

Abstract
A number of studies have reported that D-cycloserine (DCS), a partial agonist of the N-methyl-D-aspartate glutamate receptor, can facilitate the loss of conditioned fear if it is administered during an extinction trial. Here we examine the effects of DCS injected into the hippocampus or amygdala on extinction of context-evoked freezing after contextual fear conditioning in C57BL/6 mice. We find that DCS administered prior to an extinction session decreased freezing from the outset of the session regardless of which brain region was targeted. Retention tests revealed opposite effects on fear expression despite identical behavioral treatments: intra-hippocampal DCS inhibited fear expression while intra-amygdala DCS potentiated fear expression. Following post-extinction session injections of DCS, we found a similar though less pronounced effect. Closer inspection of the data revealed that the effects of DCS interacted with the behavior of the subjects during extinction. Intra-hippocampal injections of DCS enhanced extinction in those mice that showed the greatest amount of within-session extinction, but had less pronounced effects on mice that showed the least within-session extinction. Intra-amygdala injections of DCS impaired extinction in those mice that showed the least within-session, but there was some evidence that the effect in the amygdala did not depend on behavior during extinction. These findings demonstrate that even with identical extinction preparations and trial durations, the effects of DCS administered into the hippocampus and amygdala can heavily depend on the organism’s behavior during the extinction session. The broader implication of these findings is that the effects of pharmacological treatments designed to enhance extinction by targeting hippocampal or amygdalar processes may depend greatly on the responsivity of the subject to the behavioral treatment.
Introduction
The attenuation of a conditioned fear response in rodents via extinction, the process of repeated re-exposure to a conditioned fear-evoking stimulus, is a valuable model of treatments for clinical cases of anxiety and fear in humans. Indeed, treatments for fear and anxiety disorders in humans frequently involve exposure therapies that draw heavily from the findings of extinction research in rodents (Hofmann 2008; Milad et al., 2006; Milad & Quirk 2012). Although exposure-based therapies in clinical populations can be highly effective in reducing fear and anxiety levels in the short-term, patients often remain vulnerable to relapse in the long-term (Rachman, 1989). This clinical phenomenon is consistent with the well-established observation that extinction in animals involves new learning processes that largely leave the original fear memory intact and susceptible to spontaneous recovery (Rescorla, 2004), renewal (Bouton 2002) and reinstatement (Rescorla and Heth 1975). As such, one converging challenge of basic research on extinction in animals and exposure-based therapies in humans is the development of procedures that result in relatively fast and persistent inhibition of abnormal fear responses.
One way to enhance the persistence of fear inhibition is to administer pharmacological agents that target cellular and molecular processes that may be involved in extinction memory formation (e.g., Davis 2011; Griebel & Holmes, 2013; Ganasen et al., 2010; Hoffman et al., 2011; Lattal & Wood, 2013). A cellular target of particular interest is the N-methyl-D-aspartate (NMDA) glutamate receptor, which supports the process of long-term synaptic potentiation thought to underlie the new associative learning that occurs during extinction (Orsini & Maren 2012; Myers and Davis 2007). Indeed, a large body of literature indicates that administration of the partial NMDA agonist D-cycloserine (DCS) in conjunction with exposure therapy facilitates reductions in abnormal fear and anxiety in human clinical populations (Difede, et al., 2013; Ressler et al., 2004; Hoffmann et al., 2006; Guastella, Richardson et al., 2008; Nave et al., 2012). Early findings in rodents have also demonstrated that DCS can facilitate extinction when administered before or after exposure to fearful cues (Walker, Ressler, Lu & Davis 2002; Ledgerwood, Richardson, Cranney 2003), reduce the reinstatement of fear (Ledgerwood, Richardson, and Cranney 2004), and aid the generalization of extinction to a second non-extinguished cue (Ledgerwood, Richardson and Cranney 2005). Although DCS is a promising pharmacological candidate for enhancing extinction processes, there remain a number of issues concerning the drug mechanism and the consistency of results. Indeed, there have been several notable null results in the ability of DCS paired with exposure therapy to reduce fear and anxiety in humans (Guastella et al., 2007a, 2007b; Tart et al., 2013; de Kleine et al., 2012), including patients reporting greater fear levels post-treatment compared to patients in placebo conditions (Litz et al., 2012).
Research in animals has also revealed several important boundaries and caveats to the action of DCS during extinction procedures (e.g., Langton & Richardson 2008, 2010a, 2010b). Most notably, several studies have suggested that the effect of DCS may critically depend on either the extinction trial duration or the individual behavior of an animal within an extinction session. Weber, Hart and Richardson (2007) found that with a small number of extinction trials, systemic DCS only facilitated extinction of an odor-shock pairing in rats that demonstrated a loss of fear within the extinction session (post-hoc analyses of “Extinguishers” versus “Non-Extinguishers”). However, when they increased the number of extinction trials in a separate experiment to achieve a within session loss of fear among all subjects, DCS proved effective at facilitating extinction after the same odor-shock pairing.
The dependence of DCS effects on the amount of extinction that occurs was also found by Bouton, Vurbic and Woods (2008), who re-analyzed previous work from their laboratory reporting a null effect of DCS (Woods and Bouton 2006). Using the Extinguisher/Non-Extinguisher criteria of Weber et al. (2007), they found that DCS did in fact facilitate fear extinction compared to saline controls in the conditioned suppression paradigm, but only among Extinguishers that showed a decrease in fear across the extinction session. In a subsequent experimental manipulation of extinction trials—two or twelve auditory cue exposures with the amount of context exposure matched in both groups—they found a facilitation of extinction by DCS only in the 12-trial group, consistent with Weber et al. (2007).
These findings suggest that the effects of DCS may at least in part depend on the behavior of the subject during the extinction session. A similar finding was reported by Lee, Milton, and Everitt (2006), who experimentally manipulated the amount of extinction learning by re-exposing auditory fear conditioned rats either once or ten times to the fear-eliciting auditory cue. Interestingly, systemic or intra-basolateral amygdala DCS impaired extinction relative to saline with a single cue re-exposure and enhanced extinction with ten cue re-exposures. They suggested that short re-exposures engaged NMDA-dependent reconsolidation processes (Przybyslawski and Sara 1997; Suzuki et al., 2004) that were enhanced by the DCS potentiation of glutamatergic transmission, a re-exposure boundary effect replicated with DCS in several procedures (Flavell et al., 2011; Lee et al., 2009; Portero-Tesserra et al., 2013; Yamada et al., 2009), though not in all (Bouton et al., 2008).
Taken together, these findings provide strong evidence that the effect of DCS may depend on the extent to which extinction occurs within an extinction session. The correlational studies of Extinguishers and Non-Extinguishers could potentially reflect underlying neurobiological differences in extinction learning ability (Holmes and Singewald 2013). These findings are important because they demonstrate that pairing a drug with a nonreinforced exposure is not enough to promote extinction; careful attention must be paid to the organism’s behavior during the extinction session.
The goal of the following experiments was to examine the effect of DCS on contextual fear extinction when administered into two distinct brain systems – the hippocampus, which is critical for the formation of contextual memories during fear extinction (Corcoran and Maren 2001), and the amygdala, a locus for the inhibition of conditioned fear (Ehrlich et al., 2009). We attempted to separate effects of DCS on learning during extinction from effects on consolidation by administering the drug before or after extinction sessions. Previous work from our laboratory has found that the effect of pharmacological manipulations depends on the duration of the extinction session (Stafford et al., 2012), as has been reported for DCS (e.g., Lee et al., 2006). Here, we examined the effects of DCS with a single, moderate extinction session that induces individual differences in the amount of within-session extinction. Such an approach allows us to evaluate the contribution of hippocampal and amygdalar mechanisms under common behavioral conditions, as well as to examine how within-session extinction effects may interact with site-specific DCS effects.
Methods and materials
SUBJECTS
All experiments used adult male C57BL/6 mice purchased from Jackson Laboratories (Bar Harbor, ME). Prior to cannulation, mice were housed four to a cage in a temperature controlled (21°C ±1°C) colony room on a 12 hour light/dark cycle for a minimum of 1 week following arrival at Oregon Health & Science University. Mice had ad libitum access to food and water while in the colony room. Following habituation to the colony room mice ranging in age from 8 to 12-weeks old underwent surgery for implantation of cannulae targeting the dorsal hippocampus or the basolateral amygdala. All experiments took place during the light phase of the light/dark cycle. All protocols were approved by the OHSU Institutional Animal Use and Care Committee and were conducted in accordance with the National Institutes of Health (NIH) “Principles of Laboratory Animal Care.”
