1997). Among them, the function in UQ biosynthesis has been clearly defined only for CLK-1 (the orthologue of Coq7p). clk-1 mutations were identified in a genetic screen for mutations that affect developmental and behavioral timing in C. elegans (Wong et al., 1995), and the unusually long lifespan of these mutants has attracted considerable attention. C. elegans CLK-1 was later shown to be a homolog of yeast Coq7p (Ewbank et al., 1997). In yeast, mutation of the COQ7 gene results in a lack of UQ6 production. In contrast to deletions, coq7 point mutants, which result in normal levels of a defective polypeptide, were found to accumulate DMQ6, the intermediate immediately preceding the missing step, presumably due to sufficient integrity of the UQ6 biosynthetic complex provided by the normal levels of the mutant polypeptide (Padilla et al., 2004). Like the yeast mutant, clk-1 mutant worms contain essentially no UQ9 (the main UQ species in C. elegans), but instead accumulate large quantities of DMQ9 (Miyadera et al., 2001, Jonassen et al., 1998), confirming conservation of COQ7/CLK-1 function in UQ biosynthesis. As for the other seven genes (coq-1 to coq-6 and coq-8) that were identifed by their high sequence similarity to the yeast COQ genes (30%–47% identity) (Asencio et al., 2003), homozygous knockout result in larval lethality or arrest and thus stable knockout lines could not be obtained (Gavilan et al., 2005, Earls et al., 2010, Asencio et al., 2009, Hihi et al., 2002). This hinders full elucidation of the biochemical functions of these genes in worms. For coq-1 to coq-6, so far the best experimental evidence supporting their participation in UQ biosynthesis is provided by the results of an RNAi knockdown study. Placido Navas and colleagues showed that upon feeding wild-type nematodes with dsRNA of C. elegans coq genes (coq-1 to coq-8), UQ9 content in whole worm homogenates was decreased to approximately 10 to 50 percent of the control value (Asencio et al., 2003, Rodriguez-Aguilera et al., 2003). Two weeks after removal from dsRNA exposure, global UQ9 content had return to normal levels (Asencio et al., 2003). In addition, a more recent study on coq-8 knockout progeny from heterozygous hermaphrodites showed that UQ9 content in coq-8 knockout individuals (F1) is ~7% of that found in wild type controls, providing strong evidence for an important role of coq-8 in UQ biosynthesis in worms (Asencio et al., 2009). The small amount of UQ remaining in this mutant has been interpreted as indicating that the presence of the COQ-8 protein is not an absolute requirement for de novo biosynthesis of UQ, thus suggesting a non-catalytic role of COQ-8. This is in agreement with a proposed regulatory role for Coq8p on UQ6 biosynthesis in yeast. However, considering possible transfer of COQ-8 protein or mRNA from coq-8+/− hermaphrodite mothers to homozygous offspring as has been reported for CLK-1/COQ-7 (Burgess et al., 2003), one cannot rule out the possibility that trace amount of maternally-derived COQ-8 is sufficient to allow the coq-8−/− F1 worms to retain some UQ biosynthetic activity. Of the eight coq genes identified so far, only coq-5 and clk-1 cDNAs were cloned and shown to complement the UQ6 synthesis defect of their corresponding yeast coq null mutants (Rodriguez-Aguilera et al., 2003, Ewbank et al., 1997). The functional conservation of the others awaits experimental verification.
Table 1. Genes required for ubiquinone biosynthesis or function.
Yeast genes required for UQ biosynthesis (COQ1-9, ARH1 and YAH1) or UQ function in respiration (COQ10) and their homologues in higher eukaryotes are presented. ARH1 and YAH1 are found to be required for the C5-hydroxylation reaction. The functions in UQ6 biosynthesis of these two genes as well as those of three COQ genes (COQ4, COQ8 and COQ9) are not yet understood. For E. coli, only the genes that are functionally equivalent to yeast COQ genes are shown, including four genes whose products share homology with yeast Coq proteins (Okada et al., 1996, Hsu et al., 1996, Dibrov et al., 1997, Suzuki et al., 1994) and ubiF which catalyzes the same reactions as the COQ7 genes but bears no homology to them (Tran et al., 2006).
| Function | Genes
|
||||
|---|---|---|---|---|---|
| S. cerevisiae | C. elegans | M. musculus | H. sapiens | E. coli | |
| Elongation of prenyl side-chain | COQ1 | coq-1 | Pdss1/Pdss2 | PDSS1/PDSS2 | ispB* |
| Prenalytion of ring precursor | COQ2 | coq-2 | Coq2 | COQ2* | ubiA |
| O5 and O6 methylation | COQ3 | coq-3 | Coq3 | COQ3* | ubiG* |
| unknown | COQ4 | coq-4 | Coq4 | COQ4* | - |
| C2-methylation | COQ5 | coq-5* | Coq5 | COQ5 | ubiE |
| C5-hydroxylation | COQ6 | coq-6 | Coq6 | COQ6* | - |
| C6-hydroxylation | COQ7 | coq-7/clk-1* | Coq7/Mclk1 | COQ7* | ubiF* |
| unknown | COQ8 | Coq-8 | Coq8 | ADCK3* | - |
| unknown | COQ9 | - | Coq9 | COQ9 | - |
| unknown | COQ10 | - | Coq10a/Coq10b | COQ10A/10B | - |
| unknown | ARH1 | - | - | - | - |
| unknown | YAH1 | - | - | - | - |
Asterisks indicate that genes have been shown to functionally complement corresponding yeast mutants.
By sequence homology and functional complementation, mammalian homologues of all of the yeast COQ genes have also been identified. Except for the enzymes carrying out the function of Coq1p, mammalian homologues are named COQ2-COQ10, based on their yeast homologues. Human COQ2 (Mollet et al., 2007), COQ3 (Jonassen and Clarke, 2000), COQ4 (Casarin et al., 2008), COQ6 (Heeringa et al., 2011), COQ7 (Vajo et al., 1999), COQ8 (Xie et al., 2011), and COQ9 (Duncan et al., 2009) cDNAs have been shown to partially complement budding yeast mutants in corresponding genes. In the budding yeast S. cerevisiae, the side-chain length of ubiquinone is determined by the Coq1p hexaprenyl diphosphate synthase (Tran and Clarke, 2007). The human, murine and S. pombe prenyl diphosphate synthases are heterotetramers of two protein subunits rather than monomeric enzymes as in budding yeast (Coq1p) and E. coli (IspB) (Saiki et al., 2005). The genes encoding these subunits are designated Pdss1 and Pdss2 in mice, and PDSS1 and PDSS2 in humans. Amino acid sequence alignment shows Pdss1 and PDSS1 to be highly similar to the S. pombe homolog SpDps1 (30–49.0%) and the homodimeric prenyl diphosphate synthases from other organisms such as budding yeast, bacteria, and the plant A. thaliana. They also bear all seven conserved regions found in all known trans-prenyl diphosphate synthases (Saiki et al., 2005). In contrast, the subunits 2 (mouse Pdss2 and human PDSS2) have limited similarity to the S. pombe homolog SpDlp1 and the long-chain trans-prenyl diphosphate synthases from other organisms (Saiki et al., 2005). Cells expressing either subunit alone do not synthesize UQ9 or UQ10, demonstrating that both subunits are required to constitute a viable enzyme (Saiki et al., 2005, Mollet et al., 2007). It has also been reported that heterologous combination of the two subunits of prenyl diphosphoate synthase such as mouse Pdss1-human PDSS2, can produce a functional enzyme (Saiki et al., 2005).
Orthologous UQ biosynthetic genes in mice and humans share a high degree of sequence homology in coding regions (the overall identity is more than 60%). Studies have been initiated to better understand the functions of these genes and UQ biosynthesis defects in whole animals using genetic knockout mouse models. To date, three knockout mutants at different steps in the UQ biosynthetic pathway have been reported. All of them, Pdss2 (Peng et al., 2008), Coq3 (Jérôme Lapointe, 2012) and Mclk1/COQ7 (Levavasseur et al., 2001), display an embryonic lethal phenotype when both alleles are deleted. Mclk1−/− (Levavasseur et al., 2001, Nakai et al., 2001) and Pdss2−/− (Lu et al., 2011) mice fail to survive past embryonic day 12.5. Quinone analysis showed that Mclk1/Coq7-null cells and embryos make no UQ9 but instead accumulate the expected UQ precursor DMQ9, (Levavasseur et al., 2001, Nakai et al., 2001). Mouse mutants heterozygous for a mutation in Mclk1 or Coq3 were found to have a normal viability and not differ from wild type mice in overall appearance (Lapointe and Hekimi, 2008, Jérôme Lapointe, 2012). They have normal tissue levels of UQ9 despite a 2-fold reduction in protein levels (Jérôme Lapointe, 2012, Lapointe and Hekimi, 2008), which is in line with findings in other systems. For example, when whole cell lysates were analyzed, yeast diploid strains harboring heterozygous deletions of coq2 or coq6 and human fibroblasts from heterozygous carriers of a PDSS2 mutation were found to have UQ levels comparable to that of wild type controls (Salviati et al., 2012). It seems that most COQ enzymes, under normal conditions, are synthesized in excess over the amounts needed for the proper level of UQ synthesis. Surplus production that may allow for the fine regulation of an enzyme activity is not uncommon for key cellular enzymes. One proposed post-translational modification of yeast COQ proteins is phosphorylation (Xie et al., 2011, Tauche et al., 2008, Martin-Montalvo et al., 2011). However, phosphorylation of their mammalian homologues has not been reported. Interestingly, recent works revealed that haploinsufficiency of Mclk1/Coq7 or COQ4 impairs UQ biosynthesis (discussed in sections below), underscoring a more rate-limiting role in UQ production for some pathway constituents.
Advances in conditional gene targeting provide the tools to bypass the early embryonic lethality of essential genes and establish mouse models disrupting UQ biosynthetic genes in a developmental stage- and tissue-specific fashion. Detailed characterization of these models will further our understanding on the synthesis and utilization of UQ in mammals and possibly reveal unexpected roles for these genes in whole animal physiology. Peng and colleagues have described mice with liver-specific inactivation of the Pdss2 gene generated by Cre-lox technology. These animals were produced by deletion of conditional (floxed) Pdss2 alleles using Cre expression driven by the liver-specific albumin promoter. UQ9 is barely detectable in the Pdss2 mutant liver, confirming the essential function of Pdss2 for murine UQ biosynthesis (Peng et al., 2008). Other conditional knockout mice targeting different UQ biosynthesis genes can be expected to be developed in the near future and yield new insights into UQ biology.
Structural Conservation of COQ Proteins
Domain and motif identification for several COQ proteins has revealed some interesting features about them. Both yeast and human COQ2 proteins are predicted to contain six transmembrane domains suggestive of integral membrane localization (Forsgren et al., 2004, Ashby et al., 1992). In addition, putative polyprenyl diphosphate substrate-binding sites have been identified in the protein products in both species, consistent with their prenyltransferase function in UQ biosynthesis. The sequence of this site in human COQ2 is 60% identical with that of the equivalent site in yeast Coq2p (Forsgren et al., 2004). Conserved motifs identified in a large family of AdoMet(S-adenosylmethionine)-dependent methyltransferases are found in the amino acid sequences of yeast Coq3p as well as rat and human COQ3 gene products which catalyze two different O-methylation reactions (Poon et al., 1999, Jonassen and Clarke, 2000). Inspection of the amino acid sequence for CLK-1/COQ7 from a variety of species revealed the existence of a carboxylate-bridged diiron center in this enzyme (Stenmark et al., 2001), and we have demonstrated that the enzymatic activity of CLK-1/MCLK1/COQ7 is sensitive to iron availability (Wang et al., 2009). Spectroscopic characterization of purified MCLK1 protein has provided experimental data showing there exists such a diiron centre as the active site for dioxygen activation and demethoxyubiquinone hydroxylation (Behan and Lippard, 2010). For Coq4p, Coq8p and Coq10p, the functions of which have not yet been clearly defined, the analysis of protein sequences has helped understanding their roles in the UQ biosynthetic pathway. The sequence of Coq4p contains no catalytic domains indicative of an activity necessary for formation of a substituted benzoquinone ring, thus a speculation is made that Coq4p plays a structural role in the UQ6 biosynthetic pathway (Marbois et al., 2009). The crystal structure of a cyanobacteria protein homologue to Coq4p has been solved (Schwede et al., 2003). Interestingly, geranylgeranyl monophosphate, which is a polymer of four isoprenoid units, cocrystallized with the Coq4p homolog, Alr8543 from Nostoe sp. PCC7120. This information was used to propose that Coq4p functions to anchor UQ intermediates to the inner mitochondrial member by its isoprenoid tail units (Rea et al., 2010).
