

Abstract
To achieve a long lifespan, animals must be resistant to various injuries as well as be able to avoid or delay lethality from age-dependent diseases. Here we show that long-lived Mclk1+/− mutants have enhanced resistance to neurological damage following global cerebral ischemia/reperfusion (I/R) injury induced by transient bilateral common carotid artery occlusion (BCCAO). Both young (~100 days old) and relatively aged (~450 days old) mutants display increased resistance as indicated by a significant decrease in the amount of degenerating cells observed in forebrain cortex and in hippocampal areas after ischemia and reperfusion. Furthermore, less oxidative damage resulting from the procedure was measured in the brain of young Mclk1+/− animals. The finding that both young and old mutants are protected indicates that this is a basic phenotype of these mutants and not a secondary consequence of their slow rate of aging. Thus, the partial resistance to I/R injury by enhancing recovery from age-dependent vascular accidents is likely part of what allows for the increased lifespan of Mclk1+/− mutants. By relating this neuroprotective effect to previously reported characteristics of the Mclk1+/− phenotype, including altered mitochondrial metabolism and increased HIF-1α expression, this study establishes these mutants as useful models to analyze the mechanisms underlying tolerance to ischemia, particularly those associated with ischemic preconditioning, as well as to clarify the relation between aging and age-dependent diseases.
Keywords: Mclk1, Ischemia, Reperfusion, Aging, Age-related diseases, Ischemic tolerance, BCCAO, Oxidative stress
Introduction
CLK-1/MCLK1 is a mitochondrial hydroxylase that is necessary for the biosynthesis of ubiquinone (coenzyme Q or UQ), the essential electron transporter of the mitochondrial respiratory chain that is also known for its crucial antioxidant properties (Levavasseur, et al., 2001). We have previously reported that inactivation of clk-1 in Caenorhabditis elegans (Wong, et al., 1995), and partial inactivation of its orthologue in Mclk1+/− mice (Liu, et al., 2005), resulted in a significantly prolonged lifespan. By studying various aspects of the mitochondrial and metabolic phenotype of the long-lived Mclk1+/− mutants, we have found that mitochondria of young Mclk1+/− mice display slow electron transport, contain low levels of ATP and sustain high oxidative stress (Lapointe and Hekimi, 2008). Despite the early mitochondrial dysfunction, the function of Mclk1+/− mitochondria declines less rapidly with age than that of the wild type and there is a slower accumulation of global oxidative biomarkers of aging in these mutants (Lapointe, et al., 2009). Furthermore, we succeeded in demonstrating a causal link between the altered mitochondrial function of young mutants and the improved age-dependent phenotypes of aged mutants, by showing that the Mclk1+/− condition fully prevents the accelerated deterioration of mitochondrial function and the increased mitochondrial oxidative stress observed in Sod2+/− mutants (Lapointe, et al., 2009). Recently, we reported that the altered mitochondrial phenotype of Mclk1+/− mutants modulates the immune response by enhancing basal and stimulated expression of HIF-1α in liver and macrophages, in association with elevated expression of inflammatory cytokines (Wang and Hekimi, 2009).
To increase lifespan, it is generally assumed that a genetic manipulation such as the reduction of Mclk1 expression must be able to reduce the prevalence or severity of diseases that normally limit lifespan (Hekimi, 2006). This could be achieved by slowing down the development of the cellular dysfunctions that result from the aging process (e.g. loss of mitochondrial function, increased oxidative stress, or chronic inflammation), and that typically exacerbate a number of diseases including endothelial dysfunction and stroke. These kinds of diseases are frequently called age-dependent or age-associated diseases. A prolongation of lifespan could also be observed if the mutants were simply resistant to an age-dependent pathology without necessarily having induced a change in the age-dependency of the pathology. To address these questions in the Mclk1+/− mutant context, we have tested here whether both young and old animals showed some degree of tolerance to cerebral ischemic-reperfusion (I/R) injury, which is linked to severe diseases connected to aging, mitochondrial function, oxidative stress and immune function.
When blood flow to a tissue is interrupted or severely reduced as occurs during ischemia, the essential oxygen supply needed to maintain cellular homeostasis becomes insufficient. This can arise in a variety of situations, in particular as a consequence of obstruction of arteries, with the heart and the brain have been found to be the most commonly affected (Doyle, et al., 2008, Lesnefsky and Hoppel, 2003). If tissue ischemia occurs for a prolonged period, cell death is the main consequence, but, even when the ischemic episode is relatively brief, the reestablishment of blood flow at the time of reperfusion and the resulting oxygen delivery is known to induce a highly harmful process that leads to cellular damage and cell death (Warner, et al., 2004). Indeed, I/R brain injury is similar to the pathophysiology of stroke and brain trauma, which are among the leading causes of death and disability in people (Rosamond, et al., 2007). The vascular obstructions at the origin of ischemic insults are typically the consequence of age-dependent processes such as atherosclerosis and heart arrhythmias, and are also strongly favored by endothelial dysfunctions and general vascular deterioration that are hallmarks of aging (Matsumoto, et al., 2007, Park, et al., 2007, Schaller, 2007, Westrick, et al., 2007). Thus, the prevalence of I/R injury as well as its outcome have been shown to be profoundly influenced by age (Yager, et al., 2005). It is not yet well understood how exactly I/R episodes act to damage cells. However, it has been demonstrated that it involves mitochondria and particularly the toxic properties of the mitochondrial reactive oxygen species (ROS) (Fiskum, et al., 2004, Friberg, et al., 2002, Lesnefsky, et al., 2001).
To test whether the long life of Mclk1+/− mutants involved a resistance to the damaging processes underlying I/R injury we used a surgical procedure, transient bilateral common carotid occlusion (BCCAO), to induce cerebral I/R in young and old animals and scored the resultant neuronal damage. We find that the mutants have an enhanced resistance to I/R at all ages as revealed by decreased levels of degenerating cells and less oxidative damage after the procedure. This indicates that the resistance is due to the basic changes in physiology induced by the reduction in MCLK1 levels, but not to the fact that Mclk1+/− and Mclk1+/+ animals age at different rates (Lapointe, et al., 2009). Thus our study suggests that a loss-of-function phenotype can contribute to the longer survival by increasing the intrinsic resistance to injurious age-related physiological processes.