SURGERY
Hippocampus
Mice were anaesthetized using isoflurane (5% induction, 2–2.5% maintenance) in air. Following induction of anesthesia mice were secured in a stereotaxic apparatus (Kopf). The x- and y-planes were balanced and zeroed according to the location of bregma and holes were drilled bilaterally above the dorsal hippocampus (AP −1.7mm, ML ± 1.5mm). Custom purchased 23-gauge guide cannulae (Plastics One, Inc.) projecting 1.5mm below the skull surface were inserted into the holes and glued to the skull using Ketac dental cement (3 MESPE). 28-guage injector cannulae (Plastics One) were cut to project 0.5mm past the tip of the guide. 28-guage dummy cannulae (Plastics One) were cut to match the length of the guide and were inserted internally to prevent clogging. To facilitate post-operative recovery mice received 5mg/kg injections of Rimadyl (Pfizer) 10–15-min prior to surgery. Once cannulated, mice were housed individually for the remainder of all experiments to avoid damage to the guide cannulae by cagemates. Mice received between 5–7 days of recovery in their home cages before Day 1 of all experiments.
Amygdala
Induction of anesthesia and all pre- and post-operative procedures were identical as those described above. Guide cannulae (7-mm) were made in the laboratory from 26-gauge stainless steel tubing (Small Parts, Inc.) and were implanted to target the basolateral amygdala (AP −1.46, ML ± 3.1mm). Guide cannulae were placed either: (1) −3.8 DV to the skull surface and used with injector cannulae projecting 1 mm past the guide, or (2) placed −1.8 DV to the skull surface and used with injector cannulae projecting 4 mm past the guide. Neither freezing behavior nor injection tip targeting was significantly different using either set-up. Experiments employing either guide cannulae set-up were subsequently combined.
HISTOLOGY
After all experiments brains of mice with hippocampal cannulae were flash frozen with methylbutane chilled by dry ice, and then transferred to a −80°C freezer. Brains were then sectioned (50 μm) on a cryostat and mounted on gelatin-coated slides. Slides were then stained with cresyl violet and cannulae tracks were evaluated under a light microscope for correct placements targeting the dorsal hippocampus.
Mice with basolateral amygdala-targeted cannulae were put under deep anesthesia (5% isoflurane) and bilaterally injected with 0.15 μL/side of filtered thionin (.25%) at a rate 0.25 μL/min. Injectors were left in the brain for 1 min to allow diffusion. Mice were then euthanized and brains were post fixed in 4% paraformaldehyde overnight. Brains were sliced (50 μm) using a cryostat, mounted on gelatin-coated slides, and then stained with cresyl violet. Placements were confirmed under a light microscope using the central location of thionin dye, the location of the cannula track, and the presence of gliosis at the injection site as markers. Because a significant portion of tip placements were observed to be close to the border of the basolateral and central subnuclei of the amygdala, we cannot rule out that the spread of drug infusions extended beyond the basolateral amygdala complex. We therefore refer to experiments targeting DCS infusions to the basolateral amygdala as intra-amygdala DCS.
DRUG INFUSION
D-cycloserine (Sigma Aldrich) was mixed in sterile saline vehicle to obtain a concentration of 80 μg/μl. The vehicle solution consisted of sterile saline only. Both drug solution and vehicle were kept on ice prior to infusion. All subjects received bilateral microinfusions of 0.25 μl solution at a rate of 0.25 μl/min (20 μg/side DCS). The dose of DCS was based on previous work (Akirav, 2007; Akirav et al., 2009). Microinfusions were carried out in freely moving mice using 5.0 μL Hamilton syringes attached to injector cannula with polyethylene tubing and operated by a programmable dual syringe pump (Stoelting, or Chemyx, Inc.). Brief restraint prior to infusion was necessary to remove dummy cannulae and insert injectors. Cannulae targeting the basolateral amygdala were plunged with a dummy injector prior to infusion to ensure an unclogged guide path. Injectors were left in place for 30 s following microinfusions to allow diffusion of the solution.
BEHAVIORAL PROCEDURES
Apparatus
Experimental sessions were run in a separate room from the colony, which contained four identical Coulbourn Instruments (Allentown, PA) conditioning chambers (H10-11M-TC). Subject assignment to chambers was counterbalanced across groups to control for any potential differences in conditioning chambers. Chambers measured 18 cm × 18 cm and were housed in sound- and light-attenuating shells. The floor consisted of 3.2 mm wide stainless steel rods spaced 0.5 mm apart (Coulbourn Instruments H10-35M-08). Room lights remained off during sessions but a houselight provided illumination within chambers. During conditioning sessions, a scrambled shock (2 s, 0.35 mA) was delivered to the grid floor by a computer-controlled shock generator (Coulbourn Instruments, H13–15). Within each chamber, mice were confined to a Plexiglas cylinder (diameter 15 cm; height 18 cm) with an automated infrared activity monitor (Coulbourn Instruments, H24–61) mounted at its top. The cylinder aided recording of all movement and prevented mice from escaping the chambers. Video recording provided a backup to automated behavioral data. Cylinders were rinsed with water and dried in between all sessions to remove odors. Experimental events were controlled by Graphic State 3.01 software (Coulbourn Instruments).
Fear Conditioning, Extinction, and Testing
The general experimental procedure for pre- and post-extinction injection experiments ran a total of 18 days, and consisted of conditioning, extinction, extinction tests (1-day, 4-day and 14-day post-extinction), reconditioning and a reconditioning test. Following 5–7 days of post surgery recovery, mice were taken to the experiment room where they were handled for 1–2 minutes on two consecutive days prior to Day 1 of the experiment. On Day 1, following 1–2 hours of acclimation to the experiment room, mice underwent fear conditioning, which consisted of a 12-min exposure to the conditioning chamber with 4 footshocks (2s, 0.35 mA) delivered approximately every 180 s. In pre-extinction injection experiments, mice were handled 10–20 minutes prior to the conditioning session. In post-extinction injection experiments, mice were handled approximately 1–5 minutes immediately following the conditioning session. Mice were then placed into matched drug and control groups based on their mean percentage of freezing across four 180 s time blocks.
On Day 2, mice received extinction, which consisted of a 12-min exposure to the conditioning chamber with no shocks delivered. In pre-extinction injection experiments, intra-hippocampal (DCS n=22; saline n=18) or intra-amygdala (DCS n=17; saline n=18) microinfusions were performed 10–20 minutes prior to the extinction session. In post-extinction injection experiments, intra-hippocampal (DCS n=22; saline n=19) or intra-amygdala (DCS n=26; saline n=35) microinfusions were performed 1–5 minutes immediately following the extinction session.
One set of control groups were transported to the procedure room on Day 2, but did not receive exposure to the conditioning context (No Ext controls). They received injections of DCS (n=10) or saline (n=11) into the hippocampus or into the amygdala (DCS n=15; saline n=15). A different set of control groups received extinction, followed 4-hr later by injection of DCS (n=11) or saline (n=12) into the hippocampus or amygdala (DCS n=11; saline n=12).
On Days 3, 6 and 16, mice underwent 12-min nonreinforced exposures to the chamber context (1-Day, 4-Day and 14-Day post-extinction tests, respectively). As during the initial conditioning session, mice were handled 10–20 minutes prior to the test sessions in pre-extinction injection experiments and 1–5 minutes following the test sessions in post-extinction injection experiments.
On Day 17, mice were reconditioned with a 3-min exposure to the conditioning chamber paired with a single footshock.
On Day 18, reconditioning was tested in another 12-min nonreinforced exposure to the chamber context.
DATA ANALYSIS
The dependent measure of fear for all test sessions was freezing, which was defined as a lack of activity for more than 3 seconds. Coulbourn infrared activity monitors mounted above the Plexiglas cylinders in each chamber automatically scored activity. Over each session, percentage freezing was analyzed in blocks of 3 min and follow-up analyses focused on the first 3 min of the session, which is not complicated by within-session extinction that occurs during each session.