Yeast Coq8p (Leonard et al., 1998), its human homologue ADCK3 (Lagier-Tourenne et al., 2008) as well as bacterial homologue UbiB (Poon et al., 2000), belong to an atypical kinase family, harboring motifs characteristic of eukaryotic-type protein kinases (ePK). Yeast coq8 mutants were found to lack several phosphorylated forms of Coq3p, Coq5p and Coq7p (Xie et al., 2011, Tauche et al., 2008). Expression of human ADCK3 in yeast coq8 mutants rescues both UQ6 synthesis and phosphorylation of yeast COQ polypeptides, establishing a conserved function of Coq8p/ADCK3 in UQ biosynthesis (Xie et al., 2011). In yeast, as described above, deletion of any of the COQ1-COQ9 genes results in the absence of several of the other Coq polypeptides (Tran and Clarke, 2007). Over-expression of COQ8 has recently been found to stabilize the steady state levels of several Coq proteins in coq null mutants and its kinase activity seems to be important for the effect (Xie et al., 2012, Zampol et al., 2010). Thus these findings point to the possibility that Coq8p/ADCK3-dependent phosphorylation(s) might serve to regulate the stability of the UQ biosynthetic pathway constituents, and probably also their activity levels. However, to date, the phosphorylation of the mammalian COQ proteins has not been examined, nor has an effect of altered ADCK3 levels on their stability and activity been addressed. At the present time, obviously, we have more questions than answers about the function of Coq8p in yeast and ADCK3 in human UQ biosynthesis. In particular, do they directly phosphorylate COQ protein substrates? How is the stability and activity of a particular COQ protein controlled by its phosphorylation state? What is the biological significance of such regulation? Identification of phosphorylation sites and mutagenesis studies will be helpful in addressing some of these questions. Martín-Montalvo and colleagues showed that mutagenesis of putative phosphorylation sites in yeast COQ7 indeed influences the amount of Q6 and DMQ6 produced. In addition, the phosphorylation state of Coq7p appears to be affected by changes in the metabolic state of yeast cells, supporting the notion that phosphorylation is a means of regulating UQ biosynthetic activity in response to metabolic needs (Martin-Montalvo et al., 2011). Based on the homology between Coq10p and members of the steroidogenic acute regulatory protein-related transfer (START) domain superfamily, which contains a distinctive lipid-binding motif, Barros and colleagues hypothesized that Coq10p might bind to UQ and function in UQ trafficking (Barros et al., 2005). Consistent with their prediction, they found that UQ6 was present in histidine-tagged Coq10p purified from a coq10 overexpressing yeast strain (Barros et al., 2005). Surprisingly, exogenous UQ2 can fully rescue a coq10 mutant (Barros et al., 2005), indicating that Coq10p is not essential for the transport of exogenous UQ to the functional sites of UQ in the electron transport chain, at least in the case of a short side-chain form. In mammals, there are two isoforms of Coq10p homologue (Coq10a and Coq10b in mice, COQ10A and COQ10B in humans) resulting from alternative splicing. Expression of the human COQ10A cDNA rescues the respiration-deficient phenotype of yeast “ coq10 mutants, confirming conservation of function of COQ10 throughout eukaryotes (Cui and Kawamukai, 2009).
In summary, UQ biosynthetic proteins appear well-conserved across species. Except for COQ1, which is responsible for the first committed step, all the COQ genes encoding proteins involved in the final phase of UQ6 biosynthesis in the budding yeast S. cerevisiae have clear orthologues in animals. For most of these genes, genetic complementation studies have demonstrated functional conservation from budding yeast to human. Though the ring modification reactions starting with the prenylation of a benzoquinone ring precursor are similar to those in budding yeast, the order of reactions in the mammalian system has not been unambiguously established. Furthermore, the interactions between the pathway constituents and the modes of regulation of UQ biosynthesis are likely to be different. COQ genes from eukaryotes could not be readily expressed into soluble and active enzymes in E. coli (Poon et al., 1999) and thus far very few of them have been purified. This hampers the studies of exploring the chemical mechanisms of COQ-catalyzed reactions, the structural and functional relationship of COQ enzymes and the interactions of pathway constituents. In the future, further improvements in protein production and purification techniques may enable in vitro reconstitution of the UQ biosynthetic pathway and full elucidation of the enzymatic mechanisms and the sequence of events.
The UQ Biosynthetic complex
All products of the yeast COQ genes are localized to the mitochondrial inner membrane (IMM). Coq1p and Coq3p-Coq9p polypeptides are peripherally associated with the inner membrane on the matrix side, whereas Coq2p spans the IMM and Coq10p is an integral membrane protein as well (Tran and Clarke, 2007, Barros et al., 2005). Typical mitochondrial leader sequences are found at the N-terminus of most of the yeast Coq polypeptides, and it has also been demonstrated for most of them that their transport into the inner membrane requires the membrane potential “ È (Tran and Clarke, 2007). In the case of higher eukaryotic homologues, a mitochondrial leader sequence has been identified in mouse MCLK1/COQ7. The import of MCLK1 requires the leader sequence but, surprisingly, not the mitochondrial membrane potential (Jiang et al., 2001). In the N-terminal region of human COQ3 protein many basic and hydroxylated residues are found, but not acidic amino acids. These features are consistent with a classic mitochondrial targeting sequence (Jonassen and Clarke, 2000). No specific amino acid residues for a classical N-terminal import signal could be identified in human COQ2 protein, but it was shown that the first 50 amino acids from the N-terminus are required to rescue the growth defect of yeast “ coq2 mutant on glycerol respiratory medium (Forsgren et al., 2004). Advanced sequence analysis as well as more expression and functional studies can be expected in the near future which will advance our understanding of the production and processing of mature COQ proteins in animal cells. Given the high homology throughout eukaryotes, the final localization of COQ proteins in higher organisms is likely to be essentially the same as that of their yeast homologues, which is the IMM, although this has not yet been confirmed by fractionation experiments. For the proteins that have been studied, i.e. CLK-1/MCLK1 (Jiang et al., 2001) and mouse COQ3 (Jérôme Lapointe, 2012), they are found, as predicted, to be enriched in mitochondrial fractions.
The localization of COQ proteins tells us that UQ biosynthesis is carried out in mitochondria, but several studies have questioned whether mitochondria are the exclusive origin of newly synthesized UQ (Kalen et al., 1990, Teclebrhan et al., 1995, Kalen et al., 1987, Tekle et al., 2002). Although it is particularly enriched in mitochondria, UQ can be found in practically all membranes, including those of the Golgi apparatus and lysosomes, which are relatively rich in UQ, but UQ is also present in smaller amounts in other defined cellular locations including the ER, the plasma membrane and the peroxisomes (Morre and Morre, 2011). The broad localization of UQ and its differential distribution within the cell indicate that a mechanism must be in place to route UQ to various cellular membranes in a controlled manner. To date, almost no information is available on how UQ gets transported from its site of synthesis to other membranes and how the occurrence of UQ in various cellular compartments is regulated.
As previously mentioned, it has been proposed that in budding yeast several Coq polypeptides are organized into a large complex for proper activity. This kind of organization may be necessary for efficient channeling of UQ biosynthetic intermediates. Whether or not a similar complex exits in animals has not been resolved. Yeast Coq7p is a component of the UQ biosynthetic complex as evidenced by accumulation of the early intermediate HHB (the Coq2p product) in coq7 null cells cultured in the presence of 4-HB (Marbois and Clarke, 1996) rather than DMQ6 as in a mutant harboring a single amino acid point mutation (Padilla et al., 2004). Most likely the presence of mutant COQ7 protein in the point mutant prevents the UQ biosynthetic protein complex to fall apart. However, in contrast to the yeast “ coq7 mutant, clk-1/COQ7 C. elegans and mouse Mclk1 knockout mutants accumulate DMQ9, the substrate of the missing enzyme, indicating that the UQ biosynthetic pathway before the CLK-1/MCLK1 step is functional and active (Stepanyan et al., 2006). This could be an indication that UQ biosynthesis in worms and mice does not occur via a biosynthetic complex of COQ proteins. However, it remains possible that there is a complex as in yeast, but its composition is different or the complex is more robust and less susceptible to fall apart when one of its components is missing. Interestingly, a recent study on Coq8p found that yeast “ coq9 strain with Coq8p overexpression lack Coq4p and Coq7p and are capable of manufacturing DMQ6 (Xie et al., 2012). This indicates that thanks to extra Coq8p the UQ synthetic pathway in budding yeast, as in animal cells even without overexpression of COQ8, can be held in place when some constituents (e.g. Coq7p) are missing.
Substrate specificity of COQ Enzymes
COQ enzymes appear to have broad substrate specificity. As noted above, expression of COQ genes from higher eukaryotes such as hCOQ3 (Jonassen and Clarke, 2000), hCOQ6 (Heeringa et al., 2011), and hCOQ7 (Vajo et al., 1999) can restore UQ6 synthesis in the yeast mutants missing their respective homologues. In addition, expression of different types of prenyl diphosphate synthases in a yeast coq1 mutant results in formation of UQs with side chains of different lengths (5–10) (Okada et al., 1998). Clearly, ring modification enzymes are not specific in terms of the number of isoprene units on their substrates. In addition, in vitro enzymatic assays on mitochondrial membrane-enriched fractions showed that the budding yeast and human COQ3 enzymes all can methylate both eukaryotic substrates and the distinct prokaryotic substrate (Jonassen and Clarke, 2000), suggesting a low degree of specificity of the Coq3p/COQ3 enzyme. Substrate promiscuity of the product of the COQ2 gene was demonstrated with respect to both polyprenyl donor substrates and aromatic substrates. Functional complementation experiments show unequivocally that human COQ2 protein can utilize a shorter prenyl tail and thus restore UQ6 synthesis in a yeast “ coq2 mutant (Forsgren et al., 2004). Moreover, it was shown that upon incubation with decaprenyl-PP (decaprenyl diphosphate) which is the preferred substrate of human COQ2 yeast cells can synthesize a small amount of UQ10 (Forsgren et al., 2004). As discussed above, more recently, it was discovered that Coq2p prenylates both 4-HB and pABA and the resulting products, HHB and HAB respectively, are subsequently converted into UQ6 using the same enzymes that we have been discussing (Marbois et al., 2010, Pierrel et al., 2010)(Fig. 2). This low specificity of COQ enzymes has inspired searching for 4-HB-like molecules that can be processed in the UQ biosynthetic pathway. Two recent studies demonstrated that indeed biosynthetic blocks at Coq6p and Coq7p can be bypassed by providing yeast cells with hydroxylated 4-HB (Ozeir et al., 2011, Xie et al., 2012). It would be of great interest to test whether supply of non-native substrates has a similar effect in a mammalian system as such compounds could have potential therapeutic uses in treating human UQ deficiencies that involve these enzymatic steps.
UQ Biosynthetic Mutants
A number of animal mutants have been described in which different components of the UQ biosynthetic pathway are altered or eliminated (see above). In the next section, we review the phenotypes of these mutants in animal models and humans. Interestingly, mutations that disrupt different UQ biosynthetic genes give rise to different patterns of phenotypes, despite the fact that in all these mutants the same biosynthetic pathway is affected.
Caenorhabditis elegans coq mutants
From available data it appears that complete block of UQ biosynthesis in C. elegans is not viable, with the exception of the clk-1/coq-7 knockout. F2 homozygous coq-3(qm188) mutants issued from F1 coq-3(qm188)/coq-3(qm188) hermaphrodites die during larval development (Hihi et al., 2002). Gavilan and colleagues also reported that homozygous F1 coq-1(ok749) mutants arrested development during the L1/L2 larval stage and exhibited progressive paralysis associated with the malfunction of pharyngeal muscles and poor feeding (Gavilan et al., 2005). Similar phenotypes were also found in a coq-2 deletion mutant (the ok1066 allele) but to a lesser extent (Gavilan et al., 2005). In contrast, Earls and colleagues described that coq-1(ok749) mutants were slow-growing but able to develop to at least the L3 stage with a few adult escapers, and in their study a paralyzed phenotype and abnormal pharyngeal pumping were not observed (Earls et al., 2010). In the same paper, the authors also reported that the coq-1(ok749) mutant as well as coq-1 RNAi-treated worms rather developed progressive loss in motor coordination and selective death of GABA neurons. The neuron degenerative phenotype was also observed in homozygous coq-2(ok1066) and coq-3(ok506) worms and appeared responsive to UQ10 feeding. The authors thus conclude that UQ depletion is the primary pathogenic event causing GABA neuron death and the movement defect (Earls et al., 2010). Most of the progeny produced by coq-4(qm143)/coq-4(qm143) hermaphrodites die during embryogenesis. However, F1 coq-4(qm143) homozygotes issued from heterozygous mothers can grow to adulthood where they display uncoordinated movements, or even paralysis, and defective egg-laying (our unpublished data). A null mutation in the coq-6 gene is also reported to be lethal in C. elegans (http://www.shigen.nig.ac.jp/c.elegans/mutants/DetailsSearch?lang=english&seq=3369). F2 coq-8(ok840) homozygotes issued from F1 homozygote hermaphrodite mothers die during embryonic development with a few escapers which die at the first larval stage (Asencio et al., 2009). Asencio and colleagues have characterized homozygous F1 worms produced by the heterozygotes of the deletion allele coq-8(ok840) and showed that F1 knockouts have slow growth, defective gonad maturation, reduced gamete production and severe abnormalities in the hypodermis. Embryo production and development was improved most noticeably when both UQ8 and uridine were included in the food source. This indicates that pyrimidine biosynthesis, which requires ubiquinone cycling in the respiratory chain, is impaired and this may contribute to the reproduction defects (Asencio et al., 2009).
