

Abstract
The mechanisms of release, depletion, and refilling of endoplasmic reticulum (ER) Ca2+ were investigated in type I horizontal cells of the carp retina using a fluo-3-based Ca2+ imaging technique. Exogenous application of caffeine, a ryanodine receptor agonist, induced oscillatory intracellular free Ca2+ concentration ([Ca2+]i) responses in a duration- and concentration-dependent manner. In Ca2+-free Ringer’s solution, [Ca2+]i transients could also be induced by a brief caffeine application, whereas subsequent caffeine application induced no [Ca2+]i increase, which implied that extracellular Ca2+ was required for ER refilling, confirming the necessity of a Ca2+ influx pathway for ER refilling. Depletion of ER Ca2+ by thapsigargin triggered a Ca2+ influx which could be blocked by the store-operated channel inhibitor 2-APB, which proved the existence of the store-operated Ca2+ entry pathway. Taken together, these results suggested that after being depleted by caffeine, the ER was replenished by Ca2+ influx via store-operated channels. These results reveal the fine modulation of ER Ca2+ signaling, and the activation of the store-operated Ca2+ entry pathway guarantees the replenishment of the ER so that the cell can be ready for response to the subsequent stimulus.
Introduction
Ca2+ is a ubiquitous intracellular messenger that regulates numerous cellular processes including muscle contraction, transmitter release, apoptosis, and so on [1], [2]. In neurons, the basal level of intracellular free Ca2+ concentration ([Ca2+]i) is maintained very low [3]. When activated by proper stimulation, the opening of the plasma membrane Ca2+ channels or the activation of Ca2+ release channels on the intracellular Ca2+ stores (largely the endoplasmic reticulum, ER) leads to the elevation of [Ca2+]i [2].
As the primary intracellular reservoir of Ca2+ and a major source of [Ca2+]i elevation, the ER is involved in a wide range of neuronal Ca2+-dependent processes, such as synaptic transmission and plasticity [4], [5]. The ER accumulates Ca2+ by active transport of Ca2+ from the cytoplasm into the ER by Ca2+-ATPase (sarco/endoplasmic reticulum Ca2+-ATPase, SERCA) pumps expressed on the ER membrane. At the meantime, Ca2+ release from the ER is mainly mediated by two families of Ca2+ release channels, i.e., the inositol 1,4,5-trisphosphate receptor (IP3R) and ryanodine receptor (RyR) families. While IP3Rs are gated by IP3, both IP3Rs and RyRs can be activated by Ca2+, and such Ca2+-induced Ca2+ release (CICR) forms a positive feedback process. RyRs can also be activated by caffeine, which sensitizes RyR’s response to Ca2+ [6], [7].
Type I horizontal cells (H1 HCs) are interneurons in the outer retina of carp, which receive glutamate input from cone photoreceptors. It was found in the fish retina that HCs contained caffeine-sensitive ER [8], [9] which was involved in the modulation of synaptic strength [10], GABA transporter currents [11], [12], as well as voltage-gated Ca2+ channel currents [13]. Despite its functional significance, knowledge about the ER Ca2+ dynamics of H1 HCs is still limited.
The present study aims to investigate possible mechanisms of ER Ca2+ dynamics of carp H1 HCs, particularly the details of the following aspects: (1) the temporal characteristics of [Ca2+]i signals initiated by ER Ca2+ release, (2) the inter-relationship between the two major Ca2+ sources, i.e., the extracellular Ca2+ and the ER, and relevant channels underlying their interaction.
To explore the above issues, Ca2+ signals elicited by exogenously applied caffeine were recorded from freshly dissociated H1 HCs using a fluo-3-based Ca2+ imaging technique. The basic findings are: (1) caffeine induced oscillatory [Ca2+]i responses in a duration- and concentration-dependent manner, (2) removal of extracellular Ca2+ abolished the repeatability of caffeine-induced [Ca2+]i responses, (3) inhibition of L-type voltage-gated Ca2+ channels (L-VGCCs) reduced caffeine-induced [Ca2+]i oscillations, (4) inhibition of store-operated channels (SOCs) abolished caffeine-induced [Ca2+]i oscillations. These results reveal the fine modulation of ER Ca2+ signaling, and the activation of the store-operated Ca2+ entry (SOCE) pathway guarantees the replenishment of the ER so that the cell can be ready for response to the subsequent stimulus.
Materials and Methods
Ethics Statement
The animal experiments were approved by the Ethic Committee, School of Biomedical Engineering, Shanghai Jiao Tong University. All procedures strictly conformed to the humane treatment and use of animals as prescribed by the Association for Research in Vision and Ophthalmology.
Cell Isolation
H1 HCs were enzymatically dissociated from retinas of adult carp (Carassius auratus, 15–20 cm body length). After 30 min dark-adaption, the eye was enucleated and hemisected. The retina was then isolated and cut into 8–12 pieces and incubated for 30 min at room temperature of 25°C in 5 ml Hank’s solution (see below) containing 25 U/ml papain (E. Merck, Germany) and 1 mg/ml L-cysteine. The retinal pieces were then kept in normal Hank’s solution at 4°C until being used (within 4 h). To obtain dissociated H1 HCs, retinal pieces were gently triturated with fire-polished glass pipettes in normal Ringer’s solution. The cell suspension was moved to a recording chamber for Ca2+ imaging recording.
Solutions
Hank’s solution contained (in mM): 137.0 NaCl, 3.0 KCl, 1.0 MgSO4, 1.0 NaH2PO4, 0.5 NaHCO3, 2.0 CaCl2, 2.0 Na-pyruvate, 20.0 HEPES and 16.0 Glucose. Ringer’s solution contained (in mM): 145.0 NaCl, 5.0 KCl, 1.0 MgSO4, 2.0 CaCl2, 10.0 HEPES and 16.0 Glucose. Ca2+-free Ringer’s solution was prepared based on normal Ringer’s solution with CaCl2 omitted and 1 mM EGTA added. Caffeine was directly dissolved in Ringer’s solution according to the concentration required. Ryanodine (Tocris Bioscience, UK), nifedipine, thapsigargin (TG) and 2-aminoethoxydiphenyl borate (2-APB) were prepared in dimethyl sulfoxide (DMSO) and diluted to their final concentration in Ringer’s solution (DMSO <0.5%). The pH value was adjusted to 7.4 with NaOH for Ringer’s and Hank’s solutions as well as other solutions. All drugs, unless otherwise specified, were purchased from Sigma Aldrich (St. Louis, MO).
Ca2+ Imaging
[Ca2+]i changes were measured using a fluo-3 imaging system. Fluo-3/AM was dissolved in DMSO (1 mM stock solution) and added to the cell suspension at a final concentration of 5 µM (DMSO = 0.5%). Cell suspension was transferred into a perfusion chamber (RC-26, Warner Instruments, USA) with a cover glass bottom (12–548B, Thermo Fisher Scientific Inc., USA) and incubated at room temperature (25°C) for 15 min to allow for cell adherence and fluo-3 loading. The cells were then continuously perfused with Ringer’s solution at a flow rate of 1 ml/min for 10 min prior to recording. The volume of the perfusion chamber was about 500 µl. All drugs were applied by perfusion. At the perfusion rate employed, 1 min was needed for the drug to reach the perfusion chamber. Fluorescence measurements were performed on an upright fluorescence microscope (BX51WI, Olympus, Japan). H1 HCs were identified by their characteristic morphology as having a round soma with 4–8 extended, subtle dendrites [14]. Fluo-3 in its Ca2+-bound form was excited at 488 nm, and fluorescence emission at 525 nm was acquired every 2 s by a digital CCD camera (CoolSNAP ES2, Photometrics, USA). [Ca2+]i signals were presented by the fluorescence intensity F normalized to the baseline fluorescence value F0 (F/F0). For caffeine-induced [Ca2+]i responses, [Ca2+]i signals were normalized against the amplitude of the first [Ca2+]i transient. A transient peak was considered a caffeine-induced [Ca2+]i transient if it had an amplitude larger than 3×SD of data recorded in the 1 min duration prior to caffeine application.