After collecting freezing scores, all subjects were separated by drug and sorted from lowest to highest based on the Extinguisher/Non-Extinguisher measure, which was obtained by subtracting the freezing level in Block 1 of the first Extinction session by that observed in Block 4, a procedure similar to Bush, Sotres-Bayon and LeDoux (2007). This criterion was chosen in order to equivalently gauge the level of decreased within-session freezing from the start of the extinction session to the end. Mice were sorted and split within injection condition in order to avoid skewing subgroup numbers, especially in pre-extinction injection experiments where a clear drug effect on fear expression was observed. In order to further isolate extremes in within-extinction behavior, only mice in the upper third of the Extinction Block1-Block4 distribution met criterion for Extinguishers while only mice in the lower third met criterion for Non-Extinguishers. Because No-Extinction control groups did not receive extinction on Day 2, no equivalent Extinguisher/Non-Extinguisher split could be performed on these groups. Repeated measures ANOVAs were conducted on each session separately (acquisition, extinction, and subsequent test sessions) with drug (DCS or saline) and 3-min time block as factors. The effects in extinguishers and nonextinguishers were examined by comparing saline and DCS treatment within those categories. Overall effects of DCS on extinction were made by comparing extinction groups to the two control groups (no extinction or 4 hr extinction).
RESULTS
Pre-extinction hippocampal administration of DCS reduces expression of fear
Results of intra-hippocampal injections prior to extinction are shown in Figure 1A–C. Figure 1A shows the results from all subjects, regardless of behavior during extinction. As can be seen in Figure 1A, DCS disrupted the expression of freezing during extinction (main effect of drug: F(1,32)=16.2, p<.001). This effect persisted during subsequent drug-free test sessions conducted 1, 4, and 14 days later, and during a post-reconditioning test. ANOVAs conducted on the data from each test revealed a reliable main effect of drug during each test (Fs(1,32)>5.4, ps<.05).
Figure 1. Effects of DCS delivered to the hippocampus immediately before or after extinction.
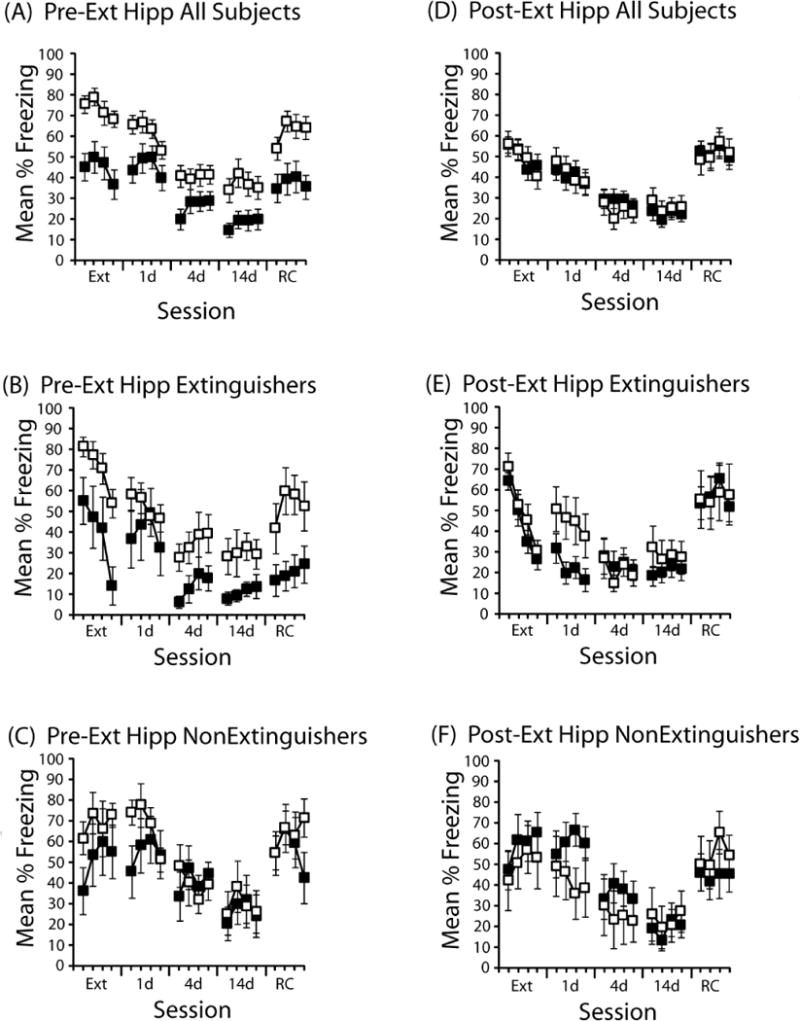
(A) DCS (black symbols; n=16) or saline (open symbols; n=18) was administered into the hippocampus immediately prior to the extinction session. Mean % time freezing is presented in 3-min time blocks during extinction (Ext), a test one day (1d), four days (4d), or 14 days (14d) after extinction, as well as one day after a reconditioning session (RC). Each session was 12 min. (B) Freezing behavior in mice categorized as Extinguishers following pre-extinction session DCS (n=5) or saline (n=6). (C) Freezing behavior in mice categorized as Non-Extinguishers following pre-extinction session DCS (n=5) or saline (n=6). (D) Freezing behavior in all mice that received DCS (n=22) or saline (n=19) immediately after the extinction session (Post-Ext). (E) Freezing behavior in mice categorized as Extinguishers following post-extinction session DCS (n=8) or saline (n=7). (F) Freezing behavior in mice categorized as Non-Extinguishers following post-extinction session DCS (n=7) or saline (n=6). Error bars indicate standard error of the mean.
The effects of hippocampal DCS generally were stronger in mice that showed the most within-session extinction (“Extinguishers” in Figure 1B). In these groups, DCS disrupted expression of freezing during extinction (F(1,9)=7.8, p<.05), with less freezing evident in DCS-treated mice during the 4-d (main effect of drug: F(1,9)=4.5, p=.06), 14-d (main effect of drug: F(1,9)=4.2, p=.07), and post-reconditioning tests (main effect of drug: F(1,9)=6.2, p<.05). However, there was some evidence that DCS caused a lasting reduction in freezing even in mice that did not show within-session extinction. Figure 1C shows the results from mice that did not show within-session extinction. During the 1-d test, an ANOVA revealed a reliable drug × block interaction (F(3,27)=3.0, p<.05). Further exploration of the interaction revealed that DCS-treated mice froze less during the first 3 min of the 1-d test than did saline-treated mice (t(9)=2.2, p=.05). This effect did not persist during the subsequent tests.
Post-extinction hippocampal administration of DCS promotes extinction only in mice that show within-session extinction
Evaluating the pre-extinction session effects of DCS are complicated by the reduction in freezing that DCS causes during the extinction session itself. It is difficult to evaluate whether the persistent changes observed with pre-session injections (Figure 1A–C) reflected enhanced extinction, or simply were residual effects of low freezing levels that occurred during extinction. To better evaluate the effects of DCS on extinction, we examined a post-session manipulation, in which DCS was administered to the hippocampus immediately after an extinction session.
The effects of post-session administration of DCS into the hippocampus are shown in Figure 1D–F. As can be seen in Figure 1D, overall, there was no effect of post-session DCS on extinction. However, it does appear that the effects of DCS interacted with the behavior of the subject during extinction. An ANOVA comparing extinguishers (Figure 1E) and nonextinguishers (Figure 1F) in performance during the 1-d post-extinction test revealed a reliable drug × extinction interaction (F(1,24)=4.8, p<.05. Further analysis of the interaction revealed that saline-treated mice did not differ at the 1d test as a function of whether extinction occurred (F(1, 11)<1.0), but DCS-treated mice showed less freezing following within-session extinction (F(1,13)=14.8, p<.005). When compared to extinguishers that received saline, DCS-treated extinguishers showed a trend in the direction of an extinction enhancement (main effect of drug: F(1,13)=4.2, p=.06). This effect did not occur in mice that showed poor within-session extinction (Figure 1F). This demonstrates that the effects of DCS delivered into the hippocampus after extinction interacted with the subjects’ behavior during extinction.