Materials and methods
Animals
Heterozygous Mclk1 mutant mice (Mclk1+/−) and their wild type littermates (Mclk1+/+) were produced as previous reported (Levavasseur, et al., 2001, Liu, et al., 2005), and were maintained in the C57BL/6J and Balb/c backgrounds by inbreeding. A total of 62 male mice in the C57BL/6J background (young animals) and 56 female mice in the Balb/c background (aged animals) were used. Experimental groups with their corresponding sample sizes are listed in Table 1. All procedures were approved by McGill’s Animal Care and Ethics committees.
Table 1.
Experimental groups and animals
| Groups | Controls (sham operated) | BCCAO operated | Average age (days) | Body weight (grams) | |
|---|---|---|---|---|---|
| Young animals (Males/C57BL/6) | Mclk1 +/+ | 11 | 20 | 107 ± 13 | 27.9 ± 2.7 |
| Mclk1 +/− | 11 | 20 | 104 ± 12 | 27.1 ± 2.7 | |
|
| |||||
| Aged animals (Females/Balb/c) | Mclk1 +/+ | 12 | 18 | 548 ± 10 | 28.6 ± 3.6 |
| Mclk1 +/− | 9 | 15 | 550 ± 16 | 28.7 ± 3.9 | |
Global cerebral ischemia protocol
Experimental animals were subjected to transient global ischemia by bilateral common carotid artery occlusion (BCCAO), a well-defined I/R procedure for mimicking cerebrovascular accidents, including in transgenic mice models (Cho, et al., 2007, Kelly, et al., 2001). Before the operation, mice were anesthetized with a specific ketamine cocktail containing 50% ketamine, 25% xylazine, 10% acepromazine and 15 % saline by intraperitoneal injection using 1μl per mg body weight. Body temperature was strictly kept at 37 ± 0.5°C by a feedback homeothermic blanket with a rectal probe (Harvard Apparatus, South Natick, MA). A midline skin incision on the ventral surface of the neck was made. The salivary glands were moved laterally, and the carotid sheath exposed bilaterally. The common carotid arteries were then carefully separated from adjacent vagus and sympathetic nerves with a microdissector and forceps. Microvascular clips (B-2, Fine Science Tools) were applied to both isolated common carotid arteries in order to block brain blood flow for the desired time: 10 minutes for young mice, 5 minutes for aged mice. After the operation, the animals were kept on the heating blanket until they awoke from anesthesia, and then transferred to their home cage at room temperature. Control animals (sham operated) underwent the same operation procedures but were not subjected to the carotid occlusion. Aged animals were treated with only 5 minutes of ischemia as it was found that longer treatment resulted in inacceptable number of deaths before 24 hours of reperfusion. Animals that died within less than 24 hours after the beginning of reperfusion were excluded.
Cerebral blood flow determination
Cerebral blood flow changes were monitored by a Doppler flow meter (BLF21, Transonic Systems Inc, Ithaca, NY). The flow meter was connected to a data acquisition systems (MP150, BIOPAC Systems Inc, Goleta, CA), which is itself connected to a computer and operated by the Acknowledge 3.8 software. A needle probe was connected to the flow meter and held by a micromanipulator. In order to measure brain blood flow, the anesthetized mouse was put in a prone position. A 0.5 cm crossline incision was then made and the skull was exposed. The calibrated flow meter was warmed up for 10 minutes and the probe was put on the skull surface at the middle right hemisphere. Brain blood flow was measured for 10 seconds at 3 time points: before ischemia, after 5 minutes arteries occlusion (young mice) or 3 minutes (aged mice), and 5 minutes post-ischemia, to record normal blood flow, blood flow reduction during occlusion, and brain blood flow resumption after ischemia, respectively. Cerebral blood flow was reported as ml/mg/min. Mean blood flow during the 10 seconds recording was used for each measurement.
Histopathology
After 24 hours reperfusion, animals were anesthetized by using the previously described ketamine cocktail and sacrificed by neck dislocation. Brain was washed by transcardiac infusion of saline, then carefully extracted from the skull and cut into two halves through the midline. The left hemisphere was snap frozen in liquid nitrogen and stored at −80 °C for further biochemical studies. The right hemisphere was fixed in 4% paraformaldehyde overnight, and then cut coronally into 4 blocks at preselected levels which are located at +1.5 mm from bregma (layer 1), bregma (layer 2), −1.5 mm from bregma (layer 3) and −3 mm bregma (layer 4), before being embedded in paraffin. Coronal sections of 4 μm thicknesses were then generated and subsequently stained with hematoxylin/eosin (HE).
For estimating brain pathological changes following I/R, we have used 4 HE stained sections per preselected level. To quantify brain damage, degenerated cells were counted using a computer assisted image analysis system (ImagePro). Degenerated neuronal cells were identified according to their pyknotic and irregular nuclei using color segmentation and size thresholding. Manual editing was performed when necessary to ensure precise cell selection. Total neuronal cells and degenerating cells were counted in fields of 1492.7μm2. Twelve non-adjacent fields were selected: 8 fields from the forehead cortex, 2 fields from the CA3 region of the hippocampus; and another 2 fields from region CA1.
Isolation of mitochondria
Non-synaptic brain mitochondria were isolated according to published methods (Lai and Clark, 1979). Briefly, the frozen cerebral tissue (about 80 mg) was homogenized in 10 volumes of homogenization buffer using an overhead electric homogenizer. The homogenization buffer contained 10 mM Hepes pH 7.4, 250 mM sucrose, 1 mM EDTA, 225 mM mannitol and 0.1% BSA. All buffers were kept at 4°C. The homogenate was centrifuged at 600 xg for 10 minutes at 4°C. The supernatant was collected, transferred to another tube and centrifuged at 13,000 xg for 10 minutes at 4°C. This supernatant was collected as cytosol. The crude mitochondria pellet was resuspended in 1 ml of 3% dialyzed Ficoll 400 (Sigma), and carefully laid on top of the 4 ml 6% Ficoll before being centrifuged at 11,500 xg for 30 minutes at 4 °C. After discarding the supernatant, the mitochondria pellet was resuspended in 1 ml of 10 mM K2HPO4 buffer pH 7.4, and centrifuged at 13,000 xg for 10 minutes at 4°C. The non-synaptic mitochondria pellet was then resuspended in an appropriate K2HPO4 buffer to adjust the final protein concentration to 2–3 mg/ml.