Statistical Analyses
Statistics were performed using SPSS software (version 17.0, SPSS Inc., Chicago, IL, USA). Values are presented as mean ± SEM. To determine statistical significance, independent-samples t test and one-way analysis of variance (ANOVA) were used for comparing the results between two groups and that among multiple groups respectively. If a significant p value was obtained for ANOVA, post hoc analyses were performed using Student-Newman-Keuls (SNK) test, p<0.05 indicates significant differences.
Results
Caffeine Induced [Ca2+]i Responses in a Duration- and Concentration-dependent Manner
Caffeine has long been used as a RyR agonist for studying RyR-mediated Ca2+ release from intracellular Ca2+ stores [15], [16]. In our present study, to investigate the temporal characteristics of [Ca2+]i signal initiated by ER Ca2+ release, caffeine was applied at six different concentrations (1, 3, 6, 10, 20, and 40 mM) in combination with four different durations (30, 60, 90, and 240 s). For each of these 6×4 = 24 combinations, one group of independent experiments was conducted (using 5–9 H1 HCs for each experimental condition).
To study the effect of duration of caffeine application, the four groups of experiments with varying durations (i.e., 30, 60, 90, and 240 s) of caffeine application at the same caffeine concentration were compared.
In our experiments, the application of 1 mM caffeine with durations of 30, 60, 90, and 240 s induced no discernible [Ca2+]i changes in all four groups of H1 HCs tested.
The application of 3 mM caffeine induced [Ca2+]i transient(s) in a duration-dependent way (Fig. 1A (a–d)). When comparing results from the four duration groups (30, 60, 90, and 240 s), the number of [Ca2+]i transients elicited by 3 mM caffeine showed an increasing tendency with the duration of caffeine application, which was increased monotonically from 0 to 5.13±0.35 (p<0.05, ANOVA, post hoc SNK test) when the duration was increased from 30 to 240 s (Fig. 1A (e)).
Figure 1. Caffeine induced [Ca2+]i increase in a duration- and concentration-dependent manner.

A–E, [Ca2+]i transients induced by the application of 3, 6, 10, 20, and 40 mM caffeine, respectively. Columns (a)-(d), [Ca2+]i transients induced by the application of caffeine for 30, 60, 90, and 240 s, respectively. Column (e), The average number of [Ca2+]i transients (transient NO.) induced by various durations of caffeine application under each concentration. F, Dependence of caffeine-induced [Ca2+]i responses (with identical application time of 90 s) on caffeine concentration. (a), Definition of amplitude and time interval between the first and the second Ca2+ peaks (ΔT1–2). The amplitude of the second [Ca2+]i transient (A2) is normalized against that of the first one (A1). (b), ΔT1–2 decreased with caffeine concentration. (c), A2/A1 decreased with caffeine concentration. Horizontal bars bellow the traces indicate the periods of caffeine applications. With our perfusion rate being 1 ml/min, 1 min was needed for the drug to reach the perfusion chamber. In this and subsequent figures, each trace is the representative of a group of independent experiments, and data are presented as mean ± SEM (with sample size in parentheses). * denotes statistical significance of p<0.05 by one-way ANOVA followed by post hoc SNK test, NS: not significant.
The application of 6 mM caffeine tended to induce more active [Ca2+]i transients (Fig. 1B (a–d)). The number of [Ca2+]i transients elicited by 6 mM caffeine was increased from 0.71±0.18 to 6.00±0.41 (p<0.05, ANOVA, post hoc SNK test), when the duration of caffeine application was increased from 30 to 240 s (Fig. 1B (e)).
When 10 mM caffeine was applied, duration-dependent [Ca2+]i transients increment was still observed (Fig. 1C (a–d)). But the increment was very much limited. The number of [Ca2+]i transients induced by 10 mM caffeine was increased from 1.50±0.19 to 2.88±0.23 (p<0.05, ANOVA, post hoc SNK test; Fig. 1C (e)) when the duration of caffeine application was increased from 30 to 60 s, and was stabilized around 3 when the duration of caffeine was further increased to 90 and 240 s (3.00±0.27 and 3.13±0.13, respectively), with no statistical significance among the 60, 90, and 240 s groups (p>0.05, post hoc SNK test; Fig. 1C (e)).
When 20 mM caffeine was applied at various durations (30, 60, 90, and 240 s), the pattern of [Ca2+]i response was almost uniform among the four groups (Fig. 1D (a–d)), the average number of [Ca2+]i transients for the four groups were 2.38±0.18, 2.75±0.16, 2.63±0.18, and 2.38±0.26, respectively, with no significant differences among groups (p>0.05, ANOVA, post hoc SNK test; Fig. 1D (e)).
During 40 mM caffeine application, only one [Ca2+]i transient could be induced no matter how long the caffeine application duration was (Fig. 1E).
The above results show that, Ca2+ oscillations could be induced by intermediate concentrations of caffeine (3 to 20 mM), with the oscillatory behavior being concentration-dependent. To examine the effect of caffeine concentration on Ca2+ oscillations, experiments conducted at varying caffeine concentrations with identical duration (90 s) of caffeine application were compared (Fig.1 column c). When the concentration of caffeine was increased from 3 to 20 mM (with identical application time of 90 s), the time interval between the first and the second Ca2+ transients (ΔT1–2; Fig. 1F (a)) decreased from 46.08±3.41 to 19.24±1.68 s (p<0.05, ANOVA, post hoc SNK test; Fig. 1F (b)).
The amplitude decrement during Ca2+ oscillations was also concentration dependent. During the application of 3 mM caffeine, the amplitudes of the oscillatory [Ca2+]i transients were changing within a limited range (Fig. 1A). However, when caffeine was applied with higher concentrations, a decreasing tendency in amplitudes was observed in the oscillatory transients (Fig. 1B–D). To quantify such amplitude decrement, we normalized the amplitude of the second transient against that of the first one (A2/A1; Fig. 1F(a)). When the concentration of caffeine was increased from 3 to 20 mM (with application time of 90 s), the normalized amplitude of the second transient (A2/A1) was decreased from 0.82±0.05 to 0.41±0.07 (p<0.05, ANOVA, post hoc SNK test; Fig. 1F(c)).
Caffeine-induced [Ca2+]i transient is composed of a rising phase and a decaying phase. In carp HCs, while the [Ca2+]i increase is caused primarily by Ca2+ release from the ER via RyRs and Ca2+ influx from the extracellular environment, the decaying phase of the transient depends on the activity of SERCA pumps [3], [8], [9], Na+/Ca2+ exchangers [17] and plasma membrane Ca2+-ATPase (PMCA) pumps [17]. The balance between these Ca2+-increasing and removal processes determines the pattern of [Ca2+]i oscillations, which is reflected in the time interval between two adjacent transients, and the amplitude of each transient. To investigate the underlying mechanisms of caffeine-induced [Ca2+]i oscillations, we looked into the involvement of RyR activation in the initiation of the [Ca2+]i oscillations.
Ryanodine Application Inhibited the Caffeine-induced [Ca2+]i Oscillations
Caffeine-induced [Ca2+]i oscillations can be initiated by Ca2+ release from the ER via RyRs. To confirm the contribution of RyRs during caffeine-induced [Ca2+]i oscillations, high concentration ryanodine (2.5 µM) was used to inhibit the Ca2+ release via RyRs [18], [19]. The group of experiments conducted with caffeine (10 mM, 60 s) in normal Ringer’s solution was taken as control. In normal Ringer’s solution, caffeine (10 mM, 60 s) induced three [Ca2+]i transients in an example HC (Fig. 2A). However, in the presence of ryanodine, the application of caffeine (10 mM, 60 s) induced a single Ca2+ transient (Fig. 2B), which is in accordance with the notion that inhibition of RyRs by ryanodine requires the activation of RyRs [20]. Besides, due to the irreversibility of RyR inhibition by ryanodine [20], subsequent application of caffeine induced no [Ca2+]i increase in the tested HC, even after washing out of ryanodine. Statistical results showed that, the number of [Ca2+]i transients induced by caffeine (10 mM, 60 s) was 1.00±0.00 for the ryanodine group, which was significantly reduced as compared with 2.88±0.23 (p<0.05, independent-samples t test) of the control group (Fig. 2C), the absence of subsequent [Ca2+]i transients after the inhibition of RyRs demonstrated that the generation of subsequent transients was also initiated by Ca2+ release via RyRs.