Unpaired delivery of DCS to the hippocampus has no effect on subsequent performance
Figure 2 shows the effects of DCS administered into the hippocampus in two control conditions. DCS administered in the absence of extinction (Figure 2A) or 4 hr after extinction (Figure 2B) did not result in a difference during any subsequent test. ANOVAs on each session revealed only reliable main effects of time block and no reliable main effect of drug or drug × time block interaction. Further analyses of the 4 hr groups did not reveal differential effects depending on whether subjects were categorized as Extinguishers or Non-Extinguishers (data not shown).
Figure 2. Effects of DCS administered to the hippocampus (A) in the absence of extinction or (B) 4 hr after extinction.
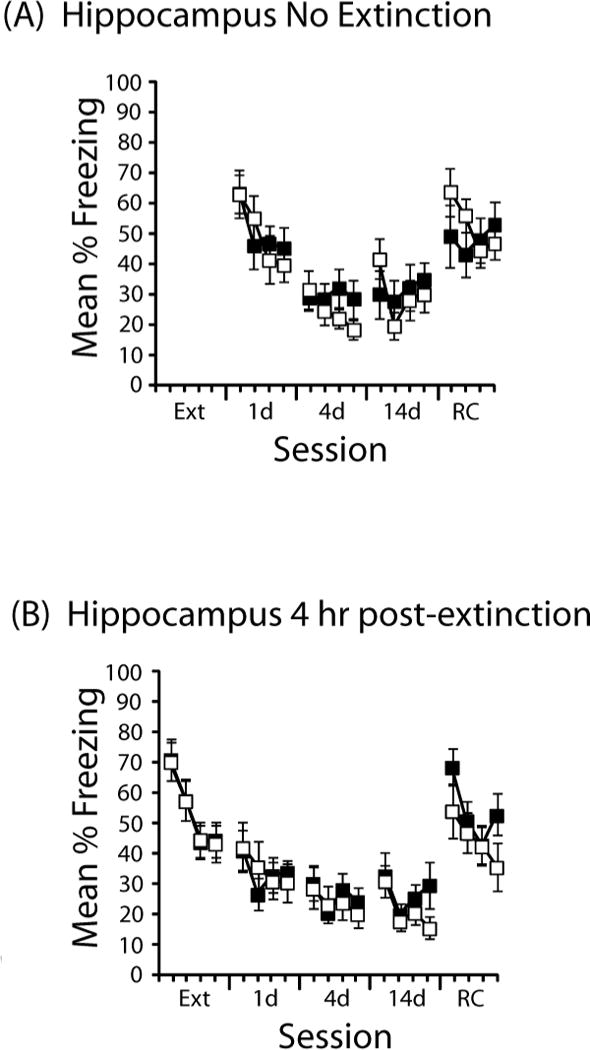
Mean % time freezing is presented in 3-min time blocks during extinction (Ext), a test one day (1d), four days (4d), or 14 days (14d) after extinction, as well as one day after a reconditioning session (RC). Each session was 12 min. DCS is depicted with black symbols; saline, with open symbols. Sample sizes in (A) were DCS (n=10) and saline (n=11) and in (B) were DCS (n=11) saline (n=12). Error bars indicate standard error of the mean.
To further evaluate the group differences induced by different treatments of DCS in the hippocampus, we conducted a two (drug) × four (extinction treatment: pre, post, no ext, 4 hr post) × four (time block) ANOVA on the data from the 1-d test. This revealed reliable main effects of extinction treatment (F(3,111)=5.0, p=.003) and block (F(3,333)=19.7, p<.001), and interactions between drug and block (F(3,9)=4.2, p=.006) and between block and extinction treatment (F(9,333)=3.7,p<.001); however, the three-way interaction was not reliable.
It is clear from Figure 2A that the animals that received DCS or saline in the absence of extinction showed high levels of freezing during the first 3 min of the 1d test, but that freezing rapidly extinguished within the session. As such, we conducted some simple planned comparisons during the first 3 min of that test session, which found that saline-treated animals did not differ as a function of whether or not extinction occurred (t(27)<1.0), but DCS before extinction resulted in less freezing compared to DCS in the absence of extinction (t(24)=2.1, p<.05), consistent with a DCS-induced enhancement in extinction.
Pre-Extinction administration of DCS in the amygdala impairs extinction only in mice that fail to show within-session extinction
Results of intra-amygdala injections prior to extinction are shown in Figure 3. Figure 3A shows the results from all subjects, regardless of behavior during extinction. As can be see in that figure, DCS disrupted expression of freezing during extinction (main effect of extinction drug: F(1,33)=14.0, p<0.001), but during the 1-d test, DCS-treated mice froze more than did saline-treated mice (drug × time block interaction: F(3,99)=3.0, p<.05). Further exploration of the interaction revealed more freezing in DCS-treated mice in the first 3 min of the test session compared to saline (t(33)=2.9, p<.005). There were no other differences during subsequent test sessions.
Figure 3. Effects of DCS delivered to the amygdala immediately before or after extinction.
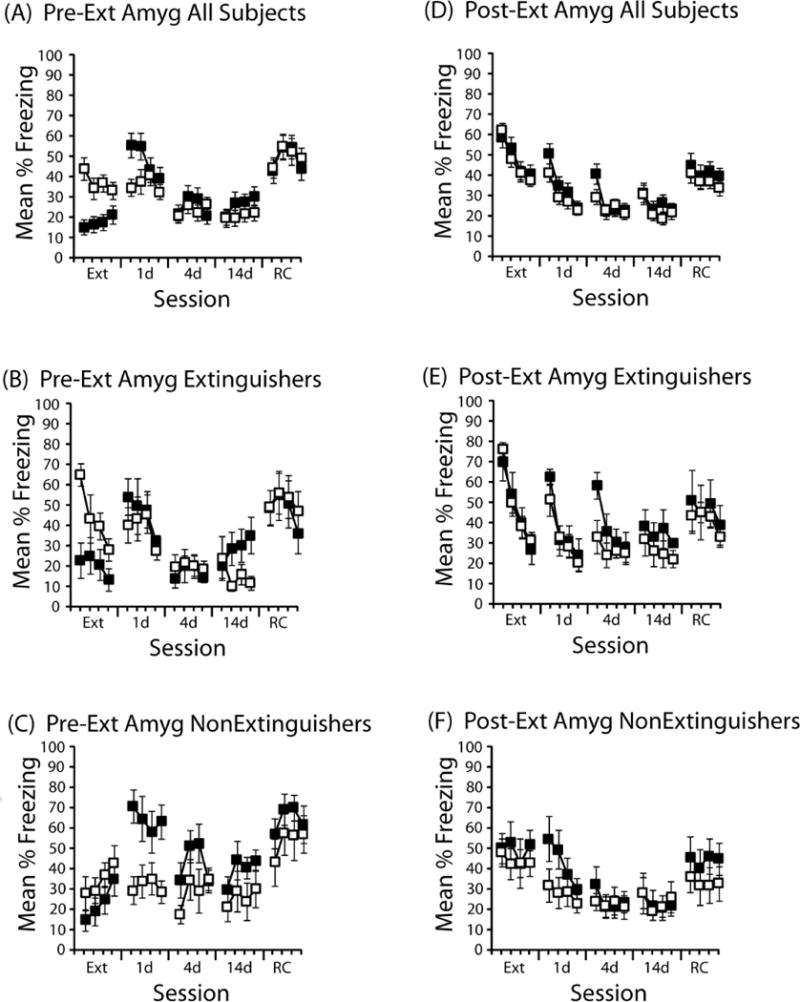
(A) DCS (black symbols; n=17) or saline (open symbols; n=18) was administered into the hippocampus immediately prior to the extinction session. Mean % time freezing is presented in 3-min time blocks during extinction (Ext), a test one day (1d), four days (4d), or 14 days (10d) after extinction, as well as one day after a reconditioning session (RC). Each session was 12 min. (B) Freezing behavior in mice categorized as Extinguishers following pre-extinction session DCS (n=5) or saline (n=6). (C) Freezing behavior in mice categorized as Non-Extinguishers following pre-extinction session DCS (n=6) or saline (n=6). (D) Freezing behavior in all mice that received DCS (n=26) or saline (n=35) immediately after the extinction session (Post-Ext). (E) Freezing behavior in mice categorized as Extinguishers following post-extinction session DCS (n=8) or saline (n=12). (F) Freezing behavior in mice categorized as Non-Extinguishers following post-extinction session DCS (n=8) or saline (n=11). Error bars indicate standard error of the mean.