Unlike the coq mutants described above, clk-1/coq-7 worms are viable, fertile and long-lived despite the fact that, like the others, they are defective in endogenous UQ biosynthesis (Wong et al., 1995, Ewbank et al., 1997). Timing of a wide range of behavioral and physiological processes is deregulated in clk-1 mutants; they exhibit a lengthening of embyronic and post-embryonic development, an extended cell cycle period, slowed rhythmic behaviors (such as defecation and pharyngeal pumping), and reduced brood dize (Felkai et al., 1999, Wong et al., 1995). clk-1 mutants need UQ in their diet in order to complete larval development and produce progeny. When fed a diet devoid of UQ, the animals no longer exhibit the pleiotropic long-lived phenotype. Instead, they arrest development as L2 larvae or develop into sterile adults when developing from dauer larvae (Hihi et al., 2002, Jonassen et al., 2001). The profound phenotype on UQ8-less bacteria is consistent with early developmental arrest and sterility of null mutations in other UQ-biosynthetic genes, indicating an essential requirement for UQ during development and reproduction. Isolation of tRNA suppressors of the missense mutation clk-1(e2519) also strongly argues for a mandatory requirement of UQ for worm survival and growth. Extensive mutagenesis screens were conducted to identify mutations that suppress the slow growth rate of clk-1 worms on UQ-replete bacteria or suppress the growth arrest and/or sterility on UQ-deficient bacteria on both the qm30 (a partial deletion) and e2519 (a Glu148Lys substitution) backgrounds. 9 suppressors were indentified and all of them suppress the mutant phenotypes on both UQ-replete and UQ-deficient bacteria. Strikingly, they are all specific to e2519, and none of them could suppress any phenotypes of clk-1(qm30) mutants. Six of them were cloned and all encode tRNAGlu genes whose anticodons were altered to read the substituted Lys codon of clk-1(e2519). Thus it appears that the growth phenotypes of clk-1 can only be repaired by restoring some CLK-1 production, which in turn is restoring at least some UQ biosynthesis activity, which shows that UQ is indeed a ‘must-have’ for animal life.
Curiously, the clk-1/coq-7 mutant is the only coq mutant that can be maintained as a homozygous line. All other coq mutants described so far are lethal, even on a UQ8-replete diet. What unique feature(s) renders the clk-1 mutant viable under these conditions still remains a mystery. The large amount of DMQ9 present in clk-1 mutants but not in the other mutants most likely determine this key aspect of the Clk-1 phenotype. On the other hand, Arroyo and colleagues reported detection of a small amount of UQ9 in purified clk-1 mitochondria and proposed that the existence of UQ9 of unknown origin could contribute to explain some aspects of the Clk-1 phenotypes such as the ability to grow and reproduce on UQ8-replete OP50 diet (Arroyo et al., 2006). DMQ differs from UQ only by missing one of the two methoxy groups (Fig. 1). Whether it is capable of fulfilling (or interfering with) some of the functions of UQ and thus underlies some of the Clk-1 phenotypes is still uncertain today. clk-1 mitochondria have mildly decreased mitochondrial respiration (Branicky et al., 2000, Kayser et al., 2004). It is likely that the dietary UQ transported to clk-1 mitochondria (Arroyo et al., 2006, Yang et al., 2009) support electron transport in the respiratory chain. However, an effect(s) of DMQ is also possible and could be significant. Cultured embryonic cells (ES) from Mclk1/COQ7 knockout mice, which produce DMQ9 in place of UQ9, are fully viable and possess significant respiratory activity, suggesting a functional role of DMQ9 in the respiratory chain (Levavasseur et al., 2001). Studies on yeast coq-7 mutants, however, concluded that DMQ6 does not support respiration (Padilla et al., 2004). Jonassen and colleagues reported that the levels of another quinone species, rhodoquinone (RQ9), were elevated in clk-1 mutants (Jonassen et al., 2002). Rhodoquinone (RQ) is structurally very similar to UQ, with the only distinction of the substituent attached to the C5-position of the quinone ring (an amino group in RQ and a methoxy group in UQ) (Brajcich et al., 2009). RQ is important for anaerobic respiration. Some parasitic species, when inside of a host where oxygen availability is limited, rely on RQ to function as an electron carrier between complex I and fumarate reductase, thus enabling cells to continue to generate ATP via oxidative phosphorylation (Tielens, 1994). The possibility that clk-1 mutants rely on RQ for respiration has been discussed (Jonassen et al., 2002). It should be noted that RQ is not found in mammals, thus the mitochondrial respiration in Mclk1−/− ES cells (Levavasseur et al., 2001) can only be explained by the ability of DMQ9 to substitute for the role of UQ9 in the electron transport chain. By having the same function as UQ, DMQ may possibly compete with UQ in the mitochondrial respiratory chain if both quinone species are present in the same mitochondria. Consistent with this thinking, a recent study (Yang et al., 2011) has found that mitochondria depleted of quinones and then replenished with exogenous UQ9 and DMQ9 displayed a lower activity of complex I–III as compared to those replenished with UQ9 alone. Complex II–III activity, however, was not significantly affected by the addition of DMQ9 which could be interpreted as indicating DMQ9 has no or poorer activity at complex II. These results agree with previous findings that clk-1 mitochondria display a decrease in complex I-dependent respiration but normal complex II respiration, leading the authors to hypothesize a causative role of inhibition by DMQ in the complex I-dependent defect (Kayser et al., 2004, Yang et al., 2009). It is worth noting that very crude pentane extracts from clk-1 mitochondria were used as DMQ9 source. Further research is needed to confirm DMQ9, rather than other clk-1 specific compounds in these extracts, accounts for the observed inhibitory effect on complex I–III. The tRNA suppressors of clk-1(e2519) described above were found to restore endogenous UQ9 biosynthesis only to a small extent. Their predominant quinone species is still DMQ9 and produce very small amounts of UQ9, much less than the amount of UQ8 obtained exogenously (Branicky et al., 2006). It would also be of interest to determine whether those suppressor mutants restore a wild type respiration. A full suppression would suggest that endogenously-synthesized UQ molecules are much more effective at driving electrons than bacterial UQ8 and can more readily outcompete DMQ. If the low respiration phenotype were not suppressed by the small amount of UQ9 it would suggest that the Clk-1 phenotypes are unrelated to the rate of respiration but to some other aspect of UQ function (see below). Investigation of other functions of DMQ9 showed that DMQ9 is not an effective antioxidant for H2O2 (Padilla et al., 2004). Outside of mitochondria, DMQ9 was detected in nematode plasma membrane where it appears inactive in redox reactions. (Arroyo et al., 2006).
Interestingly, most Clk-1 phenotypes, including defecation rate, pharyngeal pump and lifespan, are fully rescued in the tRNA suppressors of clk-1(e2519), despite a very low level of restoration of CLK-1 production and UQ biosynthesis. (Branicky et al., 2006). Therefore, a deficit of endogenous UQ rather than the presence of DMQ is important for induction of the mutant phenotypes. It remains to be determined how the Clk-1 phenotypes are related to various functions of UQ. Alternatively, the high degree of rescue could be due to restoration of other function(s) of CLK-1. Several lines of evidence have raised the possibility that in addition to the biosynthesis of UQ, CLK-1 might have other functions in C. elegans (Hihi et al., 2003, Wang et al., 2009). In particular, the missense e2519 mutant exhibits a less severe phenotype than the qm30 and qm51 null mutant, despite the fact that both mutants have complete absence of endogenous UQ9 and accumulate the same amounts of DMQ9 (Hihi et al., 2003). Analysis of various suppressors has showed many aspects of the Clk-1 phenotypes can be uncoupled from each other (Branicky et al., 2006, Branicky et al., 2001), indicating downstream mechanism underlying different phenotypes and involving different cells and tissues.
Mouse UQ biosynthesis mutants
As noted above, three knockout mutants in the UQ biosynthesis pathway have been described so far. For all three genes, Mclk1/Coq7 (Levavasseur et al., 2001), Coq3 (Jérôme Lapointe, 2012) and Pdss2 (Peng et al., 2008), homozygous knockout mutants cannot be born alive, suggesting an essential function of UQ in mouse embryonic development. Our lab has characterized Mclk1+/− and Coq3+/− mice and revealed striking phenotypic differences between them. Both mutants look superficially wild-type and show normal tissue levels of ubiquinone (Lapointe and Hekimi, 2008, Jérôme Lapointe, 2012). However, Mclk1+/− mice have been found to harbor a variety of phenotypes that do not appear in Coq3+/− mutants (Jérôme Lapointe, 2012), such as respiratory chain deficiency, decreased ATP production, higher mitochondrial oxidative stress, increased expression of HIF-1 alpha, longer life span, etc. (Lapointe and Hekimi, 2008, Wang et al., 2010). Aiming to explore a possible causal link between MCLK1 function in UQ biosynthesis and mutant phenotypes, we have evaluated the UQ levels in mitochondrial membrane fractions and discovered a decrease in the IMM (inner mitochondrial membrane) coupled with an elevation in the OMM (outer mitochondrial membrane) (Jérôme Lapointe, 2012). This local deficiency of UQ in the IMM, which is hidden from detection when UQ is quantified in crude mitochondrial fractions, is the most likely underlying cause of the phenotypic abnormalities seen in Mclk1+/− mutant mice. The same measurements were carried out with Coq3+/− mice, and all were found to be normal (Jérôme Lapointe, 2012), indicating that the MCLK1 step is more rate-limiting in UQ biosynthesis. The findings with Mclk1+/− mice also suggest a rate limiting role of UQ in mitochondrial function since a mild reduction in UQ level in the IMM appears sufficient to induce mitochondrial dysfunction which in turn leads to several phenotypic consequences. The increase in UQ levels in the OMM might be linked to the antioxidant functions of UQ. It is likely that low level of UQ in the IMM of Mclk1+/− mutants increases oxidative stress by partially inhibiting electron transport. It is well known that under oxidative stress, cells will take protective measures to ensure their survival, such as increasing the levels of an antioxidant such as UQ. Thus elevation of UQ levels in the OMM of Mclk1+/− mutants might be a protective response to reduce oxidative damage to the OMM and/or reduce ROS leakage from the mitochondria to the cytoplasm. The mechanism by which the mitochondria increase UQ levels in the OMM is unknown but could be based on slower turnover or slower export of UQ (Jérôme Lapointe, 2012).
In recent years, several conditional knockout mice of the Pdss2 gene have been generated by Cre/Lox technology. Liver specific knockout of Pdss2 generated by crossing the floxed Pdss2 (Pdss2loxP) mice with mice carry a Cre transgene driven by the liver-specific albumin promoter have been reported to look overly normal despite demonstration that their livers appear to contain no detectable UQ9. Surprisingly though, the mitochondrial respiratory capacity of Pdss2 mutant liver is only moderately impaired, suggesting that there is a high level of tolerance of liver respiratory capacity to UQ deficiency (Peng et al., 2008). Mitochondrial oxidative stress in the Pdss2 knockout livers was assayed by measuring the activity of the superoxide-sensitive enzyme aconitase and no elevation was observed as well (Falk et al., 2011). Peng and colleagues have shown that glomerular podocyte-specific deletion of Pdss2 in mice recapitulates a fatal kidney disease similar to that observed in human patients with a defective PDSS2 gene (Peng et al., 2008). Lu and colleagues have described two Pdss2 conditional knockout mouse lines in which the Pdss2 gene has been ablated specifically in the cerebellum. Pdss2loxP/-, Pax2-Cre mice, which develop a targeted deletion of Pdss2 in the midbrain-hindbrain region at embryonic day E9.5, developed severe cerebellar hypoplasia and died within the first 36h of life. However, upon targeted knockout of Pdss2 in cerebellar Purkinje cells after birth, mice gradually lose Purkinje cells and only develop cerebellar ataxia at old age (Lu et al., 2011). A more recent study has described conditional Pdss2 knockouts targeted to dopaminergic neurons (Ziegler et al., 2011). Those mice were shown to manifest some major features of Parkinson’s disease, such as severely reduced motor coordination and depletion of tyrosine hydroxylase-positive (TH+) neurons in the substantia nigra (Ziegler et al., 2011). All these models are valuable to delineate the tissue-specific pathogenesis of UQ deficiency.
Primary UQ deficiency in humans
Profound UQ10 deficiency due to a genetic defect in ubiquinone biosynthesis has been identified in a small number of human patients. This particular type of clinical disorder has been classified as primary UQ deficiency (OMIM 607426), as opposed to secondary deficiency that occur in the course of other diseases (e.g. Parkinsonism’s and Huntington’s diseases, and cancer) (Mancuso et al., 2009, Artuch et al., 2009) or is caused by mutations in genes unrelated to UQ biosynthesis (Artuch et al., 2009), for example, in the electron-transferring-flavoprotein dehydrogenase gene (ETFDH) causing myopathy (Gempel et al., 2007), or the B-Type Raf Kinase (BRAF) gene causing cardiofaciocutaneous syndrome (Aeby et al., 2007). Since a disease-causing mutation in the UQ biosynthesis pathway was first described by Quinzii in 2006 (Quinzii et al., 2006), more than 20 distinct pathogenic mutations in 7 different COQ genes have been identified worldwide. Table 2 lists all the mutations reported so far and the clinical features of the human patients carrying these lesions. These inborn errors of UQ biosynthesis are associated with a wide spectrum of clinical abnormalities, including nephrotic syndrome, encephalomyopathy, ataxia, and Leigh syndrome which is characterized by lactic acidosis, seizures, loss of motor skills, and progressive degeneration of the brain (for review, see (Quinzii and Hirano, 2010, Artuch et al., 2009, Trevisson et al., 2011, Hirano et al., 2012). Most primary UQ deficiency patients present with multisystem diseases, underscoring an essential role of UQ in normal cellular functions.