Figure 2. Ryanodine effect on the caffeine-induced [Ca2+]i oscillations.
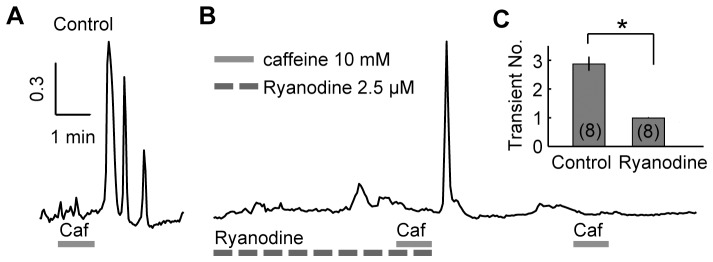
A–B, Caffeine (10 mM, 60 s) induced [Ca2+]i responses in: (A) normal Ringer’s solution (control), and (B) in the presence of ryanodine (2.5 µM). C, The average number of Ca2+ transients induced by caffeine (10 mM, 60 s) in the control group and the ryanodine group. * denotes statistical significance of p<0.05 with independent-samples t test.
Extracellular Ca2+ was Required for the Oscillatory Caffeine-induced [Ca2+]i Responses
For each [Ca2+]i transient, following Ca2+ release from the ER via RyR activation, the activation of SERCA pumps, Na+/Ca2+ exchangers, and PMCA pumps brought the elevated [Ca2+]i back to the basal level. Due to the activity of PMCA pumps and Na+/Ca2+ exchangers in the plasma membrane, Ca2+ released from the ER cannot be fully recycled back into the ER by SERCA pumps. To counterbalance this loss of ER Ca2+, Ca2+ entry from the extracellular medium is required for the oscillatory caffeine-induced [Ca2+]i responses. To investigate whether caffeine-induced [Ca2+]i responses in H1 HCs were dependent on extracellular free Ca2+ concentration ([Ca2+]o), Ca2+-free Ringer’s solution was used to abolish the putative Ca2+ influx across the plasma membrane.
The results show that, in normal Ringer’s solution (control), caffeine (10 mM, 60 s) induced three [Ca2+]i transients in the tested HC, and such response pattern was repeatable (Fig. 3A). In Ca2+-free Ringer’s solution, caffeine (10 mM, 60 s) elicited two [Ca2+]i transients, with the subsequent caffeine application inducing no measurable response (Fig. 3B), implying that the ER was depleted by the first caffeine application. After reintroduction of [Ca2+]o, caffeine (10 mM, 60 s) again induced [Ca2+]i response, confirming the necessity of Ca2+ influx for ER refilling following its depletion for the subsequent Ca2+ response.
Figure 3. Regulation of [Ca2+]o entry affects the caffeine-induced [Ca2+]i oscillations.

A–D, caffeine (10 mM, 60 s) induced [Ca2+]i oscillations in: (A) normal Ringer’s solution (control) and Ringer’s solution with (B) Ca2+-free, (C) 4 mM [Ca2+]o, and (D) nifedipine (100 µM). E–G, The average number of [Ca2+]i transients (E), ΔT1–2 (F), and A2/A1 (G) of caffeine (10 mM, 60 s) induced [Ca2+]i oscillations for the 4 mM [Ca2+]o group, the control group, the nifedipine group, and the Ca2+-free group. * denotes statistical significance of p<0.05 with independent-samples t test. Abbreviations: Ca-0: Ca2+-free; Ca-4∶4 mM [Ca2+]o.
Of all 12 cells in the Ca2+-free group, caffeine (10 mM, 60 s) induced one transient in 5 cells, and two transients in the rest 7 cells. Hence, in Ca2+-free Ringer’s solution, the number of [Ca2+]i transients induced by caffeine (10 mM, 60 s) was significantly reduced as compared with control (1.58±0.15 and 2.88±0.23, respectively, p<0.05, independent-samples t test; Fig. 3E). For the 7 cells of the Ca2+-free group in which two transients were elicited by caffeine (10 mM, 60 s), ΔT1–2 and A2/A1 were calculated. The ΔT1–2 interval was similar to that measured in control (25.33±1.10 and 25.75±1.11 for Ca2+-free and control group, respectively, p>0.05, independent-samples t test; Fig. 3F); however, the A2/A1 ratio was significantly smaller than that of control (0.18±0.04 and 0.60±0.06 for Ca2+-free and control group, respectively, p<0.05, independent-samples t test; Fig. 3G). The reduction of transients number and A2/A1 by [Ca2+]o removal suggests that Ca2+ influx contributes to caffeine-induced [Ca2+]i oscillations.
The above results show that caffeine-induced [Ca2+]i response was significantly reduced by [Ca2+]o removal. If abolishing Ca2+ entry can reduce caffeine-induced [Ca2+]i response, up-regulation of Ca2+ entry should enhance the response. To test this hypothesis, [Ca2+]o was increase from 2 mM (normal Ringer’s solution) to 4 mM to up-regulate Ca2+ entry from the extracellular space.
When caffeine (10 mM, 60 s) application was given in the presence of 4 mM [Ca2+]o, three [Ca2+]i transients were evoked in an example HC (Fig. 3C). Statistical comparison between the high-[Ca2+]o group and the control group showed that, high [Ca2+]o resulted in a modest increase in the number of caffeine-induced [Ca2+]i transients (2 mM [Ca2+]o: 2.88±0.23, 4 mM [Ca2+]o: 3.13±0.13, p>0.05, independent-samples t test; Fig. 3E); at the meantime, a significant decrease in ΔT1–2 (2 mM [Ca2+]o: 25.75±1.11, 4 mM [Ca2+]o: 21.60±0.79, p<0.05, independent-samples t test; Fig. 3F) and a modest increase in A2/A1 (2 mM [Ca2+]o: 0.60±0.06, 4 mM [Ca2+]o: 0.74±0.06, p>0.05, independent-samples t test; Fig. 3G) were also observed. Hence, up-regulation of Ca2+ entry indeed enhanced the oscillatory caffeine-induced [Ca2+]i signals.
L-VGCCs were Involved in the Caffeine-induced [Ca2+]i Oscillations
The above results show that the caffeine-induced [Ca2+]i response was significantly reduced when [Ca2+]o was removed, which indicates that Ca2+ influx is requested for the caffeine-induced [Ca2+]i oscillations. In carp HCs, Ca2+ entry from the extracellular space is known to be mediated by Ca2+-permeable glutamate receptors (GluRs) [8], [9], [21], [22], [23], [24] and L-VGCCs [23], [25], [26], [27]. In the present study, the activities of GluRs were precluded, because the activation of GluRs requires the binding of glutamate. On the other hand, the membrane potential and [Ca2+]i are strongly correlated. Oscillations in [Ca2+]i may concomitant changes of the membrane potential. Thus, L-VGCCs, which are activated by membrane depolarization, might be involved in the caffeine-induced [Ca2+]i increase. To test the possibility of L-VGCC involvement, nifedipine, an L-VGCC antagonist, was applied [28]. Previous study in carp retinal H1 HCs suggests that 100 µM nifedipine was sufficient to abolish L-VGCC-mediated Ca2+ entry [23]. Hence, 100 µM nifedipine was used for complete inhibition of L-VGCCs in the present study.