Enzymatic activities
Αlpha-Ketoglutarate dehydrogenase (α-KGDH) activity was assayed spectrophotometrically by measuring the rate of increase of absorbance at 340 nm due to NADH formation from NAD. The assay mixture contained 5.0 mM MgCL2, 40 μM rotenone, 5 mM α-ketoglutarate, 0.2 mM thymine pyrophosphate (TPP), 1.0 mM nicotinamide adenine dinucleotide (NAD), and 0.3 mM dithiothreitol (DTT) in 30 mM K2HPO4 buffer pH 7.4. Frozen-thawed mitochondria (50μg protein/ml) or cytosol (100μg protein/ml) were used in the measurements. The reaction was started by addition of 0.1 mM CoA. The initial rate corresponding to the first 3 minutes was measured. The enzyme activity was defined as the formation of NADPH from NAD in 1 mg protein for 1 minute. Aconitase activity from mitochondrial and cytosolic extracts was determined by using a standard method (Gardner, 2002) with some modifications. Prior to the analysis, 1 mM sodium citrate and 1 mM succinate were added to mitochondria and cytosol pools to protect the sensitive aconitase from inactivation. Thus, specific activity was measured by monitoring absorbance at 340 nm in a freshly prepared reaction mix containing 0.2 mM NADP+, 5 mM sodium citrate, 2 U/ml isocitrate dehydrogenase and 0.6 mM MnCl2, in 50 mM Tris-HCl pH 7.4. The assay was started by the addition of freshly thawed aliquots of mitochondria (50μg protein/ ml) or cytosol (100μg protein/ml) to the reaction mix which was previously equilibrated at 25 °C. Absorbance at 340 nm was recorded every 30 seconds for 10 minutes, and aconitase activity was calculated from the linear increase in absorbance at 340 nm. One unit of enzyme activity was defined as the amount of enzyme catalyzing the production of 1 nmole of NADH per mg protein.
The TBARS assay
Brain tissue TBARS was measured using the Oxltek TBARS assay kit (ZeptoMetrix, Buffalo, NY). The brain tissue (75–80 mg) was homogenized in 0.4 ml homogenizing buffer, and was then centrifuged at 1000 xg for 10 minutes. Protein concentration of the supernatant was measured and TBARS amount in the supernatant was assayed according to the manufacturer’s instructions.
Results
Physiological and hemodynamic effects of BCCAO and reperfusion
To assess whether reduced MCLK1 expression exerts protective effects against ischemia/ reperfusion injury, global ischemia was induced by bilateral common carotid artery occlusion (BCCAO) for either 10 minutes (young animals) or 5 minutes (old animals) by temporary occlusion of both vessels by microvascular clips. A sham BCCAO control group consisted of mice treated identically except for not being subjected to the carotid occlusion (see Materials and Methods). Most animals recovered well by 24 hours after brain ischemia and subsequent reperfusion, exhibiting normal respiration, normal movement and normal reaction to stimulation. In the experimental groups containing young animals, we have observed that a few of them were in a critical condition, exhibiting slow respiration, unsteady walking and dull reaction to stimulation. Five such critical mice were observed among the 20 Mclk1+/+ mice, but none among the 20 Mclk1+/− mice. This difference was found to be significant (p<0.05).
In the groups of young animals, the average brain blood flow was reduced to around 5% of normal after 5 minutes of BCCAO, and returned to normal or near normal levels after 5 minutes reperfusion. The brain blood flow reduction after occlusion and reperfusion was not significantly different between Mclk1+/+ and Mclk1+/− mice (Table 2). In aged mice, the blood flow was reduced to about 10% of normal level after 3 minutes BCCAO, and resumed to about 95% of normal at 5 minutes after reperfusion, with again no significant differences between genotypes (Table 2).
Table 2.
Brain blood flow changes measured after ischemia (5 minutes for young animals and 3 minutes for old animals) and after reperfusion (5 minutes).
| After ischemia (% of pre-ischemia level) | After reperfusion (% of pre-ischemia level) | ||
|---|---|---|---|
| Young animals | Mclk1 +/+ | 4.7 ± 1.7 | 93.6 ± 17 |
| Mclk1 +/− | 5.5 ± 2.2 | 97.0 ± 13 | |
|
| |||
| Aged animals | Mclk-1 +/+ | 9.7 ± 6.2 | 93.5 ± 14 |
| Mclk-1 +/− | 9.1 ± 5.6 | 95.4 ± 11 | |
Edema could increase the weight of the brains after the procedure. To check whether differences in brain edema could be a factor in any other difference observed between the experimental groups, brain weight changes after I/R were determined by weighing whole brains including cerebrum and cerebellum. The average brain/body weight ratios were similar among all groups in young and aged mice, with the average brain weight slightly above 1.6% of total body weight (not shown). Thus brain weight neither increased after ischemia injury nor differed between Mclk1+/− mutants and Mclk1+/+ controls siblings.
The Mclk1+/− genotype alleviates neuronal damage after I/R insults in several brain areas
Sham operated Mclk1+/+ and Mclk1+/− control animals were subjected to histological analysis of the different areas of the hippocampus, principally the CA1 and the CA3 regions, and the forebrain cortex following the same experimental procedure as for BCCAO but without coronary occlusion. These analyses of sham operated mice have not revealed any differences at the morphological level as well as in terms of neurological damage between genotypes in 3 or 15 months old mice (Fig. 1–4). However, both young and old animals of these control groups showed some degenerating cells in one or several areas analyzed. These cells were mostly isolated single cells scattered throughout the different brain regions (Fig. 1; Fig. 3). In contrast, after ischemia and 24 hours reperfusion (I/R), hematoxylin/eosin staining reveals many of these degenerating neurons, characterized by their irregular cell contours, acidophilic cytoplasm as well as by shrunken and triangularly shaped (pyknotic) nuclei (Fig.1; Fig. 3). As expected (Hsu, et al., 1994), we observed that the most severely affected areas were the forebrain cortex and the hippocampus, including areas CA1 and CA3, which were thus further characterized in details. In the cortex, degenerating cells were most frequently observed in layer II, IV and V cortex neuronal cells either in a scattered manner or in patches (Fig. 1-J, L; Fig. 3-J, L). Small cortex infarctions were occasionally encountered in the most severely affected animals and necrotic neuronal cells could be seen in the infarcted areas. Cells from the striatum region were normal in most animals (not shown). Marked degeneration of the sensitive pyramidal neurons of the CA1 and the CA3 regions of the hippocampus was observed in young (males in the C57BL/6 background) (Fig. 1) and old (females in the Balb/c background) animals from both genotypes after BCCAO and reperfusion episodes (Fig. 3).