Table 2.
Primary UQ10 deficiency in humans.
| Gene | Mutations | Level of UQ10 (% of mean control value) | Biochemical dysfunction | Number of patients (families) Age of onset | Family History | Symptoms | Response to UQ10 therapy | References |
|---|---|---|---|---|---|---|---|---|
| PDSS1 | hom, D308E | <5% (fibroblasts) | Decreased activities of CI+III and CII+III in either fibroblasts or muscle | 2(1) 1–2 yr |
Parents and 3 other children are healthy. | Encephaloneuropathy, deafness, cardiac valvulopathy, obesity, livedo reticularis | Yes, Active at 22 yr | (Mollet et al., 2007) |
| PDSS2 | het, Q332X+S382L | 14% (muscle) 13% (fibroblasts) |
Decreased CII+III activity (muscle +fibroblasts), deficit in ATP synthesis (fibroblasts) | 1(1) <3 mo |
Parents are healthy heterozygous carriers with normal levels of UQ10. Patient has 1 sibling who is healthy. | Nephrotic syndrome, Leigh syndrome, ataxia, seizure | No died at age 8 mo (refractory status epilepticus) | (Quinzii et al., 2008, Lopez et al., 2006, Salviati et al., 2012) |
| COQ2 | het, R197H+N228S | Severe (kidney + muscle) 36% (fibroblasts) |
Decreased CII–III activity (kidney cortex + skeletal muscle), elevated citrate synthase activity (skeletal muscle), impaired cell growth, reduced ATP level, increased ROS (fibroblasts) | 1(1) 18 mo |
1 sibling who carries a N228S mutation and appears healthy | Steroid-resistant nephritic syndrome | yes | (Diomedi-Camassei et al., 2007, Quinzii et al., 2010) |
| hom,S146N | Severe (kidney + muscle) | Decreased CII–III activity (kidney cortex + skeletal muscle), elevated citrate synthase activity (skeletal muscle) | 1(1) Birth |
1 healthy sibling, 1 dead sibling (at 2 d of life) | Acute renal failure, encephalomyopathy | No, died at age 6 mo | (Diomedi-Camassei et al., 2007) | |
| hom, Y297C | 37% (muscle) 18–30% (fibroblasts) |
Growth defect, decreased CII–III activity, deficit in ATP synthesis, impaired pyrimidine synthesis, increased ROS production (fibroblasts), decreased activities of CI+III and CII+III (muscle) | 2(1) 11–12 mo |
Parents are healthy heterozygous carriers | Male: naystagmus, encephalomyopathy, steroid-resistant NS (nephrotic syndrome), seizures, hepatopathy, cerebellar atrophy, tremor Famale: isolated NS at 12 mo of life | Yes | (Quinzii et al., 2006, Quinzii et al., 2008, Diomedi-Camassei et al., 2007) | |
| hom,N401fsX415 | 24% (fibroblasts) | Reduced complex I+III and II+III activities (liver) | 2(1) Birth |
Both parents are hererozyous for the single nuclear deletion | Infantile multiorgan failure | No died at age 1–12d | (Mollet et al., 2007, Quinzii and Hirano, 2010) | |
| COQ4 | Monoallelic deletion | 44% (fibroblasts) | Decreased CII+III activity, reduced growth rate (fibroblasts) | 1(1) 3 yr |
No sibling, parents are healthy | Dysmorphic features, metal retardation, hypotonia, encephalomyopathy | Yes | (Salviati et al., 2012) |
| COQ6 | hom, G255R | - | - | 6(2) 0.2–6.2 yr |
Parents are heterozygous carriers | SRNS (steroid-resistant nephrotic syndrome) | Yes | (Heeringa et al., 2011) |
| hom, A353D | - | - | 3(2) 2.5–6.0 yr |
Parents are heterozygous carriers | SRNS (steroid-resistant nephrotic syndrome) | Yes | (Heeringa et al., 2011) | |
| het, W447X+Q461fsX478 | - | - | 1(1) 3.0 yr |
Parents are heterozygous carriers | SRNS (steroid-resistant nephrotic syndrome) | - | (Heeringa et al., 2011) | |
| het, R162X +? * | - | - | - | Cyclosporine A-dependent nephritic syndrome | - | (Heeringa et al., 2011) | ||
| het, W188X+?* | - | - | - | Diffuse mesangial sclerosis | - | (Heeringa et al., 2011) | ||
| ADCK3 | het, R213W+G272V | 29% (muscle) | High CIV/CII+III and CII+III/CII activity ratios (muscle), elevated individual enzyme activies (CI, CII, CIII, CIV), a slight increase of CIV/CII+III (fibroblasts) | 2(1) 1 mo |
Parents are healthy. Two old siblings are heterozygous carriers for the R213W mutation and they are healthy. | Hypotonia, development delay, talus valgus, seizures, cerebellar ataxia, epilepsia partialis continua | No | (Mollet et al., 2008) |
| Hom, E551K | 8% (mucle) 100% (fibroblasts) |
Decreased oxygen consumption rate, reduced CII–III acivity (musle), a slight increase of CIV/CII+III (fibroblasts) | 1(1) 18 mo |
Parents are healthy heterozygous carriers. One old sister is healthy. | Cerebella ataxia, strabismus, muscle weakness, tonic seizure | No | (Mollet et al., 2008) | |
| het, G272D +1bp freameshit insertion c[1812_1813insG] | 5% (mucle) 100% (fibroblasts) |
Decreased CI–III and CII–III activities, elevated activities of individual complexes and citrtate synthase (muscle) | 1(1) 3 yr |
Parents are healthy. No siblings. | Exercise intolerance, cerebellar syndromes | Yes | (Mollet et al., 2008, Aure et al., 2004) | |
| hom, D420WfsX40, I467Afs X22 | 119% (fibroblasts) | Decreased CI–III activity, normal ATP levels, no ROS overproduction | 4(1) 8 – 11 yr |
One healthy sibling | Cerebellar ataxia, exercise intolerance | - | (Lagier-Tourenne et al., 2008, Quinzii et al., 2010) | |
| hom, Q167Lfsx36 | 64% (fibroblasts) | Decreased CI–III activity, normal ATP levels, no ROS overproduction | 1(1) 4 yr |
- | Cerebellar ataxia | - | (Lagier-Tourenne et al., 2008, Quinzii et al., 2010) | |
| het, Y514C+T584 del | 51% (fibroblasts) | Decreased CI–III activity, normal ATP levels, no ROS overproduction | 1(1) 5 yr |
- | Cerebellar ataxia | Yes | (Lagier-Tourenne et al., 2008, Quinzii et al., 2010) | |
| het, K314_Q360 del+ G549S | - | - | 1(1) 3 yr |
- | Cerebellar ataxia | - | (Lagier-Tourenne et al., 2008) | |
| COQ9 | hom, R244X | 15% (muscle) 18% (fibroblasts) |
Low CII–III activity (muscle) markedly decreased ATP level, no signs of ROS overproduction (fibroblasts) | 1(1) birth |
Parents and other 5 siblings are healthy. An old sister died on day one of life. | Renal tubulopathy, ventricular hypertrophy, seizure, cerebellar atrophy, development delay | No Died at 2 yr | (Rahman et al., 2001, Quinzii et al., 2010, Quinzii and Hirano, 2010, Duncan et al., 2009) |
hom: homozygous; het: heterozygous; mo: month; yr: year;
the second mutation has not been identified.
Age of onset, clinical manifestations, disease progression and response to UQ10 therapy vary greatly among these cases. This clinical heterogeneity is best illustrated by the patients that all harbor a mutation in the COQ2 gene yet are seemingly phenotypically divergent. The first patient was noted to have nystagmus at age 2 months and developed a severe steroid-resistant nephrotic syndrome, progressive encephalomyopathy, hypotonia, seizures and other symptoms at 12–18 months, whereas his young sister sharing a homozygous missense mutation in COQ2 developed nephrotic syndrome at 12 month of life without any clinical signs of neurological involvement (Diomedi-Camassei et al., 2007, Quinzii et al., 2006). Later, 4 additional patients who bore different COQ2 mutations were described and 2 of them presented with neonatal multi-organ disorders and died within 12 days after birth (Diomedi-Camassei et al., 2007, Mollet et al., 2007). The considerable heterogeneity in the clinical expression of UQ10 biosynthetic defects could be reflective of differences in the residual activities of the affected proteins and thus of variable degrees of UQ shortage. Moreover, there remains the possibility of other functions of COQ proteins in addition to the biosynthesis of UQ. Furthermore, it is reasonable to suspect that some UQ biosynthetic intermediates and defective COQ proteins may have some biological activities, which could contribute to the variation in clinical manifestations of different molecular defects. For example, Mollet and colleagues, studying one of the COQ2 patients, reported that RT-PCR on patient fibroblasts yielded an abnormal and shorter COQ2 transcript. Whether it produces a mutant protein with some residual or altered function remains to be discovered (Mollet et al., 2007).
The patients with mutations in UQ biosynthesis enzymes that are phenotypically affected are born to healthy parents and some of them have siblings with the same rare disease, suggesting an autosomal recessive mode of inheritance. Since heterozygous carriers are phenotypically normal, it is generally assumed that heterozygous deletion of a UQ biosynthetic gene does not affect de novo UQ production significantly. The UQ10 content in fibroblasts obtained from the parents of a patient carrying different mutations in the two copies of PDSS2 (compound heterozygotes) was indeed found to be normal (Salviati et al., 2012). But, interestingly, a recently reported 3-year-old patient with a 3.9MB deletion at chromosomal location 9q34.13, encompassing the COQ4 gene, was found to have a 50% reduction in COQ4 mRNA expression and a marked reduction in UQ synthesis (Salviati et al., 2012). In this patient halpoinsufficiency of COQ4 was associated with the deletion of a large number of other contiguous genes in the affected region, which makes it difficult to identify an actual causal relationship between the reduction in COQ4 and UQ synthesis and the patient’s phenotypes. However, the authors of the study have also shown that COQ4 siRNA-treated Hela cells as well as a wt/” coq4 diploid yeast strain also display a defect in cellular UQ content thus providing a compelling evidence for a significant effect of COQ4 halpoinsufficiency on the cellular amount of UQ10 (Salviati et al., 2012). It is noteworthy that the clinical symptoms of the patient with COQ4 deficiency are different from that reported in the patients with mutations in other COQ genes. After being put on oral UQ10 supplementation, the patient showed significant clinical improvements, indicating that the primary pathogenic mechanism is indeed related to UQ deficiency (Salviati et al., 2012). More definitive information on the pathogenesis of COQ4 heterozygosity awaits the characterization of animal mutants in which a single COQ4 allele is inactivated. As discussed earlier, the molecular function of Coq4p has been not been elucidated. Future studies with partial deletion mutants of COQ4 will be useful in dissecting COQ4 function in UQ biosynthesis.
Among the clinical manifestations of primary UQ deficiency, the most frequently encountered features are encephalomyopathy, nephropathy, and cerebellar ataxia (Hirano et al., 2012, Mancuso et al., 2009, Quinzii and Hirano, 2010, Rahman et al., 2011, Rotig et al., 2007). These features have been interpreted as indicating that skeletal muscle, cerebellum and kidney have a relatively higher susceptibility to damage under conditions of UQ deficiency, probably because of a relatively low safety margin of UQ content. A study of UQ distribution in rat brain showed that endogenous UQ9 level in the cerebellum is the lowest compared to seven other regions (Naini et al., 2003). If the same holds true for the human brain, it would mean that the cerebellum might be relatively more sensitive to reduced UQ biosynthesis, which would in turn explain why cerebellar ataxia appears to be common in human UQ10 deficiency, both primary and secondary (Rotig et al., 2007). Curiously, the two patients with Pdss1 mutations have no sign of kidney disease (Mollet et al., 2007). What allows them to escape renal disease is unknown.
Supplementation with oral UQ10 has been shown to be effective in the individuals with mutations in PDSS1, COQ2, COQ4, COQ6 or COQ8/ADCK3 (Mollet et al., 2007, Diomedi-Camassei et al., 2007, Heeringa et al., 2011, Mollet et al., 2008, Salviati et al., 2012). However, the patients born with defective PDSS2 or COQ9 genes did not response to UQ10 and died despite UQ10 treatment (Lopez et al., 2006, Duncan et al., 2009). One of the causes of treatment failure might have been a more advanced disease status at the time of the beginning of treatment, considering the effectiveness of a replacement treatment will depend on the percentage of salvageable cells remaining in the affected tissues. Uptake and utilization of exogenous UQ10 in various tissues have been studied in rodents. The relevance of these studies with regard to UQ10 replacement therapy for UQ deficiency diseases is uncertain, considering that the pharmacokinetics of UQ10 supplementation in the animals with normal UQ content may be very different from those in patients with UQ deficiencies.