In the presence of nifedipine (100 µM), caffeine (10 mM, 60 s) elicited two [Ca2+]i transients, the subsequent caffeine application also induced two [Ca2+]i transients (Fig. 3D). Nifedipine (100 µM) significantly reduced the number of [Ca2+]i transients induced by caffeine (10 mM, 60 s), as compared with the effect of caffeine (10 mM, 60 s) alone (2.25±0.16 and 2.88±0.23, respectively, p<0.05, independent-samples t test; Fig. 3E). The ΔT1–2 interval was slightly increased (28.00±0.98 and 25.75±1.11 for nifedipine treated and control group, respectively, p>0.05, independent-samples t test; Fig. 3F), and the A2/A1 ratio was also decreased without statistical significance (0.71±0.05 and 0.60±0.06 for nifedipine treated and control group, respectively, p>0.05, independent-samples t test; Fig. 3G).
The nifedipine-induced reduction of [Ca2+]i transient number suggested that L-VGCCs should be activated by caffeine application, and the resulting Ca2+ entry via L-VGCCs contributed to caffeine-induced [Ca2+]i oscillations. However, in the presence of nifedipine, the number of [Ca2+]i transients was significantly larger than that in Ca2+-free Ringer’s solution (p<0.05, independent-samples t test; Fig. 3E). Besides, nifedipine didn’t reduce the A2/A1 ratio, while the A2/A1 ratio was significantly reduced by Ca2+-free Ringer’s solution (Fig. 3G). More importantly, different from the result obtained with Ca2+-free Ringer’s solution (Fig. 3B), when caffeine was re-applied in the presence of nifedipine (Fig. 3D), [Ca2+]i responses could be reproduced rather than abolished, which indicated that the ER could still be refilled when L-VGCCs were blocked. These differences between the effects of nifedipine and Ca2+-free Ringer’s solution on caffeine-induced [Ca2+]i oscillations suggest that L-VGCCs weren’t the only Ca2+ entry pathway activated after caffeine application, there should be other Ca2+ entry pathway for ER refilling.
SOCs were Necessary for ER Refilling
It has been reported that in some cell types, Ca2+ influx could be triggered by the depletion of ER Ca2+, a process referred to as store-operated Ca2+ entry (SOCE) [29], [30]. The Ca2+ channels mediating SOCE are called store-operated channels (SOCs).
To examine the involvement of SOCs in ER refilling, we tested the existence of SOCs in carp H1 HCs and tried to activate SOCs by depleting the ER. To deplete the ER, HCs were incubated in Ca2+-free Ringer’s solution containing 5 µM thapsigargin, an irreversible SERCA inhibitor, which causes passive depletion of ER Ca2+ by inhibiting ER Ca2+ uptake via SERCA pumps [31]. If SOCs are expressed in carp H1 HCs and can be activated by ER depletion, re-addition of [Ca2+]o should result in an increase in [Ca2+]i, given that ER Ca2+ uptake via SERCA pumps was inhibited. As shown in Figure 4A, after being pre-incubated in thapsigargin-containing (5 µM) Ca2+-free Ringer’s solution for 20 min, re-introduction of [Ca2+]o (2 mM) caused a transient increase of [Ca2+]i in the tested HC (Fig. 4A). Similar results were obtained from 7 others HCs, demonstrating the existence of SOCE pathways in carp H1 HCs.
Figure 4. SOCs were nessicary for ER depletion-induced Ca2+ entry.

A, After pre-application of thapsigargin (TG, 5 µM) in Ca2+-free Ringer’s solution for 20 min to deplete ER, reintroduction of [Ca2+]o (2 mM) elicited a transient increase in [Ca2+]i. B, In the presence of 2-APB (a SOC inhibitor, 100 µM), the [Ca2+]i increase induced by [Ca2+]o re-addition was abolished. C, In the presence of nifedipine (a L-VGCC blocker, 100 µM), the [Ca2+]i increase after [Ca2+]o re-addition was still observed. Abbreviations: Ca-0: Ca2+-free; Ca-2∶2 mM [Ca2+]o. Traces shown are representative of three independent experiments, eight HCs were tested under each condition.
To further confirm that this [Ca2+]i increase activated by ER depletion is mediated by SOCs, 2-APB, a SOC antagonist, was applied at a concentration of 100 µM [32]. As shown in Figure 4B, in the presence of 2-APB (100 µM), [Ca2+]i increase after [Ca2+]o re-addition was abolished, however this [Ca2+]i increase was not eliminated by 100 µM nifedipine (Fig. 4C). Similar results were observed from 7 other HCs for each condition. These results indicate that SOCs rather than L-VGCCs underlay this ER depletion-induced Ca2+ entry of HCs.
During the application of 3 mM caffeine, the number of Ca2+ transients was increased as the duration of caffeine was increased. At the meantime, the amplitudes of the oscillatory [Ca2+]i transients were changing within a limited range (Fig. 1A), suggesting the occurrence of ER Ca2+ refilling between two adjacent [Ca2+]i transients. To test whether SOCs underlie ER refilling after caffeine (3 mM) application, 2-APB (100 µM) was applied.
In normal Ringer’s solution, [Ca2+]i oscillations were induced by prolonged application of 3 mM caffeine (7 min; Fig. 5A). However, such caffeine-induced [Ca2+]i oscillations were abolished when 2-APB (100 µM) was co-applied after 2 min caffeine application (Fig. 5B). For 2-APB treated group, the number of [Ca2+]i transients induced by caffeine (3 mM, 7 min) was 2.22±0.15, which was significantly reduced as compared with 8.57±0.81 in normal Ringer’s solution (control) (p<0.05, independent-samples t test; Fig. 5C).
Figure 5. 2-APB effect on the caffeine-induced [Ca2+]i oscillations.
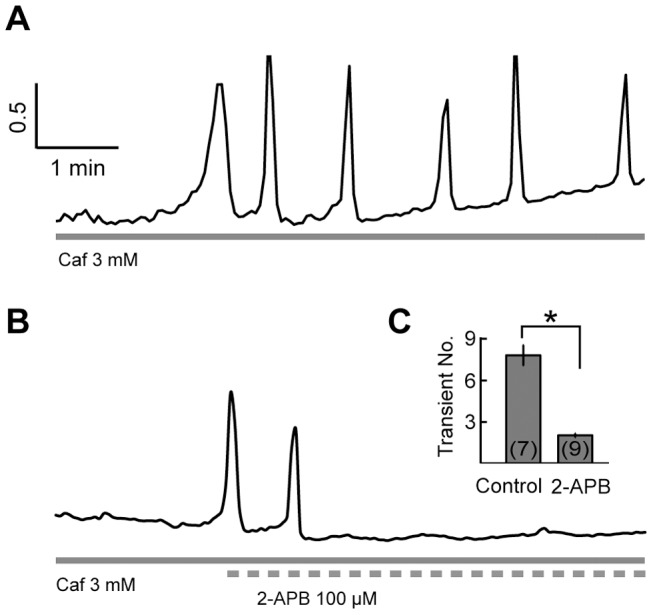
A, [Ca2+]i oscillations induced by prolonged application of caffeine (3 mM, 7 min). B, When 2-APB (100 µM) was co-applied, [Ca2+]i oscillations elicited by 3 mM caffeine was abolished. C, The average number of Ca2+ transients induced by caffeine (3 mM, 7 min) in the control group and the 2-APB group. * denotes statistical significance of p<0.05 with independent-samples t test.
Taken together, these results show that SOCs are expressed in carp H1 HCs, and can be activated by thapsigargin-induced ER depletion; when depleted by caffeine, the ER can be refilled by Ca2+ entry via SOCs.
Discussion
In the present study, we investigated the mechanisms of release, depletion, and refilling of ER Ca2+ in carp retinal HCs. H1 HCs of carp were stimulated by caffeine under various conditions, [Ca2+]i changes were recorded using a fluo-3-based Ca2+ imaging technique.