Figure 1.

Comparison of BCCAO-induced neuronal damage in hippocampus and cerebral cortex of young Mclk1+/+ and Mclk1+/− mice. The panels shown for both groups are representative hematoxylin/eosin-stained slides of the CA1 (A–D) and the CA3 (E–H) regions of the hippocampus and of a portion of the cerebral cortex (I–L). Sections of control (sham operated) and ischemia-reperfused animals were presented for each analyzed region (scale bar, 50 μm).
Figure 4.
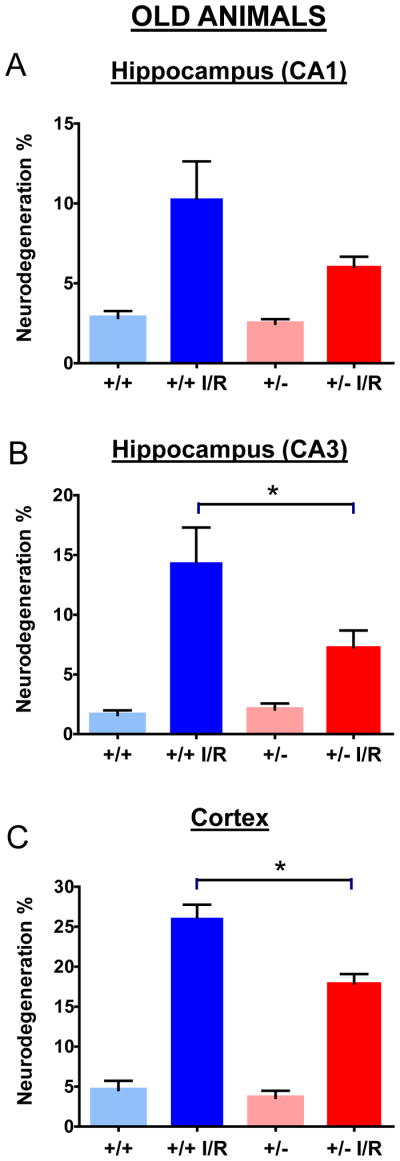
Quantification of neurodegeneration in hippocampus and cerebral cortex of old Mclk1+/+ and Mclk1+/− mice. Neurological damage was determined in CA1 (A), CA3 (B) as well as in the cortex (C) of control (sham operated) and ischemia-reperfused (I/R) animals of both groups. Data are means ± SEM of 9–18 animals. * denotes statistical significance of the differences between Mclk1+/+ and Mclk1+/− animals following BCCAO, P < 0.05.
Figure 3.

Comparison of BCCAO-induced neuronal damage in the hippocampus and cerebral cortex of old Mclk1+/+ and Mclk1+/− mice. The panels shown for both groups are representative hematoxylin/eosin-stained slides of the CA1 (A–D) and the CA3 (E–H) regions of the hippocampus and of a portion of the cerebral cortex (I–L). Sections of controls (sham operated) and ischemia-reperfused animals are presented for each region analyzed (scale bar, 50 μm).
We have thus quantified the damage in the forebrain cortex and in hippocampal CA1 and CA3 regions by counting the number of degenerating cells based on nuclear color and morphology as described in the Materials and Methods. No significant differences were found between the genotypes in sham operated animals (Fig. 2; Fig. 4). For both genotypes and ages, the I/R procedure has resulted in significant increases in degenerating cells when compared to controls (Fig. 2; Fig. 4). There was a somewhat lower level of damage in old animals, presumably because our experimental protocol with these animals involved only 5 instead of 10 minutes of ischemia to reduce animal loss because of their age (see Materials and Methods). In the cortex and CA3 areas of young Mclk1+/+ mice the I/R insult resulted in significantly higher levels of cell death than in the equivalent Mclk1+/− mutant mice (Fig. 2B-C). Interestingly, this protective effect which seems to be conferred by reduced MCLK1 levels, is conserved in old animals where less damage is also observed in the cortex and the CA3 region of the hippocampus of the mutants (Fig. 3B-C). Despite the fact that the difference in the percentage of neurodegeneration did not reach significance between genotypes, the same trend was also observed in the CA1 area of the hippocampus at both ages (Fig. 2A; Fig. 4A). Regional differences in the severity of damage after BCCAO or the level of protection conferred by particular treatments are a common observation (Cho, et al., 2007, Jeffs, et al., 2008). It is also well known that neurons from the CA1 region of the dorsal hippocampus are highly vulnerable to I/R insult while the neurons in the neighbouring CA3 region are relatively spared (Hsu, et al., 1994). We observed here that significant pyramidal cell death indeed occurred after I/R in these areas of the hippocampus from both Mclk1+/+ and Mclk1+/−, but our results rather revealed that the neurons from the CA3 region are the mostly affected at all ages (Fig. 2; Fig. 4).
Figure 2.
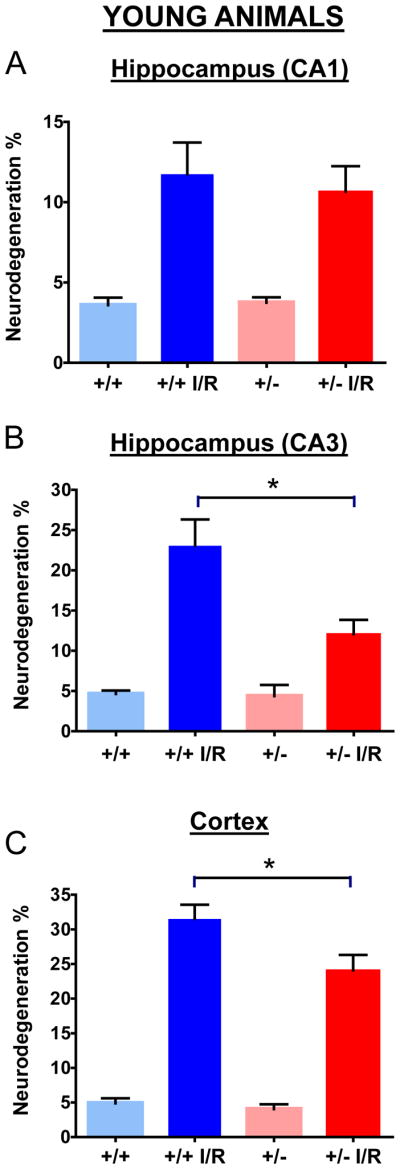
Quantification of neurodegeneration in hippocampus and cerebral cortex of young Mclk1+/+ and Mclk1+/− mice. Neurological damage was determined in CA1 (A), CA3 (B) as well as in the cortex region (C) of control (sham operated) and ischemia-reperfused (I/R) animals of both groups. Data are means ± SEM of 11–20 animals. * denotes statistical significance of the differences between Mclk1+/+ and Mclk1+/− animals following BCCAO, P < 0.05.