Pathogenesis of UQ deficiency
UQ is essential for optimal functions of several cellular processes, and its biosynthesis is complex, involving a series of steps and many genes. This may at least partially explain the phenotypic diversity and variability associated with mutations in the UQ biosynthetic pathway. The biochemical alterations that have been observed in UQ deficient mutants include respiratory chain defects, altered ROS metabolism, increased apoptosis, impaired nucleotide metabolism, and enhanced mitochondrial autophagy (Lopez-Martin et al., 2007, Rodriguez-Hernandez et al., 2009, Di Giovanni et al., 2001, Asencio et al., 2009, Lapointe and Hekimi, 2008, Kayser et al., 2004, Hirano et al., 2012). The best characterized among these is the effect on mitochondrial respiration. The current understanding is that UQ diffuses freely within the inner mitochondrial membrane and behaves as a single homogeneous pool passing electrons from donors (dehydrogenases) to acceptors (complex III) (Lenaz and Genova, 2009). Lenaz and colleagues have proposed that the size of the UQ pool also dictates the amount of quinone molecules bound within supercomplexes of the respiratory enzymes (Lenaz and Genova, 2007). Studies of the saturation kinetics of UQ have suggested that physiological UQ content is not saturating for maximal activity of complex I but possibly is for succinate oxidation by complex II (Lenaz and Genova, 2007). Thus, suboptimal ubiquinone production should impair electron transport in the respiratory chain, compromising the mitochondrial bioenergetic capacity. Indeed, as noted in a section above, we have recently revealed that Mclk1+/− mitochondria exhibit a small reduction in inner membrane UQ (Jérôme Lapointe, 2012) and this defect induces a significant impairment of mitochondrial function (Lapointe and Hekimi, 2008). In vitro studies on fibroblasts from UQ10 deficiency patients have also been informative. These studies reported decreased activities of UQ-dependent enzymes (e.g. complexes I+III or complex II+III) in several patient cell lines with varying severities of UQ deficiency (Quinzii et al., 2008, Diomedi-Camassei et al., 2007, Mollet et al., 2007, Quinzii et al., 2006, Lopez et al., 2006, Quinzii et al., 2010) and suggested a positive correlation between the severity of the bioenergetic defects and the extent of UQ deficit (Quinzii et al., 2010, Quinzii et al., 2008). As noted above, surprisingly, mouse liver seems capable of maintaining relatively robust respiratory chain activities with a substantially low level of UQ9, which can be deduced from the finding that Pdss2 knockout livers, which contain undetectable amounts of UQ9, still sustain 60% to 80% of the wild type respiratory capacity (Peng et al., 2008). A 49% average reduction in complex I-dependent respiration was described for permeabilized skeletal muscle fibers prepared from B6. Pdss2kd/kd missense mutant mice. However muscle UQ levels were not measured in these mutants (Ziegler et al., 2011). This seemingly high tolerance of the liver to UQ deficiency could be related to a relatively moderate level of respiration in this organ and thus a low requirement for UQ in hepatocyte respiration.
Reduced respiratory chain activities inhibit ATP synthesis, which could in turn compromise cellular functions. Less ATP generation was noted in Mclk1+/− mice (Lapointe and Hekimi, 2008) and patients’ fibroblasts harboring mutations in COQ genes (Lapointe and Hekimi, 2008, Quinzii et al., 2008, Quinzii et al., 2010). The other aspect of the functional consequences of UQ deficiency that has long been considered is altered oxidative stress levels. UQ plays an essential role both in ROS generation and in antioxidant defense. In animals, UQ is the only endogenously produced lipophilic antioxidant preventing oxidative damage by directly sequestering free radicals or by regenerating other antioxidants (i.e. vitamin E and C) (Navas et al., 2007). UQ also acts as a pro-oxidant mainly through the semiquinone intermediate formed during electron transport activity (Nakamura and Hayashi, 1994). Singly reduced semiquinone is believed to be capable of donating its free electron to oxygen at complex III, leading to formation of superoxide anion which is the precursor of other damaging oxygen species (Nakamura and Hayashi, 1994, Rigoulet et al., 2010, Turrens, 2003). Yeast coq null mutants were shown to have heightened sensitivity to treatment with the polyunsaturated fatty acids (PUFAs) ±-linolenic or linoleic acid, mainly owning to a lack of UQ antioxidant activity required to protect against the toxic effects of the autoxidation products of PUFAs (Poon et al., 1997, Do et al., 1996). In animal cells which have a complex and robust antioxidant system, composed of a number of interrelated antioxidant compounds and enzymes, low UQ levels would be expected to induce compensatory anti-oxidant responses that can offset a negative effect of UQ deficiency on antioxidant protection. For example, increased SOD activity has been reported in UQ biosynthetic pathway mutants Mclk1+/− mice (Lapointe and Hekimi, 2008) and patient cell lines with UQ10 deficiency (Quinzii et al., 2010). Further research is needed to dissect the potential importance of reduced UQ antioxidant capacity in the pathophysiology of UQ deficiency. There is a hypothesis that lower levels of UQ in old age and resulting decreased OXPHOS and antioxidant capacity contribute to age-dependent decline of normal tissue functions, However, there is no direct evidence yet supporting this notion and justifying routine UQ10 supplementation for combating aging and age-related diseases (Gruber et al., 2008).
The precise effect of UQ deficiency on ROS production is not well understood. Recent studies quantitating ROS production in patient fibroblasts with varying severity of UQ10 deficit have presented a very interesting relationship between UQ10 levels and ROS production (Quinzii et al., 2008, Quinzii et al., 2010, Quinzii et al., 2012). They showed that COQ2 mutant fibroblasts with moderate deficiency (30–50% UQ10) produce maximal oxidative stress (Quinzii et al., 2008, Quinzii et al., 2010), whereas PDSS2 (Quinzii et al., 2008), COQ9 mutant fibroblasts (Quinzii et al., 2010) with more severe deficiency (<20% of normal) or COQ8/ADCK3 mutant cells with residual UQ10 levels (>60%) show no overproduction of ROS or oxidative damage (Quinzii et al., 2012, Quinzii et al., 2010). Furthermore, upon treatment with 4-nitrobenzoate (4-NB), a substrate competitor of COQ2, wild-type fibroblasts produce 40–50% residual CoQ10 which is associated with increased oxidative stress and reduced viability, while COQ8/ADCK3 mutant cells show significant increases in ROS when their UQ10 levels were reduced from more than 50% to less than 50% of the wild-type level (Quinzii et al., 2012). Elucidating the mechanism of this apparently bell-shaped relationship between the degree of UQ deficiency and the level of mitochondrial ROS may shed some light on the dynamics of mitochondrial ROS generation. Unstable semiquinone, formed at the ubiquinol oxidation centre (Qo) of the complex III, is long thought to be a source of electrons for oxygen reduction, though its relative importance in generating ROS is still controversial. A reduction in UQ-pool size is expected to lead to increased UQ redox cycling, producing more pro-oxidant semiquinone. However, in a severe UQ-deficient state, significant overproduction of semiquinone is unlikely to occur and therefore possibly there is no increased superoxide production from semiquinones. Complex I is thought to be another principle point of electron leakage in the respiratory chain (Lenaz, 2001). Inefficient electron transfer would maintain complex I at a reduced state and thus create a condition that facilitate superoxide production. Along the same line of thinking, this is likely to occur when UQ deficiency is severe enough to affect the electron transport out of complex I but is not too severe to reduce the functional activity of complex I dramatically. It is worth noting that in UQ-depleted mitochondria, Complex I is able to produce oxygen radicals at a rate comparable with the enzyme in reconstituted mitochondria containing large excess of UQ10, indicating UQ is not required for ROS production at complex I (Genova et al., 2001). More recently, new developments in supercomplexes have proposed new insights into the mechanisms of ROS generation and the pathophysiology of oxidative stress. Supercomplexes are massive protein structures formed by various respiratory enzymes. As opposed to random collisions of the individual complexes, the advantage of such organization is a more efficient electron transfer and thereby restricted ROS generation during electron transfer reactions (Genova et al., 2005, Winge, 2012, Seelert et al., 2009). It is possible that under condition of severe UQ depletion, in order to sustain sufficient energy production, the mitochondrial respiratory chain may augment the electron transport via the respiratory supercomplexes, producing less ROS. Alternatively, UQ deficiency could be expected to reduce the antioxidant defense capacity in the mitochondrial membranes which could, in turn, have damaging effects on supercomplexes. As proposed by Lenaz and Genova, under condition of oxidative stress a loss of supercomplex organization occurs, which could lead to loss of efficient electron channeling and destabilization of complex I, both of which would decrease respiratory function and induce ROS generation (Genova and Lenaz, 2011). Furthermore, as noted above, UQ deficiency may induce compensatory antioxidant defense mechanism such as upregulation of superoxide dismutase (SOD). The extent of such compensation may vary depending on the severity of UQ deficiency and affects a cell’s ability to cope with increased oxidative stress.
Whether a similar bell-shaped relationship can be found in other models of UQ deficiency remains to be explored. To date, there are only few reports on ROS levels in UQ deficient animals. Previous research from our lab (Yang and Hekimi, 2010) has reported elevated production of mitochondrial ROS in clk-1 mutants and it appeared to be necessary and sufficient to increase longevity. Also, as noted in a section above, higher mitochondrial ROS was found in Mclk1+/− mice but no in Coq3+/− mice. ROS damages various cellular components and uncontrolled elevations of oxidative stress are capable of having global pathogenic effects. Besides, ROS also functions as a signaling molecule in a variety of biological processes (D’Autreaux and Toledano, 2007, Veal et al., 2007, Rhee, 2006). The question of how and in what cellular context resultant disturbance of ROS metabolism accounts for the phenotypic consequences of UQ deficiency is intriguing as well as challenging.
Besides the effects on mitochondrial function, a report from Lopez-Martin and colleagues showed that UQ10 deficiency due to COQ2 mutations also adversely affects the biosynthesis of pyrimidine presumably because of an inhibitory effect on the fourth step of the pyrimidine biosynthetic pathway (oxidation of dihydroorotate) which requires UQ as a cofactor (Lopez-Martin et al., 2007). Moreover, in the same mutant COQ2 cells plus other UQ10-less fibroblasts isolated from patients with undefined molecular defects, evidence of autophagy has been found (Rodriguez-Hernandez et al., 2009). Gene expression analysis revealed that autophagic genes are also upregulated in liver-conditional Pdss2 knockout mice (Peng et al., 2008). An important future aspect of these studies would be to demonstrate whether these alterations are common features of UQ deficiency and their possible pathogenic roles in UQ10 deficient status.
There is likely to be a great variation in tissue sensitivity to a UQ biosynthetic defect. Not only the threshold levels for UQ deficiency in individual cells required for the expression of various cellular dysfunctions are likely to vary, but also different cells may have variable sensitivity to resultant biochemical dysfunctions such as ATP deficit and elevated production of ROS. For example, it has been suggested that tissues with high energy demands (e.g. muscle tissues) might be more sensitive to OXPHOS failure. And, a high sensitivity of neuronal cells to free-radical damage has been described (Borek, 2004). The prevalence of muscle and CNS involvement in human UQ deficiency disease might in part reflect a relatively higher susceptibility of these two tissues to mitochondrial dysfunction. Tissue specific effects of UQ deficiency are best demonstrated in a study on Pdss2 conditional knockout mice showing that podocyte-specific deletion of Pdss2 recapitulates a fatal kidney disease but has no effect when targeted to renal tubular epithelium or monocytes (Rahman et al., 2011). The key factors underpinning the particularly high sensitivity of podocytes to a Pdss2 mutation are not yet clear. Last but not least, there is still an ongoing research effort to understand the unique phenotypic features of particular mutation in UQ biosynthetic pathway genes, which could yield new information on the functions of COQ proteins as well as UQ biosynthesis and metabolism. Studies on various model organisms as well as on human patients will pave the way for better understating the requirements for UQ and for developing more rational and effective UQ10 therapies.
Footnotes
Declaration of interest
We declare no conflict of interest. The work was support by grant #97869 to SH from the Canadian Institutes of Health Research and by McGill University. SH is Strathcona Professor of Zoology and Campbell Professor of Developmental Biology.