As can be seen in Figures 3B and 3C, the effects of pre-extinction DCS into the amygdala appear to be driven by mice that failed to show within-session extinction. In those subjects that showed within-session extinction (Figure 3B), DCS disrupted the expression of freezing during extinction (F(1,9)=6.4, p<0.05), but had no long-term effect during any of the subsequent test sessions. In mice that failed to show within-session extinction (Figure 3C), however, DCS caused a robust increase in freezing relative to vehicle-treated mice during the 1-d test (main effect of drug: F(1,10)=8.3, p<.02). This increase in freezing was temporary, as repeated testing revealed no differences between DCS- and saline-treated mice.
Post-Extinction administration of DCS to the amygdala impairs extinction even in mice that show within-session extinction
The effects of post-extinction session delivery of DCS are shown in Figure 3D–F. Figure 3D shows the results from all subjects that received post-session injections. There were no differences during extinction (prior to drug delivery) or during the 1-day test. However, during the 4-d test there was more early session freezing in DCS-treated mice compared to saline-treated mice (drug × time block interaction: F(3,177)=4.1, p<0.01). This difference during the 4-d test was evident in mice that showed within-session extinction (Figure 3E, 4d test; block × drug interaction: F(3, 54)<0.05), but not in mice that did not show extinction (Figure 3F). Further exploration of these interactions found a trend toward a DCS effect overall during the first 3 min of the session (t(59)=1.8, p=.07). This difference in the first 3 min was reliable in Extinguishers (Figure 3E, 4d test; first 3 min: t(18)=2.2, p<.05). Overall, these post-session data are suggestive of a DCS effect in the amygdala, but they are not as clear as are the data from pre-session delivery of DCS to the amygdala.
Figure 4. Effects of DCS administered to the amygdala (A) in the absence of extinction or (B) 4 hr after extinction.
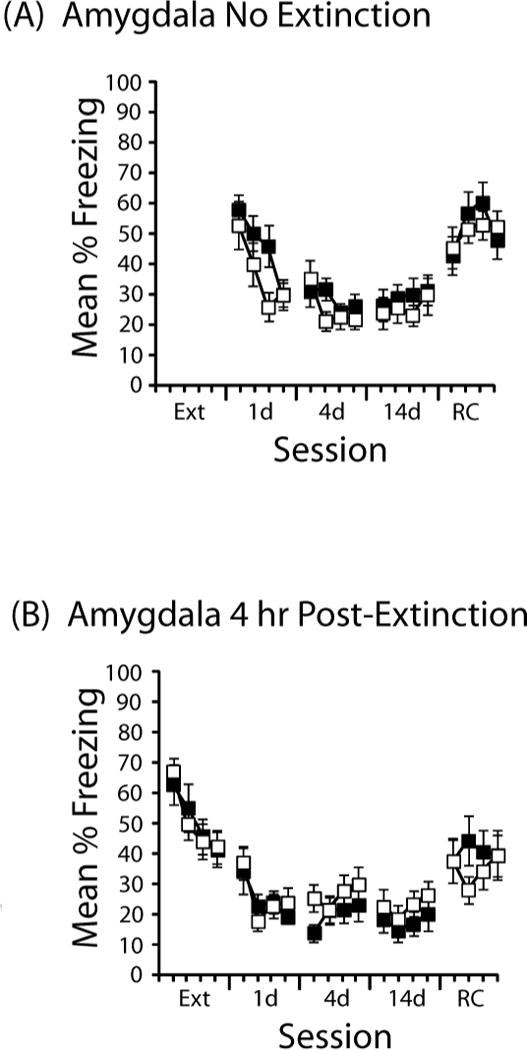
Mean % time freezing is presented in 3-min time blocks during extinction (Ext), a test one day (1d), four days (4d), or 14 days (14d) after extinction, as well as one day after a reconditioning session (RC). Each session was 12 min. Sample sizes in (A) were DCS (n=15) and saline (n=15) and in (B) were DCS (n=11) saline (n=12). Error bars indicate standard error of the mean.
Unpaired delivery of DCS to the amygdala has no effect on subsequent performance
As with the hippocampus, when DCS was administered to the amygdala unpaired with extinction, there were no effects on freezing during subsequent tests. Figure 4 shows the effects of DCS administered into the amygdala in the absence of extinction (Figure 4A) or 4 hr after extinction (Figure 4B). There was no effect of DCS in either case: ANOVAs on each session revealed only reliable main effects of time block and no reliable main effect of drug or drug × time block interaction. Further analyses of the 4 hr groups did not reveal differential effects depending on whether subjects were categorized as Extinguishers or Non-Extinguishers (data not shown).
To further evaluate the group differences induced by different treatments of DCS in the amygdala, we conducted a two (drug) × four (extinction treatment: pre, post, no ext, 4 hr post) × four (time block) ANOVA on the data from the 1-d test. This revealed reliable main effects of extinction treatment (F(3,141)=3.1, p=.03) and block (F(3,423)=37.6, p<.001), as well as a reliable three-way interaction (F(9,423)=2.8, p=.003). Further analysis of the interaction explored the difference in freezing during the first three min of the 1-d test. The high level of freezing observed in the group that received DCS prior to extinction (Figure 3A, 1d) did not differ from No Extinction DCS controls (Figure 4A, 1d; t(30)<1.0), but pre-extinction saline resulted in lower freezing compared to No Extinction saline controls (t(31)=2.1, p<.05).
Overall, these findings suggest that targeting DCS to the amygdala can impair extinction without promoting reconsolidation, and that this effect does not necessarily depend on the subject’s behavior during the extinction session.
Discussion
These findings demonstrate that the same behavioral treatment can result in different effects on extinction depending on where in the brain DCS is delivered and when it is delivered relative to extinction. In general, delivery to the hippocampus promoted extinction and delivery to the amygdala impaired extinction, but these effects were complicated by interactions with the subjects’ behavior during extinction. In the hippocampus, delivery of DCS prior to an extinction session promoted extinction by having effects in the subgroup of mice that showed the greatest within-session extinction with the drug on board. Delivery of DCS after the extinction session also resulted in an extinction enhancement in the hippocampus, but again only in those subjects that showed the greatest amount of within-session extinction immediately prior to the injection. In contrast, pre-session injections into the amygdala impaired extinction, driven by effects in mice that failed to show within-session extinction. The effects of within-session extinction in the amygdala, however, were less clear, with post-session administration having effects even in animals that showed strong within-session extinction.
These experiments extend the findings of studies showing that intra-hippocampal (Ren et al., 2013) or systemic administration of DCS can promote extinction (reviewed in Vervliet 2008; Davis 2011). Notably, as we observed with intra-hippocampal injections, systemic administration of DCS may have more pronounced effects in animals that show some evidence of learning the extinction contingencies (Weber, Hart and Richardson 2007; Bouton, Vurbic and Woods 2008). Our demonstration in these experiments that the same behavioral treatments can result in opposite long-term effects of DCS depending on where DCS is administered reinforces the idea that extinction involves the action of multiple memory and behavioral systems that may be differentially affected by perturbations of the NMDA receptor.
A potentiation of fear with intra-basolateral amygdala DCS in conjunction with a re-exposure treatment has been previously shown in several studies (e.g., Lee et al., 2006, 2009; Portero-Tesserra et al., 2013; Yamada et al., 2009). However, in those experiments, extinction exposure was designed to result in different levels of fear expression during the extinction session. With brief exposures, fear expression remained high and with long exposures, fear expression was lower. In our experiments, we found that even with longer exposures that overall result in a loss of fear, amygdala injections of DCS impaired extinction. With pre-session injections, this effect was driven by mice that did not show within-session extinction, but with post-session injections, the relation between within-session extinction and DCS was less clear.