Our experimental results demonstrated that exogenous application of caffeine induced [Ca2+]i increases in a concentration- and duration-dependent manner, with each [Ca2+]i transient initiated by RyR activation. Extracellular Ca2+ was required for ER replenishment in the caffeine-induced [Ca2+]i oscillations. Ca2+ influx via L-VGCCs contributed to the ER refilling, while SOCs expressed on H1 HCs were necessary for the ER refilling.
Temporal Patterns of the Caffeine-induced [Ca2+]i Responses
Caffeine has long been used as a RyR agonist for studying RyR-mediated Ca2+ release from the intracellular Ca2+ stores [15], [16], and caffeine-induced Ca2+ release has been observed in many types of neurons containing the ryanodine-sensitive ER [5]. In general, when the ER is filled with Ca2+, caffeine application can induce [Ca2+]i increases even in the absence of extracellular Ca2+, and caffeine-induced Ca2+ responses can be blocked by high concentration of ryanodine. Such processes were also observed in the present study. However, the temporal characteristics of caffeine-induced Ca2+ responses varied with cell types. When exposed to caffeine, transient increase in [Ca2+]i was observed in neurons such as carp retinal bipolar cells, rat spiral ganglion neurons, and rat primary sensory neurons [33], [34], [35], while sustained increase in [Ca2+]i and [Ca2+]i oscillations were observed in honeybee photoreceptors [36] and bullfrog sympathetic neurons [37], [38], respectively. Meanwhile, elevation in caffeine concentration (5–30 mM) increased the frequency of caffeine-induced Ca2+ oscillations in bullfrog sympathetic neurons. Similar to bullfrog sympathetic neurons, caffeine (3–20 mM) also induced oscillatory responses in carp H1 HCs in a concentration-dependent manner – when the concentration of caffeine was increased, the time interval between the two adjacent response peaks was decreased. However, the frequency ranges of caffeine-induced [Ca2+]i oscillations observed in bullfrog sympathetic neurons were much lower when compared with that in carp H1 HCs. On the other hand, while the amplitudes of oscillatory caffeine-induced [Ca2+]i increases were basically unaltered in bullfrog sympathetic neurons during the time course of caffeine application, a decreasing tendency was observed in the amplitudes of the oscillatory caffeine-induced [Ca2+]i increases in carp H1 HCs, such differences might result from the differences in frequency ranges. At low oscillation frequencies, depleted ER might be fully refilled, so the amplitudes of the oscillatory caffeine-induced [Ca2+]i increases were basically unaltered in bullfrog sympathetic neurons, while that measured in carp H1 HCs in our experiments had a decreasing tendency.
The Molecular Mechanisms of SOCE
SOCE is a process by which the depletion of ER Ca2+ activates Ca2+ influx across the plasma membrane (PM) [30], the concept of which was proposed by Putney in 1986 [29]. Since then, growing evidence revealed that SOCE is a ubiquitous Ca2+ influx pathway that exists in a variety of cell types, including neurons [39] and non-excitable cells [40]. In recent years, it was found that stromal interaction molecule (STIM) proteins are sensors of the ER Ca2+ content [41], [42]. The EF-hand domain of STIM residing in the ER lumen senses the free Ca2+ concentration inside the lumen of the ER ([Ca2+]ER). Two types of STIM-regulated SOCs have been described: the Orai channels [43], [44], [45] and transient receptor potential canonical (TRPC) channels [46], [47]. Upon store depletion, STIM proteins and SOCs translocate and cluster at the PM-ER junctions, leading to the formation of the STIM-Orai [44], [48] and STIM-TRPC [49] complexes and SOC activation. Activated SOCs mediateCa2+ influx to refill depleted intracellular stores and regulate cellular processes [50], [51]. The Orai channels and TRPC channels can both be inhibited by 2-APB [32], [52], [53]. So based on our experimental results, it’s hard to tell whether it is Orai channels or TRPC channels that mediated the store-operated Ca2+ entry in HCs.
SERCA is one of the proteins identified as a part of the SOC macromolecular complex [54]. Experiments performed on HEK293T cells and HeLa cells demonstrated that SERCA co-localized with STIM at the PM-ER junctions following store depletion, and co-localization of SERCA with the STIM-Orai complex resulted in a tight coupling between SOCE and ER refilling, so that Ca2+ entry via SOCs was mainly transported into the ER [55], [56], [57]. In our current study, in the process of caffeine-induced [Ca2+]i oscillations, ER refilling occurred during the interval between two adjacent [Ca2+]i transients, during which [Ca2+]i was relatively low, suggesting that the majority of Ca2+ entering the cytoplasm via SOCs was transported into the ER. This phenomenon reflected a coupling between the SOCs and SERCA pumps in carp H1 HCs.
In our experiments, 2-APB was used to inhibit SOCE (Fig. 4–5). Although 2-APB has been proven a reliable SOC inhibitor [32], it was also reported that 2-APB can affect hemi-gap-junction (HGJ) channels [58] and IP3Rs [59].
HGJ channels are expressed in fish retinal HCs, and are involved in negative feedback from HCs to cones [60], [61], [62]. These channels are gated by factors including membrane potential and [Ca2+]o [63], [64], [65]. HGJ channels mediate inward currents at negative membrane potentials and outward currents at positive membrane potentials. Both the inward and outward currents are inhibited by high [Ca2+]o in a concentration dependent manner [65]. In our experiments, isolated HCs had a resting membrane potential of about −70 mV, and [Ca2+]o was remained constant at a level of 2 mM, and a small fraction of HGJ channels might be active. However, the application of 2-APB (100 µM) had no significant effect on the basal level of [Ca2+]i (data not shown), suggesting that even if HGJ channels were active, the inward HGJ channel currents had little contribution to [Ca2+]i responses observed in the present study. Outward HGJ channel currents with amplitudes larger than the inward currents can be evoked by membrane depolarization (beyond 10 mV) [66]. However, the caffeine-induced Ca2+ oscillations were abolished by inhibiting either Ca2+ influx or ER Ca2+ release. Therefore, it is unlikely that 2-APB abolished Ca2+ oscillations by inhibiting outward HGJ channel currents.
In regard to IP3Rs, no study suggests the expression of IP3Rs in carp retinal HCs. Besides, in experiments shown in Figure 4, given that the ER was permanently depleted by thapsigargin, IP3Rs which mediate Ca2+ release from the ER were not involved in the [Ca2+]i increase observed after re-addition of extracellular Ca2+. Therefore, it is unlikely that 2-APB abolished [Ca2+]i signals observed in our experiments by inhibiting IP3Rs.
Taken together, despite the fact that 2-APB affects channels other than SOCs, we can still make an inference that there exists a SOCE pathway in carp H1 HCs, which reloads the ER after its depletion.
The Mechanism of the Caffeine-induced [Ca2+]i Oscillations in H1 HCs
Our results showed that caffeine at low concentration (1 mM) elicited no [Ca2+]i changes in H1 HCs, and a single [Ca2+]i transient was evoke by high concentration (40 mM) caffeine, while [Ca2+]i oscillations were induced by caffeine at intermediate concentrations (3 to 20 mM). The present results indicated that each [Ca2+]i transient was initiated by RyR activation, and ER refilling, which was required for the generation of subsequent [Ca2+]i transients, was depending on SOCE. Hence RyRs and SOCs are likely the two major components for caffeine-induced [Ca2+]i oscillations.
Researches on caffeine effect on single RyR channel found that RyR open probability (Po) was increased when caffeine level was elevated [6], [67]. Low dose of caffeine (<1 mM) increased the Po by increasing the opening frequency without altering the opening duration. While at high doses, caffeine elevated the Po by increasing both the frequency and the duration of opening events. The relationship between RyR Po and caffeine concentration could explain the dependence of the [Ca2+]i response pattern on caffeine concentration observed in our present study. When caffeine was applied at low level (1 mM), the RyR Po was very small [6]. With low RyR opening frequency and duration, caffeine was unable to trigger CICR from the ER. On the other hand, upon the application of high dose caffeine (40 mM), with RyR Po significantly increased and approaching 1.0 [6], ER was constantly leaky, making it impossible to accumulate Ca2+ after the initial release, therefore only one Ca2+ transient was generated.