References
- AEBY A, SZNAJER Y, CAVE H, REBUFFAT E, VAN COSTER R, RIGAL O, VAN BOGAERT P. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J Inherit Metab Dis. 2007;30:827. doi: 10.1007/s10545-007-0612-0. [DOI] [PubMed] [Google Scholar]
- ARROYO A, SANTOS-OCANA C, RUIZ-FERRER M, PADILLA S, GAVILAN A, RODRIGUEZ-AGUILERA JC, NAVAS P. Coenzyme Q is irreplaceable by demethoxy-coenzyme Q in plasma membrane of Caenorhabditis elegans. FEBS Lett. 2006;580:1740–6. doi: 10.1016/j.febslet.2006.02.025. [DOI] [PubMed] [Google Scholar]
- ARTUCH R, SALVIATI L, JACKSON S, HIRANO M, NAVAS P. Coenzyme Q10 deficiencies in neuromuscular diseases. Adv Exp Med Biol. 2009;652:117–28. doi: 10.1007/978-90-481-2813-6_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASENCIO C, NAVAS P, CABELLO J, SCHNABEL R, CYPSER JR, JOHNSON TE, RODRIGUEZ-AGUILERA JC. Coenzyme Q supports distinct developmental processes in Caenorhabditis elegans. Mechanisms of ageing and development. 2009;130:145–53. doi: 10.1016/j.mad.2008.10.004. [DOI] [PubMed] [Google Scholar]
- ASENCIO C, RODRIGUEZ-AGUILERA JC, RUIZ-FERRER M, VELA J, NAVAS P. Silencing of ubiquinone biosynthesis genes extends life span in Caenorhabditis elegans. FASEB J. 2003;17:1135–7. doi: 10.1096/fj.02-1022fje. [DOI] [PubMed] [Google Scholar]
- ASHBY MN, KUTSUNAI SY, ACKERMAN S, TZAGOLOFF A, EDWARDS PA. COQ2 is a candidate for the structural gene encoding para-hydroxybenzoate:polyprenyltransferase. The Journal of biological chemistry. 1992;267:4128–36. [PubMed] [Google Scholar]
- AURE K, BENOIST JF, OGIER DE BAULNY H, ROMERO NB, RIGAL O, LOMBES A. Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology. 2004;63:727–9. doi: 10.1212/01.wnl.0000134607.76780.b2. [DOI] [PubMed] [Google Scholar]
- BARROS MH, JOHNSON A, GIN P, MARBOIS BN, CLARKE CF, TZAGOLOFF A. The Saccharomyces cerevisiae COQ10 gene encodes a START domain protein required for function of coenzyme Q in respiration. J Biol Chem. 2005;280:42627–35. doi: 10.1074/jbc.M510768200. [DOI] [PubMed] [Google Scholar]
- BEHAN RK, LIPPARD SJ. The aging-associated enzyme CLK-1 is a member of thecarboxylate-bridged diiron family of proteins. Biochemistry. 2010;49:9679–81. doi: 10.1021/bi101475z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENTINGER M, BRISMAR K, DALLNER G. The antioxidant role of coenzyme Q. Mitochondrion. 2007;7(Suppl):S41–50. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- BOREK C. Antioxidants and radiation therapy. J Nutr. 2004;134:3207S–3209S. doi: 10.1093/jn/134.11.3207S. [DOI] [PubMed] [Google Scholar]
- BRAJCICH BC, IAROCCI AL, JOHNSTONE LA, MORGAN RK, LONJERS ZT, HOTCHKO MJ, MUHS JD, KIEFFER A, REYNOLDS BJ, MANDEL SM, MARBOIS BN, CLARKE CF, SHEPHERD JN. Evidence that ubiquinone is a required intermediate for rhodoquinone biosynthesis in Rhodospirillum rubrum. J Bacteriol. 2009;192:436–45. doi: 10.1128/JB.01040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRANICKY R, BENARD C, HEKIMI S. clk-1, mitochondria, and physiological rates. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22:48–56. doi: 10.1002/(SICI)1521-1878(200001)22:1<48::AID-BIES9>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- BRANICKY R, NGUYEN PA, HEKIMI S. Uncoupling the pleiotropic phenotypes of clk-1 with tRNA missense suppressors in Caenorhabditis elegans. Mol Cell Biol. 2006;26:3976–85. doi: 10.1128/MCB.26.10.3976-3985.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRANICKY R, SHIBATA Y, FENG J, HEKIMI S. Phenotypic and suppressor analysis of defecation in clk-1 mutants reveals that reaction to changes in temperature is an active process in Caenorhabditis elegans. Genetics. 2001;159:997–1006. doi: 10.1093/genetics/159.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURGESS J, HIHI AK, BENARD CY, BRANICKY R, HEKIMI S. Molecular mechanism of maternal rescue in the clk-1 mutants of Caenorhabditis elegans. J Biol Chem. 2003;278:49555–62. doi: 10.1074/jbc.M308507200. [DOI] [PubMed] [Google Scholar]
- CASARIN A, JIMENEZ-ORTEGA JC, TREVISSON E, PERTEGATO V, DOIMO M, FERRERO-GOMEZ ML, ABBADI S, ARTUCH R, QUINZII C, HIRANO M, BASSO G, OCANA CS, NAVAS P, SALVIATI L. Functional characterization of human COQ4, a gene required for Coenzyme Q10 biosynthesis. Biochem Biophys Res Commun. 2008;372:35–9. doi: 10.1016/j.bbrc.2008.04.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRANE FL. Biochemical functions of coenzyme Q10. J Am Coll Nutr. 2001;20:591–8. doi: 10.1080/07315724.2001.10719063. [DOI] [PubMed] [Google Scholar]
- CRANE FL. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion. 2007;7(Suppl):S2–7. doi: 10.1016/j.mito.2007.02.011. [DOI] [PubMed] [Google Scholar]
- CUI TZ, KAWAMUKAI M. Coq10, a mitochondrial coenzyme Q binding protein, is required for proper respiration in Schizosaccharomyces pombe. FEBS J. 2009;276:748–59. doi: 10.1111/j.1742-4658.2008.06821.x. [DOI] [PubMed] [Google Scholar]
- D’AUTREAUX B, TOLEDANO MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–24. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- DI GIOVANNI S, MIRABELLA M, SPINAZZOLA A, CROCIANI P, SILVESTRI G, BROCCOLINI A, TONALI P, DI MAURO S, SERVIDEI S. Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology. 2001;57:515–8. doi: 10.1212/wnl.57.3.515. [DOI] [PubMed] [Google Scholar]
- DIBROV E, ROBINSON KM, LEMIRE BD. The COQ5 gene encodes a yeast mitochondrial protein necessary for ubiquinone biosynthesis and the assembly of the respiratory chain. J Biol Chem. 1997;272:9175–81. doi: 10.1074/jbc.272.14.9175. [DOI] [PubMed] [Google Scholar]
- DIOMEDI-CAMASSEI F, DI GIANDOMENICO S, SANTORELLI FM, CARIDI G, PIEMONTE F, MONTINI G, GHIGGERI GM, MURER L, BARISONI L, PASTORE A, MUDA AO, VALENTE ML, BERTINI E, EMMA F. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18:2773–80. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- DO TQ, SCHULTZ JR, CLARKE CF. Enhanced sensitivity of ubiquinone-deficient mutants of Saccharomyces cerevisiae to products of autoxidized polyunsaturated fatty acids. Proc Natl Acad Sci U S A. 1996;93:7534–9. doi: 10.1073/pnas.93.15.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNCAN AJ, BITNER-GLINDZICZ M, MEUNIER B, COSTELLO H, HARGREAVES IP, LOPEZ LC, HIRANO M, QUINZII CM, SADOWSKI MI, HARDY J, SINGLETON A, CLAYTON PT, RAHMAN S. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet. 2009;84:558–66. doi: 10.1016/j.ajhg.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EARLS LR, HACKER ML, WATSON JD, MILLER DM., 3RD Coenzyme Q protects Caenorhabditis elegans GABA neurons from calcium-dependent degeneration. Proc Natl Acad Sci U S A. 2010;107:14460–5. doi: 10.1073/pnas.0910630107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERNSTER L, DALLNER G. Biochemical, physiological and medical aspects of ubiquinone function. Biochimica et biophysica acta. 1995;1271:195–204. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- ERNSTER L, FORSMARK-ANDREE P. Ubiquinol: an endogenous antioxidant in aerobic organisms. Clin Investig. 1993;71:S60–5. doi: 10.1007/BF00226842. [DOI] [PubMed] [Google Scholar]
- EVANS DR, GUY HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem. 2004;279:33035–8. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- EWBANK JJ, BARNES TM, LAKOWSKI B, LUSSIER M, BUSSEY H, HEKIMI S. Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science. 1997;275:980–3. doi: 10.1126/science.275.5302.980. [DOI] [PubMed] [Google Scholar]
- FALK MJ, POLYAK E, ZHANG Z, PENG M, KING R, MALTZMAN JS, OKWUEGO E, HORYN O, NAKAMARU-OGISO E, OSTROVSKY J, XIE LX, CHEN JY, MARBOIS B, NISSIM I, CLARKE CF, GASSER DL. Probucol ameliorates renal and metabolic sequelae of primary CoQ deficiency in Pdss2 mutant mice. EMBO Mol Med. 2011;3:410–27. doi: 10.1002/emmm.201100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELKAI S, EWBANK JJ, LEMIEUX J, LABBE JC, BROWN GG, HEKIMI S. CLK-1 controls respiration, behavior and aging in the nematode Caenorhabditis elegans. The EMBO journal. 1999;18:1783–92. doi: 10.1093/emboj/18.7.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FONTAINE E, ICHAS F, BERNARDI P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J Biol Chem. 1998;273:25734–40. doi: 10.1074/jbc.273.40.25734. [DOI] [PubMed] [Google Scholar]
- FORSGREN M, ATTERSAND A, LAKE S, GRUNLER J, SWIEZEWSKA E, DALLNER G, CLIMENT I. Isolation and functional expression of human COQ2, a gene encoding apolyprenyl transferase involved in the synthesis of CoQ. Biochem J. 2004;382:519–26. doi: 10.1042/BJ20040261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAVILAN A, ASENCIO C, CABELLO J, RODRIGUEZ-AGUILERA JC, SCHNABEL R, NAVAS P. C. elegans knockouts in ubiquinone biosynthesis genes result in different phenotypes during larval development. Biofactors. 2005;25:21–9. doi: 10.1002/biof.5520250104. [DOI] [PubMed] [Google Scholar]
- GEMPEL K, TOPALOGLU H, TALIM B, SCHNEIDERAT P, SCHOSER BG, HANS VH, PALMAFY B, KALE G, TOKATLI A, QUINZII C, HIRANO M, NAINI A, DIMAURO S, PROKISCH H, LOCHMULLER H, HORVATH R. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130:2037–44. doi: 10.1093/brain/awm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GENOVA ML, BIANCHI C, LENAZ G. Supercomplex organization of the mitochondrial respiratory chain and the role of the Coenzyme Q pool: pathophysiological implications. Biofactors. 2005;25:5–20. doi: 10.1002/biof.5520250103. [DOI] [PubMed] [Google Scholar]
- GENOVA ML, LENAZ G. New developments on the functions of coenzyme Q in mitochondria. Biofactors. 2011;37:330–54. doi: 10.1002/biof.168. [DOI] [PubMed] [Google Scholar]
- GENOVA ML, VENTURA B, GIULIANO G, BOVINA C, FORMIGGINI G, PARENTI CASTELLI G, LENAZ G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 2001;505:364–8. doi: 10.1016/s0014-5793(01)02850-2. [DOI] [PubMed] [Google Scholar]
- GIBSON F, YOUNG IG. Isolation and characterization of intermediates in ubiquinone biosynthesis. Methods Enzymol. 1978;53:600–9. doi: 10.1016/s0076-6879(78)53061-9. [DOI] [PubMed] [Google Scholar]
- GRUBER J, SCHAFFER S, HALLIWELL B. The mitochondrial free radical theory of ageing--where do we stand? Front Biosci. 2008;13:6554–79. doi: 10.2741/3174. [DOI] [PubMed] [Google Scholar]
- HEERINGA SF, CHERNIN G, CHAKI M, ZHOU W, SLOAN AJ, JI Z, XIE LX, SALVIATI L, HURD TW, VEGA-WARNER V, KILLEN PD, RAPHAEL Y, ASHRAF S, OVUNC B, SCHOEB DS, MCLAUGHLIN HM, AIRIK R, VLANGOS CN, GBADEGESIN R, HINKES B, SAISAWAT P, TREVISSON E, DOIMO M, CASARIN A, PERTEGATO V, GIORGI G, PROKISCH H, ROTIG A, NURNBERG G, BECKER C, WANG S, OZALTIN F, TOPALOGLU R, BAKKALOGLU A, BAKKALOGLU SA, MULLER D, BEISSERT A, MIR S, BERDELI A, VARPIZEN S, ZENKER M, MATEJAS V, SANTOS-OCANA C, NAVAS P, KUSAKABE T, KISPERT A, AKMAN S, SOLIMAN NA, KRICK S, MUNDEL P, REISER J, NURNBERG P, CLARKE CF, WIGGINS RC, FAUL C, HILDEBRANDT F. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 2011;121:2013–24. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIHI AK, GAO Y, HEKIMI S. Ubiquinone is necessary for Caenorhabditis elegans development at mitochondrial and non-mitochondrial sites. J Biol Chem. 2002;277:2202–6. doi: 10.1074/jbc.M109034200. [DOI] [PubMed] [Google Scholar]