Even though intra-amygdala injection of DCS resulted in higher fear compared to saline injections, this effect did not appear to reflect the strengthening of the memory. Instead, DCS impaired extinction, as DCS-treated mice froze at similar levels at test, regardless of whether DCS was delivered before extinction or in the absence of extinction. The large increase in freezing in Non-extinguishers in Figure 3C may be consistent with a potentiation of reconsolidation, but such an interpretation must be made with caution because comparisons cannot be made with the No Extinction control group, which cannot be categorized based on extinction performance.
Whether DCS in the amygdala promotes reconsolidation or impairs extinction through other means, these findings add to a complicated literature with DCS and the amygdala. Some studies have found that DCS can promote extinction (e.g., Walker et al., 2002) or impair extinction, as described above. It is becoming increasingly clear that the circuits that mediate fear expression and fear extinction are highly interconnected within the amygdala (Erhlich et al., 2009; Stafford et al., 2013) and that these circuits can become differentially engaged by experience (Holmes and Singewald 2013). It is thus interesting to speculate that extinction-inducing conditions may excite certain fear extinction circuits, such as those recruiting amygdala intercalated cells (Likhtik et al., 2008), whereas extinction-impairing conditions may excite certain fear expression circuits, such as those involving particular basolateral amygdala neurons (Herry et al., 2008). That we did not observe a consistent effect of within-session extinction with amygdala injections demonstrates that much more work is needed to characterize the relation between behavior, pharmacological manipulations, and amygdala activation.
Despite the different long-term effects after injections into the hippocampus or amygdala, a consistent finding from both studies was that DCS disrupted the expression of freezing relative to vehicle-treated animals. This suggests that the effects of DCS on expression of fear do not by themselves predict the long-term effects. Although sometimes unreported in the literature, there have been several previous demonstrations of a suppression of fear expression with pre-extinction injections of DCS. Systemic injections of DCS 30-minutes prior to an initial extinction session in a contextual fear conditioning experiment in Yamada et al. (2009; Fig. 4) revealed freezing deficits compared to vehicle controls during extinction that persisted through a second test and a reinstatement test. Interestingly, Ledgerwood et al. (2003) reported a significant inhibition of freezing to the conditioning context during a brief 2-min pre-cue exposure in rats injected with DCS compared with those given vehicle 15 min prior to an initial extinction session, but by the second cue presentation DCS-treated rats had risen to the freezing levels of vehicle-treated rats.
The reduction in freezing at the outset of the extinction session induced by DCS could have several effects on the impact of the extinction experience in our experiments. First, the low levels of freezing may result in greater context exploration, which would allow the organism to associate more of the components of the context with the absence of shock, thereby promoting the retrieval of extinction during subsequent tests (e.g., Stafford & Lattal, 2009). This pattern of results occurred with pre-session delivery into the hippocampus, which may be involved in forming contextual representations (e.g., Fanselow, 1999). Second, these low levels of freezing may reflect a reduced shock expectation, which would mean that the prediction error would be smaller than that of saline-treated animals, which showed higher levels of freezing at the outset of extinction. Many studies have shown that this predictive error is a major determinant of the learning that occurs during a fear extinction trial (e.g., Leung & Westbrook, 2008) and NMDA receptors may be particularly important for modulating the impact of that predictive error (e.g., Cole & McNally, 2007, 2009; reviewed in McNally et al., 2011). The reduced freezing that we observed may have been expected to weaken the impact of the behavioral experience during extinction, resulting in enhanced spontaneous recovery (e.g., Leung & Westbrook, 2008). This pattern of results occurred with pre-session delivery into the amygdala in our experiments, which may be involved in regulating the impact of prediction error (e.g., Cole & McNally, 2009).
One popular idea for DCS-induced extinction enhancements has been that DCS potentiates the acquisition of inhibitory properties to the extinction context. Evidence for this idea comes from decreased reinstatement (Woods and Bouton 2006, Mao, Lin, & Gean 2008, Ledgerwood, Richardson, & Cranney 2004), intact contextual renewal (Woods and Bouton 2006) and rapid reacquisition (Ledgerwood, Richardson, & Cranney 2005), all of which can be explained by a DCS enhanced contextual inhibitory learning (see also Vervliet 2008). Under this model, DCS enhances inhibitory learning to the context, but allows extinction to remain vulnerable to behavioral manipulations that affect the context memory, including the contextual renewal and reconditioning effects. In a direct test of the context inhibition idea, Baker et al. (2012) found that DCS promoted extinction, but the extinction context could be conditioned just as readily as a control context; i.e., there was no evidence for a retardation of acquisition effect that is often used as evidence for inhibition.
Other accounts have suggested that DCS may promote extinction not by enhancing inhibition that may develop during extinction, but instead by erasing aspects of the original memory. Based on in vitro molecular studies of amygdala slices, Mao et al. (2006) and (2008) have suggested that DCS might affect in vivo impairments in fear expression 24 hours after an extinction session by erasing the original fear memory. They base this conclusion on the observation that D-cycloserine paired with Low Frequency Stimulation (LFS) in basolateral amygdala neural cultures, but not DCS or LFS alone, causes the internalization of surface Glutamate receptor subunit 1 (Glu1) and subunit 2 (Glu2) which had previously undergone exocytosis during fear conditioning. Mao et al. (2008) point out this conclusion is consistent with findings that DCS administered during extinction blocks the reinstatement of fear when given a US alone reminder treatment (Ledgerwood et al., 2005; Mao et al., 2008). However, these kinds of effects are consistent with accounts that do not appeal to erasure processes (reviewed in Lattal & Stafford, 2010; Lattal & Wood, 2013). Further, DCS as a memory eraser is inconsistent with behavioral studies showing intact contextual renewal of fear when rats were tested in a context other than that of extinction (Woods and Bouton 2006), and intact rapid reacquisition of fear following a small number of CS-US pairings after extinction learning (Ledgerwood et al., 2005). Further, the fact that intra-amygdala injections of the transcription blocker actinomycin D and the protein synthesis inhibitor anisomycin are both capable of blocking DCS-facilitation of extinction adds molecular support to the evidence arguing for an extinction consolidation effect of DCS (Yang and Lu 2005).
Adding a further level of complexity to the DCS literature are the findings that traditional tests conducted after extinction sometimes do not reveal DCS effects, but subsequent testing does. For example, Groblewski et al. (2009) failed to observe effects of DCS on rate of extinction or on post-extinction testing, but they did find that DCS administered during extinction resulted in impairments in post-extinction reconditioning, relative to a saline-treated group. Similarly, Chang and Maren (2011) also failed to observe DCS effects during extinction or post-extinction testing, but did observe a savings effect when extinction was conducted again following reconditioning. Findings like these illustrate that just as there are multiple possibilities for positive effects of DCS on extinction, null effects do not necessarily mean that DCS did not affect extinction.
Finally, our experiments are consistent with the idea that individual differences during conditioning and extinction may lead to different effects in response to identical neurobiological manipulations. Individual differences in behavior have been recognized as a contributor to extinction effects from the time of Pavlov’s early work: “under the same set of external conditions some animals will have the conditioned reflexes rapidly extinguished, while in others the whole process will be much delayed” (Pavlov, 1927, pp.51). Many others since then have observed that the effects of an identical extinction session can be quite different depending on the behavior that occurs during extinction (Hefner et al., 2008; Lehner et al., 2010; Smits, et al., 2013; Sotres-Bayon et al., 2008). Regarding potential causes, recent work has highlighted both genetic variation among mouse strains (MacPherson et al., 2013) and the possibility of epigenetic changes due to experience (Whittle et al., 2013). These causal factors appear to particularly affect local circuitry and interconnections between hippocampal-prefrontal (Stafford et al., 2012) and prefrontal-amygdala pathways (Busti et al., 2011; Likhtik et al., 2013; Maren, 2005). Together, these results suggest that effects of a manipulation on extinction can be modulated by the nature of the behavioral experience (how much extinction occurs), the genetic predispositions of individuals to respond to extinction (how genes interact with extinction experiences), and by individual variability that is not obviously due to genetics or experience. Understanding the causes, the neural mechanisms, and defining the behavioral processes that underlie individual and genetic differences in fear learning are therefore important areas for future research.