Reduced mitochondrial oxidative stress following I\R in Mclk1+/− mutants
Ischemia/reperfusion injury to cells does not necessarily result only in cellular mortality but might transiently or permanently damage cells without leading to their death. MCLK1 is a mitochondrial protein (Jiang, et al., 2001, Levavasseur, et al., 2001) and reduction in its levels profoundly affects mitochondrial function and oxidative stress (Lapointe and Hekimi, 2008). We therefore tested a number of parameters linked to oxidative stress in the brain after the I/R procedure. Due to the limited amount of material available, we decided to primarily evaluate recognized oxidative stress markers: the enzymatic activities of cytoplasmic and mitochondrial aconitase, mitochondrial α-ketoglutarate dehydrogenase (α-KGDH), and the level of thiobarbituric acid reactive substances (TBARS). The activity of aconitase is considered a particularly sensitive measure of oxidative stress (Gardner, 2002). α-KGDH also is susceptible to oxidative stress and compensatory increases in its activity can result from oxidative stress (Tretter and Adam-Vizi, 2005). TBARS are a reliable measure of lipid oxidation products (Del Rio, et al., 2005). In young Mclk1+/+ animals, the enzymatic activities of both aconitases (cytoplasmic and mitochondrial) and of α-KGDH were reduced whereas TBARS levels were found to be increased after the I/R episode, thus indicating that the I/R protocol resulted in significant level of oxidative stress, presumably in surviving cells (Table 3). The Mclk1+/− genotype however appeared to lessen or abolish these deleterious changes in young animals (Table 3). In old animals, I/R had an identical effect on both genotypes (Table 3). Based on these data, the differences between the observations in the two age groups suggest that any increased resistance to oxidative stress is not the basis of the mutant’s increased resistance to cerebral I/R. Additional measures will however be needed to confirm this hypothesis.
Table 3.
Mitochondrial oxidative stress-related measurements
| Control (sham operated) | Ischemia/Reperfusion | |||||||
|---|---|---|---|---|---|---|---|---|
| Mclk1+/+ | Mclk1+/− | Mclk1+/+ | Mclk1+/− | |||||
| Young | Old | Young | Old | Young | Old | Young | Old | |
|
| ||||||||
| Mitochondrial Aconitase activity (nmol/mg/min) | 4.7a ± 0.6 | 7.5 ± 1.1 | 4.8 ± 0.6 | 8.4 ± 1.1 | 3.1 ± 0.6 | 3.6 ± 0.7 | 5.6 ±0.7* | 2.5 ± 0.5 |
|
| ||||||||
| Cytosolic Aconitase activity (nmol/mg/min) | 19.3 ± 2.1 | 16.1 ± 0.9 | 22.1 ± 2.5 | 15.7 ± 1 | 14.9 ±1.3 | 14.6 ±1.1 | 19.2±1.5* | 15.4 ±0.8 |
|
| ||||||||
| Mitochondrial α-KGDH activity (nmol/mg/min) | 19.6 ± 1.4 | 18.1 ± 2 | 21.5 ± 1.9 | 19.4 ± 1.6 | 17.3 ±1.6 | 15.2 ± 1.9 | 23.4±1.4* | 16.4 ± 2 |
|
| ||||||||
| Whole brain TBARS (nmol/g) | 2.7 ± 0.3 | n.d. | 2.4 ± 0.3 | n.d. | 4.4 ± 0.5 | n.d. | 3.2± 0.2* | n.d. |
data are means ± SEM of 10–18 samples
significantly different from the corresponding controls (Mclk1+/+) at P < 0.05
Discussion
The incidence and the severity of a large number of important human diseases are known to increase sharply with age (Yager, et al., 2005). These diseases include many neurodegenerative pathologies and cerebral vascular accidents such as stroke and brain trauma. Thus the physiological changes that accompany aging are risk factors for these diseases. For example, aging is accompanied by mitochondrial dysfunction and an increase in oxidative stress that are believed to strongly participate in the development of neurodegenerative (Lin and Beal, 2006) and cerebrovascular diseases (Fiskum, et al., 2004, Soane, et al., 2007, Warner, et al., 2004). However, an alternative view of the relation between aging and age-dependent diseases is possible. In this view, every disease develops independently, and aging is but the collection of all age-dependent diseases and their interactions. This later interpretation would mean that biomarkers of aging, such as oxidative stress damage, might only exist because oxidative stress is a kind of imbalance that is common to several age-dependent diseases. Thus, increased oxidative stress would only be part of what characterizes these diseases, and would not result from aging independently of diseases. It is fair to say that the question of which view provides a more useful paradigm for understanding aging and age-dependent diseases is unresolved (Hekimi, 2006). The same questions arise for long-lived genetic mutants or animals that are long-lived thanks to a particular treatment or diet: does the mutation or the intervention slow down the rate of aging, thus delaying the appearance of age-dependent diseases and consequently increasing lifespan, or does the mutation or intervention protect from one, or a subset, of diseases that reduce survival at any age and prolong lifespan in this way. For example, it was shown that caloric restriction, which is well known to increased lifespan in many species, delays the onset of a variety of age-associated diseases and mortality in rodents and primates (Colman, et al., 2009). In fact, if a mutation in a particular gene or a treatment were to protect from all or most known causes of death it might make the second view, the idea that aging is but a collection of independent age-dependent diseases, rather unlikely, as we could define aging as the process affected by the mutation or the treatment. On the other hand, the finding that a mutation protects from one, or a few related, causes of death is not incompatible with either view. It is particularly true if the mutation protects young as well as old animals from similar injury, as we have found here for Mclk1+/− mutants subjected to I/R.