- HIHI AK, KEBIR H, HEKIMI S. Sensitivity of Caenorhabditis elegans clk-1 mutants to ubiquinone side-chain length reveals multiple ubiquinone-dependent processes. J Biol Chem. 2003;278:41013–8. doi: 10.1074/jbc.M305034200. [DOI] [PubMed] [Google Scholar]
- HIRANO M, GARONE C, QUINZII CM. CoQ(10) deficiencies and MNGIE: Two treatable mitochondrial disorders. Biochim Biophys Acta. 2012;1820:625–31. doi: 10.1016/j.bbagen.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HSU AY, POON WW, SHEPHERD JA, MYLES DC, CLARKE CF. Complementation of coq3 mutant yeast by mitochondrial targeting of the Escherichia coli UbiG polypeptide: evidence that UbiG catalyzes both O-methylation steps in ubiquinone biosynthesis. Biochemistry. 1996;35:9797–806. doi: 10.1021/bi9602932. [DOI] [PubMed] [Google Scholar]
- JÉRÔME LAPOINTE YW, EVE BIGRAS, SIEGFRIED HEKIMI. Thesub-mitochondrial distribution of ubiquinone affects respiration in long-lived Mclk1+/−mice. The Journal of Cell Biology. 2012 doi: 10.1083/jcb.201203090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIANG N, LEVAVASSEUR F, MCCRIGHT B, SHOUBRIDGE EA, HEKIMI S. Mouse CLK-1 is imported into mitochondria by an unusual process that requires a leader sequence but no membrane potential. J Biol Chem. 2001;276:29218–25. doi: 10.1074/jbc.M103686200. [DOI] [PubMed] [Google Scholar]
- JOHNSON A, GIN P, MARBOIS BN, HSIEH EJ, WU M, BARROS MH, CLARKE CF, TZAGOLOFF A. COQ9, a new gene required for the biosynthesis of coenzyme Q in Saccharomyces cerevisiae. J Biol Chem. 2005;280:31397–404. doi: 10.1074/jbc.M503277200. [DOI] [PubMed] [Google Scholar]
- JONASSEN T, CLARKE CF. Isolation and functional expression of human COQ3, a gene encoding a methyltransferase required for ubiquinone biosynthesis. J Biol Chem. 2000;275:12381–7. doi: 10.1074/jbc.275.17.12381. [DOI] [PubMed] [Google Scholar]
- JONASSEN T, LARSEN PL, CLARKE CF. A dietary source of coenzyme Q is essential for growth of long-lived Caenorhabditis elegans clk-1 mutants. Proc Natl Acad Sci U S A. 2001;98:421–6. doi: 10.1073/pnas.021337498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONASSEN T, MARBOIS BN, FAULL KF, CLARKE CF, LARSEN PL. Development and fertility in Caenorhabditis elegans clk-1 mutants depend upon transport of dietary coenzyme Q8 to mitochondria. J Biol Chem. 2002;277:45020–7. doi: 10.1074/jbc.M204758200. [DOI] [PubMed] [Google Scholar]
- JONASSEN T, PROFT M, RANDEZ-GIL F, SCHULTZ JR, MARBOIS BN, ENTIAN KD, CLARKE CF. Yeast Clk-1 homologue (Coq7/Cat5) is a mitochondrial protein in coenzyme Q synthesis. J Biol Chem. 1998;273:3351–7. doi: 10.1074/jbc.273.6.3351. [DOI] [PubMed] [Google Scholar]
- KALEN A, APPELKVIST EL, CHOJNACKI T, DALLNER G. Nonaprenyl-4-hydroxybenzoate transferase, an enzyme involved in ubiquinone biosynthesis, in the endoplasmic reticulum-Golgi system of rat liver. The Journal of biological chemistry. 1990;265:1158–64. [PubMed] [Google Scholar]
- KALEN A, NORLING B, APPELKVIST EL, DALLNER G. Ubiquinone biosynthesis by the microsomal fraction from rat liver. Biochimica et biophysica acta. 1987;926:70–8. doi: 10.1016/0304-4165(87)90183-8. [DOI] [PubMed] [Google Scholar]
- KAYSER EB, SEDENSKY MM, MORGAN PG, HOPPEL CL. Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J Biol Chem. 2004;279:54479–86. doi: 10.1074/jbc.M403066200. [DOI] [PubMed] [Google Scholar]
- LAGIER-TOURENNE C, TAZIR M, LOPEZ LC, QUINZII CM, ASSOUM M, DROUOT N, BUSSO C, MAKRI S, ALI-PACHA L, BENHASSINE T, ANHEIM M, LYNCH DR, THIBAULT C, PLEWNIAK F, BIANCHETTI L, TRANCHANT C, POCH O, DIMAURO S, MANDEL JL, BARROS MH, HIRANO M, KOENIG M. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82:661–72. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAPOINTE J, HEKIMI S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. The Journal of biological chemistry. 2008;283:26217–27. doi: 10.1074/jbc.M803287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LENAZ G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52:159–64. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- LENAZ G, GENOVA ML. Kinetics of integrated electron transfer in the mitochondrial respiratory chain: random collisions vs. solid state electron channeling. Am J Physiol Cell Physiol. 2007;292:C1221–39. doi: 10.1152/ajpcell.00263.2006. [DOI] [PubMed] [Google Scholar]
- LENAZ G, GENOVA ML. Mobility and function of coenzyme Q(ubiquinone) in the mitochondrial respiratory chain. Biochim Biophys Acta. 2009;1787:563–73. doi: 10.1016/j.bbabio.2009.02.019. [DOI] [PubMed] [Google Scholar]
- LEONARD CJ, ARAVIND L, KOONIN EV. Novel families of putative protein kinases in bacteria and archaea: evolution of the “eukaryotic” protein kinase superfamily. Genome Res. 1998;8:1038–47. doi: 10.1101/gr.8.10.1038. [DOI] [PubMed] [Google Scholar]
- LEVAVASSEUR F, MIYADERA H, SIROIS J, TREMBLAY ML, KITA K, SHOUBRIDGE E, HEKIMI S. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J Biol Chem. 2001;276:46160–4. doi: 10.1074/jbc.M108980200. [DOI] [PubMed] [Google Scholar]
- LOPEZ-MARTIN JM, SALVIATI L, TREVISSON E, MONTINI G, DIMAURO S, QUINZII C, HIRANO M, RODRIGUEZ-HERNANDEZ A, CORDERO MD, SANCHEZ-ALCAZAR JA, SANTOS-OCANA C, NAVAS P. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum Mol Genet. 2007;16:1091–7. doi: 10.1093/hmg/ddm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOPEZ LC, SCHUELKE M, QUINZII CM, KANKI T, RODENBURG RJ, NAINI A, DIMAURO S, HIRANO M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125–9. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LU S, LU LY, LIU MF, YUAN QJ, SHAM MH, GUAN XY, HUANG JD. Cerebellar defects in Pdss2 conditional knockout mice during embryonic development and in adulthood. Neurobiol Dis. 2011;45:219–33. doi: 10.1016/j.nbd.2011.08.006. [DOI] [PubMed] [Google Scholar]
- MANCUSO M, ORSUCCI D, VOLPI L, CALSOLARO V, SICILIANO G. Coenzyme Q10 in neuromuscular and neurodegenerative disorders. Curr Drug Targets. 2009;11:111–21. doi: 10.2174/138945010790031018. [DOI] [PubMed] [Google Scholar]
- MARBOIS B, GIN P, FAULL KF, POON WW, LEE PT, STRAHAN J, SHEPHERD JN, CLARKE CF. Coq3 and Coq4 define a polypeptide complex in yeast mitochondria for the biosynthesis of coenzyme Q. J Biol Chem. 2005;280:20231–8. doi: 10.1074/jbc.M501315200. [DOI] [PubMed] [Google Scholar]
- MARBOIS B, GIN P, GULMEZIAN M, CLARKE CF. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim Biophys Acta. 2009;1791:69–75. doi: 10.1016/j.bbalip.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARBOIS B, XIE LX, CHOI S, HIRANO K, HYMAN K, CLARKE CF. para-Aminobenzoic acid is a precursor in coenzyme Q6 biosynthesis in Saccharomyces cerevisiae. The Journal of biological chemistry. 2010;285:27827–38. doi: 10.1074/jbc.M110.151894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARBOIS BN, CLARKE CF. The COQ7 gene encodes a protein in saccharomyces cerevisiae necessary for ubiquinone biosynthesis. J Biol Chem. 1996;271:2995–3004. doi: 10.1074/jbc.271.6.2995. [DOI] [PubMed] [Google Scholar]
- MARTIN-MONTALVO A, GONZALEZ-MARISCAL I, PADILLA S, BALLESTEROS M, BRAUTIGAN DL, NAVAS P, SANTOS-OCANA C. Respiratory-induced coenzyme Q biosynthesis is regulated by a phosphorylation cycle of Cat5p/Coq7p. Biochem J. 2011;440:107–14. doi: 10.1042/BJ20101422. [DOI] [PubMed] [Google Scholar]
- MEGANATHAN R. Ubiquinone biosynthesis in microorganisms. FEMS Microbiol Lett. 2001;203:131–9. doi: 10.1111/j.1574-6968.2001.tb10831.x. [DOI] [PubMed] [Google Scholar]
- MIYADERA H, AMINO H, HIRAISHI A, TAKA H, MURAYAMA K, MIYOSHI H, SAKAMOTO K, ISHII N, HEKIMI S, KITA K. Altered quinone biosynthesis in the long-lived clk-1 mutants of Caenorhabditis elegans. J Biol Chem. 2001;276:7713–6. doi: 10.1074/jbc.C000889200. [DOI] [PubMed] [Google Scholar]
- MOLLET J, DELAHODDE A, SERRE V, CHRETIEN D, SCHLEMMER D, LOMBES A, BODDAERT N, DESGUERRE I, DE LONLAY P, DE BAULNY HO, MUNNICH A, ROTIG A. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;82:623–30. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLLET J, GIURGEA I, SCHLEMMER D, DALLNER G, CHRETIEN D, DELAHODDE A, BACQ D, DE LONLAY P, MUNNICH A, ROTIG A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 2007;117:765–72. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORRE DJ, MORRE DM. Non-mitochondrial coenzyme Q. Biofactors. 2011;37:355–60. doi: 10.1002/biof.156. [DOI] [PubMed] [Google Scholar]
- NAINI A, LEWIS VJ, HIRANO M, DIMAURO S. Primary coenzyme Q10 deficiency and the brain. Biofactors. 2003;18:145–52. doi: 10.1002/biof.5520180217. [DOI] [PubMed] [Google Scholar]
- NAKAI D, YUASA S, TAKAHASHI M, SHIMIZU T, ASAUMI S, ISONO K, TAKAO T, SUZUKI Y, KUROYANAGI H, HIROKAWA K, KOSEKI H, SHIRSAWA T. Mouse homologue of coq7/clk-1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem Biophys Res Commun. 2001;289:463–71. doi: 10.1006/bbrc.2001.5977. [DOI] [PubMed] [Google Scholar]
- NAKAMURA M, HAYASHI T. One-and two-electron reduction of quinones by rat liver subcellular fractions. J Biochem. 1994;115:1141–7. doi: 10.1093/oxfordjournals.jbchem.a124470. [DOI] [PubMed] [Google Scholar]
- NAVAS P, VILLALBA JM, DE CABO R. The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion. 2007;7(Suppl):S34–40. doi: 10.1016/j.mito.2007.02.010. [DOI] [PubMed] [Google Scholar]
- NOHL H, GILLE L. Lysosomal ROS formation. Redox Rep. 2005;10:199–205. doi: 10.1179/135100005X70170. [DOI] [PubMed] [Google Scholar]
- OKADA K, KAINOU T, MATSUDA H, KAWAMUKAI M. Biological significance of the side chain length of ubiquinone in Saccharomyces cerevisiae. FEBS letters. 1998;431:241–4. doi: 10.1016/s0014-5793(98)00753-4. [DOI] [PubMed] [Google Scholar]
- OKADA K, SUZUKI K, KAMIYA Y, ZHU X, FUJISAKI S, NISHIMURA Y, NISHINO T, NAKAGAWA T, KAWAMUKAI M, MATSUDA H. Polyprenyl diphosphate synthase essentially defines the length of the side chain of ubiquinone. Biochim Biophys Acta. 1996;1302:217–23. doi: 10.1016/0005-2760(96)00064-1. [DOI] [PubMed] [Google Scholar]
- OZEIR M, MUHLENHOFF U, WEBERT H, LILL R, FONTECAVE M, PIERREL F. Coenzyme Q biosynthesis: Coq6 is required for the C5-hydroxylation reaction and substrate analogs rescue Coq6 deficiency. Chem Biol. 2011;18:1134–42. doi: 10.1016/j.chembiol.2011.07.008. [DOI] [PubMed] [Google Scholar]
- PADILLA S, JONASSEN T, JIMENEZ-HIDALGO MA, FERNANDEZ-AYALA DJ, LOPEZ-LLUCH G, MARBOIS B, NAVAS P, CLARKE CF, SANTOS-OCANA C. Demethoxy-Q, an intermediateof coenzyme Q biosynthesis, fails to support respiration in Saccharomyces cerevisiae and lacks antioxidant activity. J Biol Chem. 2004;279:25995–6004. doi: 10.1074/jbc.M400001200. [DOI] [PubMed] [Google Scholar]
- PADILLA S, TRAN UC, JIMENEZ-HIDALGO M, LOPEZ-MARTIN JM, MARTIN-MONTALVO A, CLARKE CF, NAVAS P, SANTOS-OCANA C. Hydroxylation of demethoxy-Q6 constitutes a control point in yeast coenzyme Q6 biosynthesis. Cell Mol Life Sci. 2009;66:173–86. doi: 10.1007/s00018-008-8547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENG M, FALK MJ, HAASE VH, KING R, POLYAK E, SELAK M, YUDKOFF M, HANCOCK WW, MEADE R, SAIKI R, LUNCEFORD AL, CLARKE CF, GASSER DL. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008;4:e1000061. doi: 10.1371/journal.pgen.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIERREL F, HAMELIN O, DOUKI T, KIEFFER-JAQUINOD S, MUHLENHOFF U, OZEIR M, LILL R, FONTECAVE M. Involvement of mitochondrial ferredoxin and para-aminobenzoic acid in yeast coenzyme Q biosynthesis. Chemistry & biology. 2010;17:449–59. doi: 10.1016/j.chembiol.2010.03.014. [DOI] [PubMed] [Google Scholar]