Acknowledgments
This work was supported by NIH grant MH077111.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akirav I. NMDA partial agonist reverses blocking of extinction of aversive memory by GABAA agonist in the amygdala. Neuropsychopharmacology. 2007;32(3):542–550. doi: 10.1038/sj.npp.1301050. [DOI] [PubMed] [Google Scholar]
- Akirav I, Segev A, Motanis H, Maroun M. d-Cycloserine into the BLA reverses the impairing effects of exposure to stress on the extinction of contextual fear, but not conditioned taste aversion. Learn Mem. 2009;16(11):682–686. doi: 10.1101/lm.1565109. [DOI] [PubMed] [Google Scholar]
- Baker KD, McNally GP, Richardson R. D-cycloserine does not facilitate fear extinction by reducing conditioned stimulus processing or promoting conditioned inhibition to contextual cues. Learn Mem. 2012;19(10):461–9. doi: 10.1101/lm.026674.112. [DOI] [PubMed] [Google Scholar]
- Bouton ME. Context, ambiguity, and unlearning: sources of relapse after behavioral extinction. Biol Psychiatry. 2002;52:976–86. doi: 10.1016/s0006-3223(02)01546-9. [DOI] [PubMed] [Google Scholar]
- Bouton ME, Vurbic D, Woods AM. D-cycloserine facilitates context-specific fear extinction learning. Neurobiol Learn Mem. 2008;90(3):504–10. doi: 10.1016/j.nlm.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush DE, Sotres-Bayon F, LeDoux JE. Individual differences in fear: isolating fear reactivity and fear recovery phenotypes. J Trauma Stress. 2007;20:413–422. doi: 10.1002/jts.20261. [DOI] [PubMed] [Google Scholar]
- Busti D, Geracitano R, Whittle N, Dalezios Y, Manko M, Kaufmann W, Satzler K, Singewald N, Capogna M, Ferraguti F. Different fear states engage distinct networks within the intercalated cell clusters of the amygdala. J Neurosci. 2011;31:5131–44. doi: 10.1523/JNEUROSCI.6100-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Maren S. Medial prefrontal cortex activation facilitates re-extinction of fear in rats. Learning & Memory. 2011;18:221–225. doi: 10.1101/lm.2070111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S, McNally GP. Temporal-difference prediction errors and Pavlovian fear conditioning: role of NMDA and opioid receptors. Behav Neurosci. 2007;121:1043–52. doi: 10.1037/0735-7044.121.5.1043. [DOI] [PubMed] [Google Scholar]
- Cole S, McNally GP. Complementary roles for amygdala and periaqueductal gray in temporal-difference fear learning. Learn Mem. 2009;16:1–7. doi: 10.1101/lm.1120509. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, MAren S. Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J Neurosci. 2001;21:1720–1726. doi: 10.1523/JNEUROSCI.21-05-01720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. NMDA receptors and fear extinction: implications for cognitive behavioral therapy. Dialogues Clin Neurosci. 2011;13(4):463–74. doi: 10.31887/DCNS.2011.13.4/mdavis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difede J, Cukor J, Wyka K, Olden M, Hoffman H, Lee FS, Altemus M. D-cycloserine Augmentation of Exposure Therapy for Posttraumatic Stress Disorder: A Pilot Randomized Clinical Trial. Neuropsychopharmacology. 2013 doi: 10.1038/npp.2013.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Fanselow MS. Learning theory and neuropsychology: Configuring their disparate elements in the hippocampus. Journal of Experimental Psychology: Animal Behavior Processes. 1999;25:275–283. [Google Scholar]
- Flavell CR, Barber DJ, Lee JL. Behavioural memory reconsolidation of food and fear memories. Nat Commun. 2011;2:504. doi: 10.1038/ncomms1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganasen KA, Ipser JC, Stein DJ. Augmentation of cognitive behavioral therapy with pharmacotherapy. Psychiatr Clin North Am. 2010;33(3):687–99. doi: 10.1016/j.psc.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Griebel G, Holmes A. 50 years of hurdles and hope in anxiolytic drug discovery. Nature Reviews Drug Discovery. 2013;12:1–21. doi: 10.1038/nrd4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groblewski PA, Lattal KM, Cunningham CL. Effects of D-cycloserine on extinction and reconditioning of ethanol-seeking behavior in mice. Alcohol Clin Exp Res. 2009;33(5):772–82. doi: 10.1111/j.1530-0277.2009.00895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guastella AJ, Dadds MR, Lovibond PF, Mitchell P, Richardson GA. A randomized controlled trial of the effect of D-cycloserine on exposure therapy for spider fear. J Psych Res. 2007;41:466–471. doi: 10.1016/j.jpsychires.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Guastella AJ, Lovibond PF, Dadds MR, Mitchell P, Richardson R. A randomized controlled trial of the effect of D-cycloserine on extinction and fear conditioning in humans. Behaviour Research and Therapy. 2007;45(4):663–672. doi: 10.1016/j.brat.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Guastella AJ, Richardson R, Lovibond PF, Rapee RM, Gaston JE, Mitchell P, Dadds MR. A randomized controlled trial of D-Cylcoserine enhancement of exposure therapy for social anxiety disorder. Biol Psychiatry. 2008;63:544–549. doi: 10.1016/j.biopsych.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Hefner K, Whittle N, Juhasz J, Norcross M, Karlsson RM, Saksida LM, Bussey TJ, Singewald N, Holmes A. Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J Neurosci. 2008;28:8074–85. doi: 10.1523/JNEUROSCI.4904-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454(7204):600–6. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Hofmann SG, Meuret AE, Smits JA, Simon NM, Pollack MH, Eisenmenger K, Sheikh M, Otto MW. Augmentation of exposure therapy for social anxiety disorder with D-cycloserine. Arch Gen Psychiatry. 2006;63:298–304. doi: 10.1001/archpsyc.63.3.298. [DOI] [PubMed] [Google Scholar]
- Hofmann SG. Cognitive processes during fear acquisition and extinction in animals and humans: Implications for exposure therapy of anxiety disorders. Clinical Psychology Review. 2008;28(2):199–210. doi: 10.1016/j.cpr.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann SG, Smits JAJ, Asnaani A, Gutner CA, Otto MW. Cognitive enhancers for anxiety disorders. Pharmacol Biochem Be 2011. 2011;99:275–284. doi: 10.1016/j.pbb.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Singewald N. Individual differences in recovery from traumatic fear. Trends in Neuroscience. 2013;36(1):23–31. doi: 10.1016/j.tins.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kleine RA, Hendriks GJ, Kusters WJ, Broekman TG, van Minnen A. A randomized placebo-controlled trial of D-cycloserine to enhance exposure therapy for posttraumatic stress disorder. Biol Psychiatry 2012. 2012;71:962–968. doi: 10.1016/j.biopsych.2012.02.033. [DOI] [PubMed] [Google Scholar]
- Langton JM, Richardson R. D-Cycloserine Facilitates Extinction the First Time but not the Second Time: An Examination of the Role of NMDA Across the Course of Repeated Extinction Sessions. Neuropsychopharmacology. 2008;33:3096–3102. doi: 10.1038/npp.2008.32. [DOI] [PubMed] [Google Scholar]
- Langton JM, Richardson R. The effect of D-cycloserine on immediate vs. delayed extinction of learned fear. Learning & Memory. 2010a;17:547–551. doi: 10.1101/lm.1927310. [DOI] [PubMed] [Google Scholar]
- Langton JM, Richardson R. The temporal specificity of the switch from NMDAr-dependent extinction to NMDAr-independent re-extinction. Behavioural Brain Research. 2010b;208:646–649. doi: 10.1016/j.bbr.2009.12.018. [DOI] [PubMed] [Google Scholar]
- Lattal KM, Stafford JM. What does it take to demonstrate memory erasure? Theoretical comment on Norrholm et al (2008) Behavioral Neuroscience. 2008;122:1186–1190. doi: 10.1037/a0012993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Wood MA. Epigenetics and persistent memory: implications for reconsolidation and silent extinction beyond zero. Nat Neurosci. 2013;16(2):124–9. doi: 10.1038/nn.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledgerwood L, Richardson R, Cranney J. effects of d-cycloserine on extinction of conditioned freezing. Behave Neurosci. 2003;117:341–349. doi: 10.1037/0735-7044.117.2.341. [DOI] [PubMed] [Google Scholar]
- Ledgerwood L, Richardson R, Cranney J. D-cycloserine and the facilitation of extinction of conditioned fear: consequences for reinstatement. Behav Neurosci. 2004;118:505–513. doi: 10.1037/0735-7044.118.3.505. [DOI] [PubMed] [Google Scholar]