The level of neuronal degeneration in response to I/R has been shown to be influenced by age, sex and background. Lesser susceptibility to post-ischemic and post-traumatic brain injury in females has been observed in experimental models (Roof and Hall, 2000). Backgrounds can also be different quantitatively in their sensitivity to I/R including BCCAO (Kelly, et al., 2001, Majid, et al., 2000). We have shown here that the partial resistance to I/R induced by the Mclk1+/− genotype is quite broad as it protects both young and aged, male and females, and is independent of genetic background, at least for the two backgrounds tested (C57BL6 and Balb/c). Because the length of the ischemic period was different in the two groups (5 minutes for old animals but 10 minutes for the young ones) we cannot interpret our findings in an exact quantitative manner beyond the observation that all conditions are at least partially protected.
Significantly reduced levels of degenerating cells were observed in the cortex and in the CA3 region of the hippocampus of the Mclk1+/− mice while the reduction that was also observed in the CA1 region did not reach the significance level. Pyramidal neurons of the CA1 region are among the cells most vulnerable to loss of blood supply, but death frequently occurs days after the initial ischemic insult (Kirino, 1982). This phenomenon is termed delayed neuronal death and could explain why no difference between genotype was observed with our experimental protocol, as all the animals were sacrificed 24 hours after reperfusion.
The observation that reduced levels of MCLK1 protect against I/R injuries at different ages is relevant as to the link that exist between mitochondria and cerebral damage following ischemia as well as on the phenomenon of ischemic tolerance. It is indeed now well recognized that mitochondria are the principal targets, and effectors, of the progression of the neurodegenerative process that occurs in the brain with aging as well as after I/R injury (Bertoni-Freddari, et al., 2004, Friberg, et al., 2002, Lesnefsky, et al., 2001). The occurrence of many mitochondrial alterations after I/R but prior to neuronal death have been reported in vivo, including decrease respiration, impairment of the electron transport chain, decrease ATP synthesis, increase ROS production and associated oxidative damage, altered calcium homeostasis, opening of the permeability transition pore and release of proapoptotic factors (Chan, 2001, Nakka, et al., 2008, Rouslin, et al., 1990). Therefore, the majority of the neuroprotective mechanisms that have been studied to enhance brain resistance against I/R actually have been attempts to regulate and protect mitochondrial function. So far, drug therapies that have been elaborated to counteract the deleterious effects of I/R insult have only been modestly effective and the only experimental treatment that truly protects against this kind of injury and induces ischemic tolerance is brain preconditioning (Schaller, 2007). The concept of preconditioning is based on the observation that repeated sublethal ischemic insults are accompanied by the upregulation of protective mechanisms and the concomitant downregulation of pro-degenerative pathways that ultimately confer a partial resistance to ischemic episodes (Schurr, et al., 1986). Of course, this phenomenon will likely never be of clinical use, but it provides insight into the cellular and molecular events implicated in brain protection and is intensely studied with the objective of developing more efficient therapies. In fact, mitochondria were quickly identified as important effectors of preconditioning (Christophe and Nicolas, 2006, Dirnagl and Meisel, 2008). Thus, it seems that repeated short ischemic episodes induce a specific mitochondrial state that contribute to neuroprotection by inducing beneficial cellular mechanisms. Strikingly, the mitochondrial dysfunction phenotype which is normally induced by I/R is highly similar to the one that has been observed in many tissues of the long-lived Mclk1+/− mutant mice, which are also characterized by decreased respiration rate, low ATP levels, and increased oxidative stress (Lapointe and Hekimi, 2008). Interestingly, it has been proposed that blockage of the electron transport chain and the production of ROS are essential to obtain an efficient preconditioning response in experimental models (Lesnefsky, et al., 2004, Ravati, et al., 2001). The mitochondrial dysfunction phenotype observed in many tissues of Mclk1+/− mutants that have been analyzed has not yet been observed in the brain (Lapointe and Hekimi, 2008), which could suggest that it is different or much milder in this organ. Indeed, in contrast to other organs, the brain’s mitochondrial function and oxidative status in Mclk1+/− animals has not been explored in depth, and, to date, only a small, non-significant, decrease in mitochondrial oxygen consumption was reported (Lapointe and Hekimi, 2008, Lapointe, et al., 2009). The evaluation of additional markers, expecially those related to oxidative stress, will be needed to confirm the notion of a milder phenotype. A chronic phenotype as induced by a mutation might have to be mild for it to function as preconditioning and still be favorable. Another way to explore this question will be to study the response to I/R in tissues that are more severely affected in the mutants, such as the heart.
The hypothesis that a cerebral preconditioning-like effect is involved in the resistance of the Mclk1+/− mice against I/R insult is strengthen by our previous observation of the upregulation of several protective mechanisms in these animals. Indeed, the liver mitochondria of Mclk1+/− mice exhibit higher GPx and SOD2 activities, which are the main mitochondrial enzymatic antioxidants. The raise in SOD2 activity is particularly relevant because it was reported that a decrease in SOD2 activity is associated to I/R injury and that SOD2 deficiency exacerbates cerebral infarction and worsens neurological deficits after I/R in Sod2+/− mice (Jung, et al., 2009, Kim, et al., 2002). In contrast, mice overexpressing the same enzyme developed smaller infarcts in response to cerebral artery occlusion (Keller, et al., 1998). Moreover, SOD2 expression is enhanced by preconditioning treatment in rodents (Kato, et al., 1995). Taken together, all these data concerning the principal mitochondrial defence against superoxide suggested that elevated SOD2 expression in Mclk1+/− mutants could contribute to their neuroprotection and might consequently contribute to increase their lifespan. Recently, we have also demonstrated that the Mclk1+/− condition prevents the deterioration of mitochondrial function and the associated increase of global oxidative stress that is normally observed in Sod2+/− mutants (Lapointe, et al., 2009).