In addition to SOCs, Ca2+ influx via L-VGCCs also contributed to ER refilling. The involvement of L-VGCCs in caffeine-induced [Ca2+]i oscillations suggests that the membrane potential of H1 HCs should be depolarized upon caffeine application. The application of caffeine triggered ER Ca2+ release and subsequent Ca2+ influx. In some cell types, the increase in [Ca2+]i leads to the activation of several kinds of plasma membrane ion channels, including Ca2+-activated chloride channels, potassium channels, and non-selective channels [68], [69], [70], [71]. Since no evidence suggests that these channels are expressed in retinal horizontal cells [26], we infer that the main reason for membrane depolarization upon caffeine application was Ca2+ currents.
Taking all factors into consideration, we propose the following mechanisms underlying the caffeine-induced [Ca2+]i oscillations (Fig. 6). When caffeine was applied at intermediate concentrations (3 to 20 mM), RyR Po was at intermediate levels, which was sufficient for CICR. Upon application, caffeine increased the Po of the RyR, leading to CICR from the ER. This resulted in an increase in [Ca2+]i and ER depletion, forming the rapid increasing phase of the [Ca2+]i transient. The elevation of [Ca2+]i and the decreasing of [Ca2+]ER further caused the activation of PMCA pumps, Na+/Ca2+ exchangers and SERCA pumps. PMCA pumps and Na+/Ca2+ exchangers extruded Ca2+ into the extracellular space, while SERCA pumps transported Ca2+ into the ER. The activities of these pumps and exchangers brought [Ca2+]i back to the baseline level, forming the decay phase of the [Ca2+]i transient, thus the first Ca2+ transient was generated.
Figure 6. Schematic illustration of Ca2+ pathways relevant to the caffeine-induced [Ca2+]i oscillations in carp retinal HCs.
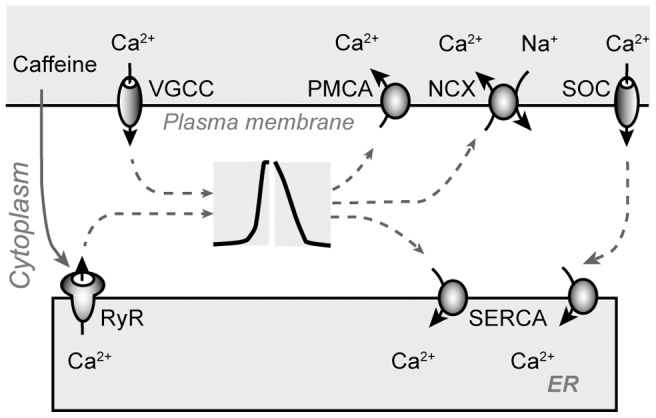
SOC: store-operated channel; VGCC: voltage-gated Ca2+ channel; PMCA: plasma membrane Ca2+-ATPase; NCX: Na+/Ca2+ exchanger; RyR: ryanodine receptor; SERCA: sarco/endoplasmic reticulum Ca2+-ATPase; ER: endoplasmic reticulum.
Meanwhile, ER depletion activated SOCs on the PM, activated SOC mediated Ca2+ influx from the extracellular space. With a tight coupling between SOCE and ER refilling, Ca2+ influx via SOCs was mainly transported into the ER by SERCA pumps without disturbing [Ca2+]i.
Due to the activity of SERCA pumps and SOCs, the ER was refilled after the first [Ca2+]i transient, partially or fully. At this moment, if caffeine was continuously applied, CICR could be re-activated, and a second or more [Ca2+]i transients could be generated, thereby forming [Ca2+]i oscillations. The higher the caffeine concentration, the larger the RyR Po, and the shorter it took for CICR initiation, thus ΔT1–2 was decreased when the caffeine concentration was increased.
The amplitudes of the subsequent [Ca2+]i transients depend on the [Ca2+]ER level. ΔT1–2 was decreased when the concentration of caffeine was increased, which means that time duration for ER refilling was decreased when the caffeine concentration was increased, therefore the A2/A1 ratio was decreased when the caffeine concentration was increased. For [Ca2+]i oscillations induced by 3 mM caffeine, the time interval between two adjacent [Ca2+]i transients was relatively long, which might result in the full refilling of the ER. Thus the amplitudes of Ca2+ transients generated during the time course of caffeine application were remained within a limited range. For [Ca2+]i oscillations induced by higher concentrations of caffeine (6–20 mM), short interval between two adjacent [Ca2+]i transients resulted in inadequate ER replenishment, thus the amplitudes of the oscillatory Ca2+ transients were gradually decreased until the ER was finally running out of Ca2+.
Ca2+ Signaling in Carp HCs
Similar to other cell types, Ca2+ is an important intracellular messenger in carp HCs. [Ca2+]i increase is involved in many HCs cellular functions, including modulation of GABA transporter currents [11], [12], modulation of VGCC currents [13], modulation of synaptic connections [10], as well as the plasticity of spinules at dendrites [21], [72], gating of HGJ channels [63], and maintenance of electrical coupling between HCs [73], etc. The universality of Ca2+ signaling requires the expression of many Ca2+-related components to create a wide range of spatially and temporally distributed signals [2].
In the vertebrate retina, glutamate is continuously released from photoreceptors in the dark, thus GluRs on HCs can be tonically activated. Activated ionotropic GluRs mediated cation influx (including Ca2+), leading to HC depolarization, VGCC activation, CICR from the ER and an increase in [Ca2+]i [8], [9], [23]. A sustained high [Ca2+]i is cytotoxic [74]. Besides, after carrying out its signaling functions, elevated [Ca2+]i must be brought back to the basal level so that the cell can be ready for response to the subsequent stimulus. After the initial [Ca2+]i increase, AMPA receptor desensitization [75], VGCC inhibition via store depletion [13], [76], and VGCC inhibition via AMPA receptor activation [13] might help to restrict Ca2+ influx, regulating [Ca2+]i increases and CICR from the ER. Hence, oscillatory [Ca2+]i responses, such as that observed in the present study, might be one of the strategies that HCs adopt in exposure to tonic glutamate input, so as to prevent cytotoxicity, as well as regulating [Ca2+]i by the rate and duration of photoreceptor glutamate release.
Oscillations of the membrane potential in response to a brief light flash have been recorded from HCs in the intact retina [77], [78]. Since the membrane potential and [Ca2+]i are strongly correlated, we infer that [Ca2+]i oscillations can be induced under physiological conditions. But the frequency ranges of the membrane potential oscillations were between 1.5 and 3.5 Hz, much higher than the [Ca2+]i oscillations recorded in our experiments. In our experiments, bath application of caffeine affects RyRs in isolated H1 HCs, leading to the generation of global intracellular Ca2+ signals. While in the intact retina, HCs receive input from synapses of rods and cones, producing localized Ca2+ signals. Depletion of the ER in a restricted area takes shorter time to refill, while depletion of the whole ER as observe in the current study takes longer to refill, this might account for the differences in oscillation frequencies observed in HCs in the intact retina and isolated HCs.
Generally, caffeine-induced [Ca2+]i oscillations in carp H1 HCs is the repetition of the following two steps: ER Ca2+ release and subsequent ER refilling. Ca2+ release from the ER is mediated by RyRs, while ER refilling depends primarily on Ca2+ influx via SOCs. Hence the SOC is an essential component for ER refilling, and its activation is required for the maintenance of [Ca2+]i oscillations. The ER is the primary intracellular reservoir of Ca2+ and a major source of [Ca2+]i elevation, and it is involved in many cellular processes. The existence of SOCs in H1 HCs and the coupling between SOCE and ER refilling guarantee the efficient replenishment of the ER so that the cell can be ready for response to the subsequent stimulus. Thus SOCs are essential for the fulfilling of normal physiological functions of carp H1 HCs.