- POON WW, BARKOVICH RJ, HSU AY, FRANKEL A, LEE PT, SHEPHERD JN, MYLES DC, CLARKE CF. Yeast and rat Coq3 and Escherichia coli UbiG polypeptides catalyze both O-methyltransferase steps in coenzyme Q biosynthesis. J Biol Chem. 1999;274:21665–72. doi: 10.1074/jbc.274.31.21665. [DOI] [PubMed] [Google Scholar]
- POON WW, DAVIS DE, HA HT, JONASSEN T, RATHER PN, CLARKE CF. Identification of Escherichia coli ubiB, a gene required for the first monooxygenase step in ubiquinone biosynthesis. J Bacteriol. 2000;182:5139–46. doi: 10.1128/jb.182.18.5139-5146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POON WW, DO TQ, MARBOIS BN, CLARKE CF. Sensitivity to treatment with polyunsaturated fatty acids is a general characteristic of the ubiquinone-deficient yeast coq mutants. Mol Aspects Med. 1997;18(Suppl):S121–7. doi: 10.1016/s0098-2997(97)00004-6. [DOI] [PubMed] [Google Scholar]
- QUINZII C, NAINI A, SALVIATI L, TREVISSON E, NAVAS P, DIMAURO S, HIRANO M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006;78:345–9. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUINZII CM, HIRANO M. Coenzyme Q and mitochondrial disease. Dev Disabil Res Rev. 2010;16:183–8. doi: 10.1002/ddrr.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUINZII CM, LOPEZ LC, GILKERSON RW, DORADO B, COKU J, NAINI AB, LAGIER-TOURENNE C, SCHUELKE M, SALVIATI L, CARROZZO R, SANTORELLI F, RAHMAN S, TAZIR M, KOENIG M, DIMAURO S, HIRANO M. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 2010;24:3733–43. doi: 10.1096/fj.09-152728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUINZII CM, LOPEZ LC, VON-MOLTKE J, NAINI A, KRISHNA S, SCHUELKE M, SALVIATI L, NAVAS P, DIMAURO S, HIRANO M. Respiratory chain dysfunction and oxidative stress correlate with severity of primary CoQ10 deficiency. FASEB J. 2008;22:1874–85. doi: 10.1096/fj.07-100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUINZII CM, TADESSE S, NAINI A, HIRANO M. Effects of inhibiting CoQ10 biosynthesis with 4-nitrobenzoate in human fibroblasts. PLoS One. 2012;7:e30606. doi: 10.1371/journal.pone.0030606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAHMAN S, CLARKE CF, HIRANO M. 176th ENMC International Workshop: diagnosis and treatment of coenzyme Q(1)(0) deficiency. Neuromuscul Disord. 2011;22:76–86. doi: 10.1016/j.nmd.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAHMAN S, HARGREAVES I, CLAYTON P, HEALES S. Neonatal presentation of coenzyme Q10 deficiency. J Pediatr. 2001;139:456–8. doi: 10.1067/mpd.2001.117575. [DOI] [PubMed] [Google Scholar]
- REA SL, GRAHAM BH, NAKAMARU-OGISO E, KAR A, FALK MJ. Bacteria, yeast, worms, and flies: exploiting simple model organisms to investigate human mitochondrial diseases. Dev Disabil Res Rev. 2010;16:200–18. doi: 10.1002/ddrr.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RHEE SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–3. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- RIGOULET M, YOBOUE ED, DEVIN A. Mitochondrial ROS generation and its regulation: mechanisms involved in H(2)O(2) signaling. Antioxidants & redox signaling. 2010;14:459–68. doi: 10.1089/ars.2010.3363. [DOI] [PubMed] [Google Scholar]
- RODRIGUEZ-AGUILERA JC, ASENCIO C, RUIZ-FERRER M, VELA J, NAVAS P. Caenorhabditis elegans ubiquinone biosynthesis genes. Biofactors. 2003;18:237–44. doi: 10.1002/biof.5520180226. [DOI] [PubMed] [Google Scholar]
- RODRIGUEZ-HERNANDEZ A, CORDERO MD, SALVIATI L, ARTUCH R, PINEDA M, BRIONES P, GOMEZ IZQUIERDO L, COTAN D, NAVAS P, SANCHEZ-ALCAZAR JA. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009;5:19–32. doi: 10.4161/auto.5.1.7174. [DOI] [PubMed] [Google Scholar]
- ROTIG A, MOLLET J, RIO M, MUNNICH A. Infantile and pediatric quinone deficiency diseases. Mitochondrion. 2007;7(Suppl):S112–21. doi: 10.1016/j.mito.2007.02.008. [DOI] [PubMed] [Google Scholar]
- SAIKI R, NAGATA A, KAINOU T, MATSUDA H, KAWAMUKAI M. Characterization of solanesyl and decaprenyl diphosphate synthases in mice and humans. FEBS J. 2005;272:5606–22. doi: 10.1111/j.1742-4658.2005.04956.x. [DOI] [PubMed] [Google Scholar]
- SALVIATI L, TREVISSON E, RODRIGUEZ HERNANDEZ MA, CASARIN A, PERTEGATO V, DOIMO M, CASSINA M, AGOSTO C, DESBATS MA, SARTORI G, SACCONI S, MEMO L, ZUFFARDI O, ARTUCH R, QUINZII C, DIMAURO S, HIRANO M, SANTOS-OCANA C, NAVAS P. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J Med Genet. 2012;49:187–91. doi: 10.1136/jmedgenet-2011-100394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHWEDE T, KOPP J, GUEX N, PEITSCH MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEELERT H, DANI DN, DANTE S, HAUSS T, KRAUSE F, SCHAFER E, FRENZEL M, POETSCH A, REXROTH S, SCHWASSMANN HJ, SUHAI T, VONCK J, DENCHER NA. From protons to OXPHOS supercomplexes and Alzheimer’s disease: structure-dynamics-function relationships of energy-transducing membranes. Biochim Biophys Acta. 2009;1787:657–71. doi: 10.1016/j.bbabio.2009.02.028. [DOI] [PubMed] [Google Scholar]
- STENMARK P, GRUNLER J, MATTSSON J, SINDELAR PJ, NORDLUND P, BERTHOLD DA. A new member of the family of di-iron carboxylate proteins. Coq7 (clk-1), a membrane-bound hydroxylase involved in ubiquinone biosynthesis. J Biol Chem. 2001;276:33297–300. doi: 10.1074/jbc.C100346200. [DOI] [PubMed] [Google Scholar]
- STEPANYAN Z, HUGHES B, CLICHE DO, CAMP D, HEKIMI S. Genetic and molecular characterization of CLK-1/mCLK1, a conserved determinant of the rate of aging. Exp Gerontol. 2006;41:940–51. doi: 10.1016/j.exger.2006.06.041. [DOI] [PubMed] [Google Scholar]
- SUZUKI K, UEDA M, YUASA M, NAKAGAWA T, KAWAMUKAI M, MATSUDA H. Evidence that Escherichia coli ubiA product is a functional homolog of yeast COQ2, and the regulation of ubiA gene expression. Biosci Biotechnol Biochem. 1994;58:1814–9. doi: 10.1271/bbb.58.1814. [DOI] [PubMed] [Google Scholar]
- TAUCHE A, KRAUSE-BUCHHOLZ U, RODEL G. Ubiquinone biosynthesis in Saccharomyces cerevisiae: the molecular organization of O-methylase Coq3p depends on Abc1p/Coq8p. FEMS Yeast Res. 2008;8:1263–75. doi: 10.1111/j.1567-1364.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- TECLEBRHAN H, JAKOBSSON-BORIN A, BRUNK U, DALLNER G. Relationship between the endoplasmic reticulum-Golgi membrane system and ubiquinone biosynthesis. Biochimica et biophysica acta. 1995;1256:157–65. doi: 10.1016/0005-2760(95)00016-6. [DOI] [PubMed] [Google Scholar]
- TEKLE M, BENTINGER M, NORDMAN T, APPELKVIST EL, CHOJNACKI T, OLSSON JM. Ubiquinone biosynthesis in rat liver peroxisomes. Biochemical and biophysical research communications. 2002;291:1128–33. doi: 10.1006/bbrc.2002.6537. [DOI] [PubMed] [Google Scholar]
- TIELENS AG. Energy generation in parasitic helminths. Parasitol Today. 1994;10:346–52. doi: 10.1016/0169-4758(94)90245-3. [DOI] [PubMed] [Google Scholar]
- TRAN UC, CLARKE CF. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion. 2007;7(Suppl):S62–71. doi: 10.1016/j.mito.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRAN UC, MARBOIS B, GIN P, GULMEZIAN M, JONASSEN T, CLARKE CF. Complementation of Saccharomyces cerevisiae coq7 mutants by mitochondrial targeting of the Escherichia coli UbiF polypeptide: two functions of yeast Coq7 polypeptide in coenzyme Q biosynthesis. J Biol Chem. 2006;281:16401–9. doi: 10.1074/jbc.M513267200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TREVISSON E, DIMAURO S, NAVAS P, SALVIATI L. Coenzyme Q deficiency in muscle. Curr Opin Neurol. 2011;24:449–56. doi: 10.1097/WCO.0b013e32834ab528. [DOI] [PubMed] [Google Scholar]
- TURRENS JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURUNEN M, OLSSON J, DALLNER G. Metabolism and function of coenzyme Q. Biochimica et biophysica acta. 2004;1660:171–99. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- VAJO Z, KING LM, JONASSEN T, WILKIN DJ, HO N, MUNNICH A, CLARKE CF, FRANCOMANO CA. Conservation of the Caenorhabditis elegans timing gene clk-1 from yeast to human: a gene required for ubiquinone biosynthesis with potential implications for aging. Mamm Genome. 1999;10:1000–4. doi: 10.1007/s003359901147. [DOI] [PubMed] [Google Scholar]
- VEAL EA, DAY AM, MORGAN BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- WANG D, MALO D, HEKIMI S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/− mouse mutants. Journal of immunology. 2010;184:582–90. doi: 10.4049/jimmunol.0902352. [DOI] [PubMed] [Google Scholar]
- WANG Y, BRANICKY R, STEPANYAN Z, CARROLL M, GUIMOND MP, HIHI A, HAYES S, MCBRIDE K, HEKIMI S. The anti-neurodegeneration drug clioquinol inhibits the aging-associated protein CLK-1. J Biol Chem. 2009;284:314–23. doi: 10.1074/jbc.M807579200. [DOI] [PubMed] [Google Scholar]
- WINGE DR. Sealing the mitochondrial respirasome. Mol Cell Biol. 2012;32:2647–52. doi: 10.1128/MCB.00573-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WONG A, BOUTIS P, HEKIMI S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics. 1995;139:1247–59. doi: 10.1093/genetics/139.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIE LX, HSIEH EJ, WATANABE S, ALLAN CM, CHEN JY, TRAN UC, CLARKE CF. Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants. Biochim Biophys Acta. 2011;1811:348–60. doi: 10.1016/j.bbalip.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIE LX, OZEIR M, TANG JY, CHEN JY, KIEFFER-JAQUINOD S, FONTECAVE M, CLARKE CF, PIERREL F. Over-expression of the Coq8 kinase in Saccharomyces cerevisiae coq null mutants allows for accumulation of diagnostic intermediates of the Coenzyme Q6 biosynthetic pathway. J Biol Chem. 2012 doi: 10.1074/jbc.M112.360354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG W, HEKIMI S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG YY, GANGOITI JA, SEDENSKY MM, MORGAN PG. The effect of different ubiquinones on lifespan in Caenorhabditis elegans. Mechanisms of ageing and development. 2009;130:370–6. doi: 10.1016/j.mad.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG YY, VASTA V, HAHN S, GANGOITI JA, OPHEIM E, SEDENSKY MM, MORGAN PG. The role of DMQ(9) in the long-lived mutant clk-1. Mechanisms of ageing and development. 2011;132:331–9. doi: 10.1016/j.mad.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAMPOL MA, BUSSO C, GOMES F, FERREIRA-JUNIOR JR, TZAGOLOFF A, BARROS MH. Over-expression of COQ10 in Saccharomyces cerevisiae inhibits mitochondrial respiration. Biochem Biophys Res Commun. 2010;402:82–7. doi: 10.1016/j.bbrc.2010.09.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZIEGLER CG, PENG M, FALK MJ, POLYAK E, TSIKA E, ISCHIROPOULOS H, BAKALAR D, BLENDY JA, GASSER DL. Parkinson’s disease-like neuromuscular defects occur in prenyl diphosphate synthase subunit 2 (Pdss2) mutant mice. Mitochondrion. 2011;12:248–57. doi: 10.1016/j.mito.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]