After reperfusion, blood supply is not immediately fully recovered and brain cells are still under hypoxia because many microvessels are damaged during ischemia. Under these conditions, the affected brain initiates physiological responses such as induction of protective and angiogenic factors to ensure cellular adaptation to lower oxygen. Several genes encoding such factors have been shown to be induced at the transcriptional level in response to cerebral hypoxia (Kim, et al., 2009). It is now well recognized that the key regulator of this response is the hypoxia-inducible factor-1 (HIF-1α) (Chandel and Budinger, 2007, Huang, et al., 1998, Schumacker, 2005). Indeed, during hypoxia, HIF-1α regulates the expression of many genes notably involved in oxygen transport, energy metabolism, apoptosis, cell proliferation, and other processes that are directly related to cell survival (Hu, et al., 2003, Semenza, 2000). Interestingly, we have recently reported that Mclk1+/− mice display increased basal and induced expression of HIF-1α in liver and macrophages (Wang and Hekimi, 2009). It is therefore possible that the ischemic tolerance of Mclk1 mutants is mediated by increased HIF-1α expresssion. Notably, it was suggested that since low levels of HIF-1α remain active even under normoxic conditions, basal HIF-1α activity can affect cellular resistance to acute stresses that do not permit sufficient time to establish a de novo transcription–translation response (Loor and Schumacker, 2008). If this assumption is correct, the increased basal levels of HIF-1α that was observed in tissues and macrophages of Mclk1+/− mice could likely contributed to neuroprotection. Moreover, ischemic episodes are also generally associated with an inflammatory response mainly triggered by HIF-1α and in which several inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), are released or activated in order to promote infiltration of leukocytes and activation of resident microglial and astroglial cells. In fact these two cytokines were shown to be up-regulated in the Mclk1+/− mice (Wang and Hekimi, 2009). These molecules are also implicated in brain preconditioning and neuroprotection against various toxic insults such as I/R (Saha, et al., 2009, Wang, et al., 2000, Westberg, et al., 2007).
In summary, despite the fact that is difficult to evaluate by how much cerebral accidents really contribute to death in Mclk1+/− mutants, we surmise that the ischemic tolerance phenotype revealed in the present study is likely implicated in their increased lifespan. Moreover, previously characterized aspects of the Mclk1+/− phenotype, such as reduced mitochondrial function, increased HIF-1α expression, and an enhanced inflammatory response, could be linked to this neuroprotective effect.
References
- 1.Bertoni-Freddari C, Fattoretti P, Giorgetti B, Solazzi M, Balietti M, Meier-Ruge W. Role of mitochondrial deterioration in physiological and pathological brain aging. Gerontology. 2004;50:187–192. doi: 10.1159/000076779. [DOI] [PubMed] [Google Scholar]
- 2.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Chandel NS, Budinger GR. The cellular basis for diverse responses to oxygen. Free Radic Biol Med. 2007;42:165–174. doi: 10.1016/j.freeradbiomed.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 4.Cho KO, Kim SK, Cho YJ, Sung KW, Kim SY. Regional differences in the neuroprotective effect of minocycline in a mouse model of global forebrain ischemia. Life Sci. 2007;80:2030–2035. doi: 10.1016/j.lfs.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Christophe M, Nicolas S. Mitochondria: a target for neuroprotective interventions in cerebral ischemia-reperfusion. Curr Pharm Des. 2006;12:739–757. doi: 10.2174/138161206775474242. [DOI] [PubMed] [Google Scholar]
- 6.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis. 2005;15:316–328. doi: 10.1016/j.numecd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Dirnagl U, Meisel A. Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology. 2008;55:334–344. doi: 10.1016/j.neuropharm.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 9.Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55:310–318. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiskum G, Rosenthal RE, Vereczki V, Martin E, Hoffman GE, Chinopoulos C, Kowaltowski A. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J Bioenerg Biomembr. 2004;36:347–352. doi: 10.1023/B:JOBB.0000041766.71376.81. [DOI] [PubMed] [Google Scholar]
- 11.Friberg H, Wieloch T, Castilho RF. Mitochondrial oxidative stress after global brain ischemia in rats. Neurosci Lett. 2002;334:111–114. doi: 10.1016/s0304-3940(02)01116-3. [DOI] [PubMed] [Google Scholar]
- 12.Gardner PR. Aconitase: sensitive target and measure of superoxide. Methods Enzymol. 2002;349:9–23. doi: 10.1016/s0076-6879(02)49317-2. [DOI] [PubMed] [Google Scholar]
- 13.Hekimi S. How genetic analysis tests theories of animal aging. Nat Genet. 2006;38:985–991. doi: 10.1038/ng1881. [DOI] [PubMed] [Google Scholar]
- 14.Hsu M, Sik A, Gallyas F, Horvath Z, Buzsaki G. Short-term and long-term changes in the postischemic hippocampus. Ann N Y Acad Sci. 1994;743:121–139. doi: 10.1111/j.1749-6632.1994.tb55790.x. discussion 139–140. [DOI] [PubMed] [Google Scholar]
- 15.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeffs GJ, Meloni BP, Sokolow S, Herchuelz A, Schurmans S, Knuckey NW. NCX3 knockout mice exhibit increased hippocampal CA1 and CA2 neuronal damage compared to wild-type mice following global cerebral ischemia. Exp Neurol. 2008;210:268–273. doi: 10.1016/j.expneurol.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Jiang N, Levavasseur F, McCright B, Shoubridge EA, Hekimi S. Mouse CLK-1 is imported into mitochondria by an unusual process that requires a leader sequence but no membrane potential. J Biol Chem. 2001;276:29218–29225. doi: 10.1074/jbc.M103686200. [DOI] [PubMed] [Google Scholar]
- 19.Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci. 2009;29:7003–7014. doi: 10.1523/JNEUROSCI.1110-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato H, Kogure K, Araki T, Liu XH, Kato K, Itoyama Y. Immunohistochemical localization of superoxide dismutase in the hippocampus following ischemia in a gerbil model of ischemic tolerance. J Cereb Blood Flow Metab. 1995;15:60–70. doi: 10.1038/jcbfm.1995.7. [DOI] [PubMed] [Google Scholar]