Acknowledgments
The authors thank the Instrumental Analysis Center of SJTU for providing the fluo-3 imaging system. The authors also thank Lei Xiao for important technical contributions.
Funding Statement
This work was supported by the National Natural Science Foundation of China (30870836, http://www.nsfc.gov.cn/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Berridge MJ, Bootman MD, Lipp P (1998) Calcium-a life and death signal. Nature 395: 645–648. [DOI] [PubMed] [Google Scholar]
- 2. Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1: 11–21. [DOI] [PubMed] [Google Scholar]
- 3. Hayashida Y, Yagi T (2002) On the interaction between voltage-gated conductances and Ca2+ regulation mechanisms in retinal horizontal cells. J Neurophysiol 87: 172–182. [DOI] [PubMed] [Google Scholar]
- 4. Verkhratsky A (2002) The endoplasmic reticulum and neuronal calcium signalling. Cell Calcium 32: 393–404. [DOI] [PubMed] [Google Scholar]
- 5. Verkhratsky A (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 85: 201–279. [DOI] [PubMed] [Google Scholar]
- 6. Porta M, Zima AV, Nani A, Diaz-Sylvester PL, Copello JA, et al. (2011) Single ryanodine receptor channel basis of caffeine’s action on Ca2+ sparks. Biophys J 100: 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomas NL, Williams AJ (2012) Pharmacology of ryanodine receptors and Ca2+-induced Ca2+ release. Wiley Interdiscip Rev Membr Transp Signal 1: 383–397. [Google Scholar]
- 8. Huang SY, Liu Y, Liang PJ (2004) Role of Ca2+ store in AMPA-triggered Ca2+ dynamics in retinal horizontal cells. Neuroreport 15: 2311–2315. [DOI] [PubMed] [Google Scholar]
- 9. Wang XL, Jiang XD, Liang PJ (2008) Intracellular calcium concentration changes initiated by N-methyl-D-aspartic acid receptors in retinal horizontal cells. Neuroreport 19: 675–678. [DOI] [PubMed] [Google Scholar]
- 10. Huang SY, Hu JF, Gong HQ, Liang PJ (2006) Postsynaptic calcium pathway contributes to synaptic plasticity between retinal cones and luminosity-type horizontal cells. Sheng Li Xue Bao 58: 407–414. [PubMed] [Google Scholar]
- 11. Jiang XD, Wang XL, Sun Y, Gong HQ, Liang PJ (2008) NMDA modulation of GABA transporter current in carp retinal horizontal cells. Brain Res 1240: 105–110. [DOI] [PubMed] [Google Scholar]
- 12. Kreitzer MA, Andersen KA, Malchow RP (2003) Glutamate modulation of GABA transport in retinal horizontal cells of the skate. J Physiol 546: 717–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Linn CL, Gafka AC (2001) Modulation of a voltage-gated calcium channel linked to activation of glutamate receptors and calcium-induced calcium release in the catfish retina. J Physiol 535: 47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu T, Yang XL (1994) Carp retinal horizontal cells: dissociation, morphology and physiological characteristics. Chinese Journal of Neuroanatomy 11: 299–306. [Google Scholar]
- 15. Fabiato A, Fabiato F (1977) Calcium release from the sarcoplasmic reticulum. Circ Res 40: 119–129. [DOI] [PubMed] [Google Scholar]
- 16. Neering IR, McBurney RN (1984) Role for microsomal Ca storage in mammalian neurones? Nature 309: 158–160. [DOI] [PubMed] [Google Scholar]
- 17. Hayashida Y, Yagi T, Yasui S (1998) Ca2+ regulation by the Na+/Ca2+ exchanger in retinal horizontal cells depolarized by L-glutamate. Neurosci Res 31: 189–199. [DOI] [PubMed] [Google Scholar]
- 18. Jenden DJ, Fairhurst AS (1969) The pharmacology of ryanodine. Pharmacol Rev 21: 1–25. [PubMed] [Google Scholar]
- 19. Sutko JL, Airey JA, Welch W, Ruest L (1997) The pharmacology of ryanodine and related compounds. Pharmacol Rev 49: 53–98. [PubMed] [Google Scholar]
- 20. Meissner G (1986) Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J Biol Chem 261: 6300–6306. [PubMed] [Google Scholar]
- 21. Okada T, Schultz K, Geurtz W, Hatt H, Weiler R (1999) AMPA-preferring receptors with high Ca2+ permeability mediate dendritic plasticity of retinal horizontal cells. Eur J Neurosci 11: 1085–1095. [DOI] [PubMed] [Google Scholar]
- 22. Yang XL (2004) Characterization of receptors for glutamate and GABA in retinal neurons. Prog Neurobiol 73: 127–150. [DOI] [PubMed] [Google Scholar]
- 23. Huang SY, Liang PJ (2005) Ca2+-permeable and Ca2+-impermeable AMPA receptors coexist on horizontal cells. Neuroreport 16: 263–266. [DOI] [PubMed] [Google Scholar]
- 24. Shen Y, Zhang M, Jin Y, Yang XL (2006) Functional N-methyl-D-aspartate receptors are expressed in cone-driven horizontal cells in carp retina. Neurosignals 15: 174–179. [DOI] [PubMed] [Google Scholar]
- 25. Tachibana M (1981) Membrane properties of solitary horizontal cells isolated from goldfish retina. J Physiol 321: 141–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tachibana M (1983) Ionic currents of solitary horizontal cells isolated from goldfish retina. J Physiol 345: 329–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yagi T, Kaneko A (1988) The axon terminal of goldfish retinal horizontal cells: a low membrane conductance measured in solitary preparations and its implication to the signal conduction from the soma. J Neurophysiol 59: 482–494. [DOI] [PubMed] [Google Scholar]
- 28. Tachibana M, Okada T, Arimura T, Kobayashi K, Piccolino M (1993) Dihydropyridine-sensitive calcium current mediates neurotransmitter release from bipolar cells of the goldfish retina. J Neurosci 13: 2898–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Putney JW Jr (1986) A model for receptor-regulated calcium entry. Cell Calcium 7: 1–12. [DOI] [PubMed] [Google Scholar]
- 30. Parekh AB, Putney JW (2005) Store-operated calcium channels. Physiol Rev 85: 757–810. [DOI] [PubMed] [Google Scholar]
- 31. Thastrup O, Cullen PJ, Drøbak B, Hanley MR, Dawson AP (1990) Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci U S A 87: 2466–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bootman MD, Collins TJ, MacKenzie L, Roderick HL, Berridge MJ, et al. (2002) 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J 16: 1145–1150. [DOI] [PubMed] [Google Scholar]
- 33. Wu D, Zhu PH (1999) Caffeine-sensitive Ca2+ stores in carp retinal bipolar cells. Neuroreport 10: 3897–3901. [DOI] [PubMed] [Google Scholar]
- 34. Morton-Jones RT, Cannell MB, Housley GD (2008) Ca2+ entry via AMPA-type glutamate receptors triggers Ca2+-induced Ca2+ release from ryanodine receptors in rat spiral ganglion neurons. Cell Calcium 43: 356–366. [DOI] [PubMed] [Google Scholar]
- 35. Daher JPL, Gover TD, Moreira THV, Lopes VGS, Weinreich D (2009) The identification of a caffeine-induced Ca2+ influx pathway in rat primary sensory neurons. Mol Cell Biochem 327: 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walz B, Baumann O, Zimmermann B, Ciriacy-Wantrup EV (1995) Caffeine- and ryanodine-sensitive Ca2+-induced Ca2+ release from the endoplasmic reticulum in honeybee photoreceptors. J Gen Physiol 105: 537–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nohmi M, Hua SY, Kuba K (1992) Basal Ca2+ and the oscillation of Ca2+ in caffeine-treated bullfrog sympathetic neurones. J Physiol 450: 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Friel D (1995) [Ca2+]i oscillations in sympathetic neurons: an experimental test of a theoretical model. Biophys J 68: 1752–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Putney JW Jr (2003) Capacitative calcium entry in the nervous system. Cell Calcium 34: 339–344. [DOI] [PubMed] [Google Scholar]
- 40. Elliott AC (2001) Recent developments in non-excitable cell calcium entry. Cell Calcium 30: 73–93. [DOI] [PubMed] [Google Scholar]
- 41. Liou J, Kim ML, Do Heo W, Jones JT, Myers JW, et al. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15: 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, et al. (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169: 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vig M, Peinelt C, Beck A, Koomoa D, Rabah D, et al. (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312: 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang SL, Yeromin AV, Zhang XHF, Yu Y, Safrina O, et al. (2006) Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci U S A 103: 9357–9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, et al. (2006) Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443: 226–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smyth JT, DeHaven WI, Jones BF, Mercer JC, Trebak M, et al. (2006) Emerging perspectives in store-operated Ca2+ entry: Roles of Orai, Stim and TRP. Biochim Biophys Acta 1763: 1147–1160. [DOI] [PubMed] [Google Scholar]
- 47. Huang GN, Zeng W, Kim JY, Yuan JP, Han L, et al. (2006) STIM1 carboxyl-terminus activates native SOC, Icrac and TRPC1 channels. Nat Cell Biol 8: 1003–1010. [DOI] [PubMed] [Google Scholar]
- 48. Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, et al. (2006) Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat Cell Biol 8: 771–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S (2007) STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol 9: 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carrasco S, Meyer T (2010) Cracking CRAC. Nat Cell Biol 12: 416–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong J, Kim M, Lee K, Yuan J, Muallem S (2012) STIM-TRP pathways. In: Groschner K, Graier WF, Romanin C, editors. Store-operated Ca2+ entry (SOCE) pathways. Vienna: Springer. 57–72.