- 21.Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly S, McCulloch J, Horsburgh K. Minimal ischaemic neuronal damage and HSP70 expression in MF1 strain mice following bilateral common carotid artery occlusion. Brain Res. 2001;914:185–195. doi: 10.1016/s0006-8993(01)02801-3. [DOI] [PubMed] [Google Scholar]
- 23.Kim GW, Kondo T, Noshita N, Chan PH. Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice: implications for the production and role of superoxide radicals. Stroke. 2002;33:809–815. doi: 10.1161/hs0302.103745. [DOI] [PubMed] [Google Scholar]
- 24.Kim HA, Mahato RI, Lee M. Hypoxia-specific gene expression for ischemic disease gene therapy. Adv Drug Deliv Rev. 2009;61:614–622. doi: 10.1016/j.addr.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 26.Lai JC, Clark JB. Preparation of synaptic and nonsynaptic mitochondria from mammalian brain. Methods Enzymol. 1979;55:51–60. doi: 10.1016/0076-6879(79)55008-3. [DOI] [PubMed] [Google Scholar]
- 27.Lapointe J, Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J Biol Chem. 2008;283:26217–26227. doi: 10.1074/jbc.M803287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lapointe J, Stepanyan Z, Bigras E, Hekimi S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/− mice. J Biol Chem. 2009;284:20364–20374. doi: 10.1074/jbc.M109.006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 30.Lesnefsky EJ, Hoppel CL. Ischemia-reperfusion injury in the aged heart: role of mitochondria. Arch Biochem Biophys. 2003;420:287–297. doi: 10.1016/j.abb.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 31.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia--reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 32.Levavasseur F, Miyadera H, Sirois J, Tremblay ML, Kita K, Shoubridge E, Hekimi S. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J Biol Chem. 2001;276:46160–46164. doi: 10.1074/jbc.M108980200. [DOI] [PubMed] [Google Scholar]
- 33.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19:2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008;15:686–690. doi: 10.1038/cdd.2008.13. [DOI] [PubMed] [Google Scholar]
- 36.Majid A, He YY, Gidday JM, Kaplan SS, Gonzales ER, Park TS, Fenstermacher JD, Wei L, Choi DW, Hsu CY. Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke. 2000;31:2707–2714. doi: 10.1161/01.str.31.11.2707. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto T, Baker DJ, d’Uscio LV, Mozammel G, Katusic ZS, van Deursen JM. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke. 2007;38:1050–1056. doi: 10.1161/01.STR.0000257967.86132.01. [DOI] [PubMed] [Google Scholar]
- 38.Nakka VP, Gusain A, Mehta SL, Raghubir R. Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Mol Neurobiol. 2008;37:7–38. doi: 10.1007/s12035-007-8013-9. [DOI] [PubMed] [Google Scholar]
- 39.Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- 40.Ravati A, Ahlemeyer B, Becker A, Klumpp S, Krieglstein J. Preconditioning-induced neuroprotection is mediated by reactive oxygen species and activation of the transcription factor nuclear factor-kappaB. J Neurochem. 2001;78:909–919. doi: 10.1046/j.1471-4159.2001.00463.x. [DOI] [PubMed] [Google Scholar]
- 41.Roof RL, Hall ED. Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. 2000;17:367–388. doi: 10.1089/neu.2000.17.367. [DOI] [PubMed] [Google Scholar]
- 42.Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell CJ, Roger V, Rumsfeld J, Sorlie P, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y. Heart disease and stroke statistics--2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 43.Rouslin W, Broge CW, Grupp IL. ATP depletion and mitochondrial functional loss during ischemia in slow and fast heart-rate hearts. Am J Physiol. 1990;259:H1759–1766. doi: 10.1152/ajpheart.1990.259.6.H1759. [DOI] [PubMed] [Google Scholar]
- 44.Saha RN, Ghosh A, Palencia CA, Fung YK, Dudek SM, Pahan K. TNF-alpha preconditioning protects neurons via neuron-specific up-regulation of CREB-binding protein. J Immunol. 2009;183:2068–2078. doi: 10.4049/jimmunol.0801892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaller BJ. Influence of age on stroke and preconditioning-induced ischemic tolerance in the brain. Exp Neurol. 2007;205:9–19. doi: 10.1016/j.expneurol.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 46.Schumacker PT. Hypoxia-inducible factor-1 (HIF-1) Crit Care Med. 2005;33:S423–425. doi: 10.1097/01.ccm.0000191716.38566.e0. [DOI] [PubMed] [Google Scholar]
- 47.Schurr A, Reid KH, Tseng MT, West C, Rigor BM. Adaptation of adult brain tissue to anoxia and hypoxia in vitro. Brain Res. 1986;374:244–248. doi: 10.1016/0006-8993(86)90418-x. [DOI] [PubMed] [Google Scholar]
- 48.Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- 49.Soane L, Kahraman S, Kristian T, Fiskum G. Mechanisms of impaired mitochondrial energy metabolism in acute and chronic neurodegenerative disorders. J Neurosci Res. 2007;85:3407–3415. doi: 10.1002/jnr.21498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang D, Hekimi S. Elevated mitochondrial ROS generation affects the immune response via HIF-1α in long-lived Mclk1+/− mouse mutants. J Immunol. 2009 doi: 10.4049/jimmunol.0902352. submitted. [DOI] [PubMed] [Google Scholar]
- 52.Wang X, Li X, Erhardt JA, Barone FC, Feuerstein GZ. Detection of tumor necrosis factor-alpha mRNA induction in ischemic brain tolerance by means of real-time polymerase chain reaction. J Cereb Blood Flow Metab. 2000;20:15–20. doi: 10.1097/00004647-200001000-00004. [DOI] [PubMed] [Google Scholar]
- 53.Warner DS, Sheng H, Batinic-Haberle I. Oxidants, antioxidants and the ischemic brain. J Exp Biol. 2004;207:3221–3231. doi: 10.1242/jeb.01022. [DOI] [PubMed] [Google Scholar]
- 54.Westberg JA, Serlachius M, Lankila P, Penkowa M, Hidalgo J, Andersson LC. Hypoxic preconditioning induces neuroprotective stanniocalcin-1 in brain via IL-6 signaling. Stroke. 2007;38:1025–1030. doi: 10.1161/01.STR.0000258113.67252.fa. [DOI] [PubMed] [Google Scholar]
- 55.Westrick RJ, Winn ME, Eitzman DT. Murine models of vascular thrombosis (Eitzman series) Arterioscler Thromb Vasc Biol. 2007;27:2079–2093. doi: 10.1161/ATVBAHA.107.142810. [DOI] [PubMed] [Google Scholar]
- 56.Wong A, Boutis P, Hekimi S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics. 1995;139:1247–1259. doi: 10.1093/genetics/139.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yager JY, Wright S, Armstrong EA, Jahraus CM, Saucier DM. A new model for determining the influence of age and sex on functional recovery following hypoxic-ischemic brain damage. Dev Neurosci. 2005;27:112–120. doi: 10.1159/000085982. [DOI] [PubMed] [Google Scholar]