- 52.Flockerzi V, Nilius B (2007) Transient receptor potential (TRP) Channels. Verlag Berlin Heidelberg: Springer. 623 p. [Google Scholar]
- 53. Ramsey IS, Delling M, Clapham DE (2006) An introduction to TRP channels. Annu Rev Physiol 68: 619–647. [DOI] [PubMed] [Google Scholar]
- 54. Vaca L (2010) SOCIC: the store-operated calcium influx complex. Cell Calcium 47: 199–209. [DOI] [PubMed] [Google Scholar]
- 55. Manjarrés IM, Rodríguez-García A, Alonso MT, García-Sancho J (2010) The sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) is the third element in capacitative calcium entry. Cell Calcium 47: 412–418. [DOI] [PubMed] [Google Scholar]
- 56. Manjarrés IM, Alonso MT, García-Sancho J (2011) Calcium entry-calcium refilling (CECR) coupling between store-operated Ca2+ entry and sarco/endoplasmic reticulum Ca2+-ATPase. Cell Calcium 49: 153–161. [DOI] [PubMed] [Google Scholar]
- 57. Alonso MT, Manjarrés IM, García-Sancho J (2012) Privileged coupling between Ca2+ entry through plasma membrane store-operated Ca2+ channels and the endoplasmic reticulum Ca2+ pump. Mol Cell Endocrinol 353: 37–44. [DOI] [PubMed] [Google Scholar]
- 58. Bai D, del Corsso C, Srinivas M, Spray DC (2006) Block of specific gap junction channel subtypes by 2-aminoethoxydiphenyl borate (2-APB). J Pharmacol Exp Ther 319: 1452–1458. [DOI] [PubMed] [Google Scholar]
- 59. Mignen O, Brink C, Enfissi A, Nadkarni A, Shuttleworth TJ, et al. (2005) Carboxyamidotriazole-induced inhibition of mitochondrial calcium import blocks capacitative calcium entry and cell proliferation in HEK-293 cells. J Cell Sci 118: 5615–5623. [DOI] [PubMed] [Google Scholar]
- 60. Kamermans M, Fahrenfort I, Schultz K, Janssen-Bienhold U, Sjoerdsma T, et al. (2001) Hemichannel-mediated inhibition in the outer retina. Science 292: 1178–1180. [DOI] [PubMed] [Google Scholar]
- 61. Klaassen LJ, Fahrenfort I, Kamermans M (2012) Connexin hemichannel mediated ephaptic inhibition in the retina. Brain Res 1487: 25–38. [DOI] [PubMed] [Google Scholar]
- 62. DeVries S, Schwartz E (1992) Hemi-gap-junction channels in solitary horizontal cells of the catfish retina. J Physiol 445: 201–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang DQ, McMahon DG (2001) Gating of retinal horizontal cell hemi gap junction channels by voltage, Ca2+, and retinoic acid. Mol Vis 7: 247–252. [PubMed] [Google Scholar]
- 64. Sun Z, Zhang DQ, McMahon DG (2009) Zinc modulation of hemi-gap-junction channel currents in retinal horizontal cells. J Neurophysiol 101: 1774–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun Z, Risner ML, van Asselt JB, Zhang DQ, Kamermans M, et al. (2012) Physiological and molecular characterization of connexin hemichannels in zebrafish retinal horizontal cells. J Neurophysiol 107: 2624–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Malchow R, Qian H, Ripps H (1993) Evidence for hemi-gap junctional channels in isolated horizontal cells of the skate retina. J Neurosci Res 35: 237–245. [DOI] [PubMed] [Google Scholar]
- 67. Sitsapesan R, Williams AJ (1990) Mechanisms of caffeine activation of single calcium-release channels of sheep cardiac sarcoplasmic reticulum. J Physiol 423: 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Byrne N, Large W (1988) Membrane ionic mechanisms activated by noradrenaline in cells isolated from the rabbit portal vein. J Physiol 404: 557–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Currie KP, Scott RH (1992) Calcium-activated currents in cultured neurones from rat dorsal root ganglia. Br J Pharmacol 106: 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Marrion NV, Adams PR (1992) Release of intracellular calcium and modulation of membrane currents by caffeine in bull-frog sympathetic neurones. J Physiol 445: 515–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schoppe J, Hochstrete P, Schlue WR (1997) Caffeine mediates cation influx and intracellular Ca2+ release in leech P neurones. Cell Calcium 22: 385–397. [DOI] [PubMed] [Google Scholar]
- 72. Weiler R, Schultz K, Janssen-Bienhold U (1996) Ca2+-dependency of spinule plasticity at dendrites of retinal horizontal cells and its possible implication for the functional role of spinules. Vision Res 36: 3891–3900. [DOI] [PubMed] [Google Scholar]
- 73. McMahon DG, Mattson MP (1996) Horizontal cell electrical coupling in the giant danio: synaptic modulation by dopamine and synaptic maintenance by calcium. Brain Res 718: 89–96. [DOI] [PubMed] [Google Scholar]
- 74. Kass G, Orrenius S (1999) Calcium signaling and cytotoxicity. Environ Health Perspect 107: 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sun Y, Olson R, Horning M, Armstrong N, Mayer M, et al. (2002) Mechanism of glutamate receptor desensitization. Nature 417: 245–253. [DOI] [PubMed] [Google Scholar]
- 76. Park CY, Shcheglovitov A, Dolmetsch R (2010) The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330: 101–105. [DOI] [PubMed] [Google Scholar]
- 77. Akopian A, Krizaj D, Witkovsky P (1997) Both high- and low voltage-activated calcium currents contribute to the light-evoked responses of luminosity horizontal cells in the Xenopus retina. Brain Res 762: 121–130. [DOI] [PubMed] [Google Scholar]
- 78. Normann R, Pochobradský J (1976) Oscillations in rod and horizontal cell membrane potential: evidence for feed-back to rods in the vertebrate retina. J Physiol 261: 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]