- Ledgerwood L, Richardson R, Cranney J. D-cycloserine facilitates extinction of learned fear: effects on reacquisition and generalized extinction. Biol Psychiatry. 2005;57:841–847. doi: 10.1016/j.biopsych.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. reconsolidation and extinction of conditioned fear: inhibition and potentiation. J neurosci. 2006;26:10051–10056. doi: 10.1523/JNEUROSCI.2466-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Gardner RJ, Butler VJ, Everitt BJ. D-cycloserine potentiates the reconsolidation of cocaine-associated memories. Learn Mem. 2009;16:82–5. doi: 10.1101/lm.1186609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung HT, Westbrook RF. Spontaneous recovery of extinguished fear responses deepens their extinction: a role for error-correction mechanisms. J Exp Psychol Anim Behav Process. 2008;34:461–74. doi: 10.1037/0097-7403.34.4.461. [DOI] [PubMed] [Google Scholar]
- Lehner M, Wislowska-Stanek A, Taracha E, Maciejak P, Szyndler J, Skorzewska A, Turzynska D, Sobolewska A, Hamed A, Bidzinski A, Plaznik A. The effects of midazolam and D-cycloserine on the release of glutamate and GABA in the basolateral amygdala of low and high anxiety rats during extinction trial of a conditioned fear test. Neurobiol Learn Mem. 2010;94(4):468–80. doi: 10.1016/j.nlm.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Pare D. Amygdala intercalated neurons are required for expression of fear extinction. Nature. 2008;454(7204):642–5. doi: 10.1038/nature07167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Stujenske JM, Topiwala MA, Harris AZ, Gordon JA. Prefrontal entrainment of amygdala activity signals safety in learned fear and innate anxiety. Nature Neuroscience. 2013 doi: 10.1038/nn.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litz BT, Salters-Pedneault K, Steenkamp MM, Hermos JA, Bryant RA, Otto MW, Hofmann SG. A randomized placebo-controlled trial of D-cycloserine and exposure therapy for posttraumatic stress disorder. J Psychiatr Res. 2012;46:1184–1190. doi: 10.1016/j.jpsychires.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Mao SC, Hsiao YH, Gean PW. Extinction training in conjunction with a partial agonist of the glycine site on the NMDA receptor erases memory trace. J Neurosci. 2006;26(35):8892–9. doi: 10.1523/JNEUROSCI.0365-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao SC, Lin HC, Gean PW. Augmentation of fear extinction by D-cycloserine is blocked by proteasome inhibitors. Neuropsychopharmacology. 2008;33(13):3085–95. doi: 10.1038/npp.2008.30. [DOI] [PubMed] [Google Scholar]
- Maren S. Synaptic mechanisms of associative memory in the amygdala. Neuron. 2005;47(6):783–6. doi: 10.1016/j.neuron.2005.08.009. 15. [DOI] [PubMed] [Google Scholar]
- Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. 2006;73:61–71. doi: 10.1016/j.biopsycho.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirck GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–51. doi: 10.1146/annurev.psych.121208.131631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers K, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12(2):120–50. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- Nave AM, Tolin DF, Stevens MC. Exposure therapy, D-Cycloserine, and functional magnetic resonance imaging in patients with snake phobia: a randomized pilot study. J Clin Psychiatry. 2012;73(9):1179–1186. doi: 10.4088/JCP.11m07564. [DOI] [PubMed] [Google Scholar]
- Orsini CA, Maren S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev. 2012;36:1773–802. doi: 10.1016/j.neubiorev.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portero-Tresserra M, Marti-Nicolovius M, Guillazo-Blanch G, Boadas-Vaello P, Vale-Martinez A. D-cycloserine in the basolateral amygdala prevents extinction and enhances reconsolidation of odor-reward associative learning in rats. Neurobiol Learn Mem. 2013;100:1–11. doi: 10.1016/j.nlm.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Przybyslawski J, Sara SJ. Reconsolidation of memory after its reactivation. Behav Brain Res. 1997;84:241–246. doi: 10.1016/s0166-4328(96)00153-2. [DOI] [PubMed] [Google Scholar]
- Quirk GJ. Memory for extinction of conditioned fear is long-lasting and persists following spontaneous recovery. Learn Mem. 2002;9:402–7. doi: 10.1101/lm.49602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachman S. The return of fear: review and prospect. Clinical Psychology Review. 1989;9:147–168. [Google Scholar]
- Ren J, Li X, Zhang X, Li M, Wang Y, Ma Y. The effects of intra-hippocampal microinfusion of D-cycloserine on fear extinction, and the expression of NMDA receptor subunit NR2B and neurogenesis in the hippocampus in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2013;44:257–264. doi: 10.1016/j.pnpbp.2013.02.017. [DOI] [PubMed] [Google Scholar]
- Rescorla RA. Spontaneous recovery. Learn Mem. 2004;11:501–9. doi: 10.1101/lm.77504. [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Heth D. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol: Anim Behav Process. 1975;104:88–96. [PubMed] [Google Scholar]
- Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. Cognitive enhancers as adjuncts to psychotherapy: Use of D-Cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- Smits JA, Rosenfield D, Otto MW, Powers MB, Hofmann SG, Telch MJ, Pollack MH, Tart CD. D-Cycloserine Enhancement of Fear Extinction is Specific to Successful Exposure Sessions: Evidence from the Treatment of Height Phobia. Biological Psychiatry. 2013;73:1054–1058. doi: 10.1016/j.biopsych.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Corcoran KA, Peters J, Sierra-Mercado D. Neural correlates of individual variability in fear extinction. J Neurosci. 2008;28:12147–9. doi: 10.1523/JNEUROSCI.4373-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Maughan DK, Ilioi EC, Lattal KM. Exposure to a fearful context during periods of memory plasticity impairs extinction via hyperactivation of frontal-amygdalar circuits. Learn Mem. 2013;20:156–63. doi: 10.1101/lm.029801.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM. Increasing histone-acetylation the hippocampus-infralimbic network enhances fear extinction. Biol Psychiatry. 2012;72(1):25–33. doi: 10.1016/j.biopsych.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tart CD, Handelsman PR, Deboer LB, Rosenfield D, Pollack MH, Hofmann SG, Powers MB, Otto MW, Smits JAJ. Augmentation of exposure therapy with post-session administration of D-cycloserine. J Psychiatr Res. 2013;47:168–174. doi: 10.1016/j.jpsychires.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervliet B. Learning and memory in conditioned fear extinction: Effects of D-cycloserine. Acta Psychologica. 2008;127:601–613. doi: 10.1016/j.actpsy.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration of intra-amygdala infusions of d-cycloserine as assessed with fear-poteniated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hart J, Richardson R. Effects of D-cycloserine on extinction of learned fear to an olfactory cue. Neurobiology of Learning and Memory. 2007;87:476–482. doi: 10.1016/j.nlm.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Whittle N, Schmuckermair C, Gunduz Cinar O, Hauschild M, Ferraguti F, Holmes A, Singewald N. Deep brain stimulation, histone deacetylase inhibitors and glutamatergic drugs rescue resistance to fear extinction in a genetic mouse model. Neuropharmacology. 2013;64:414–423. doi: 10.1016/j.neuropharm.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods AM, Bouton ME. d-cycloserine facilitates extinction but does not eliminate renewal of the conditioned emotional response. Behave neurosci. 2006;120:1159–1162. doi: 10.1037/0735-7044.120.5.1159. [DOI] [PubMed] [Google Scholar]
- Yamada D, Zushida K, Wada K, Sekiguchi M. Pharmacological discrimination of extinction and reconsolidation of contextual fear memory by a potentiator of AMPA receptors. Neuropsychopharmacology. 2009;34:2574–2584. doi: 10.1038/npp.2009.86. [DOI] [PubMed] [Google Scholar]
- Yang YL, Lu KT. Facilitation of conditioned fear extinction by D-cycloserine is mediated by mitogen-activated protein kinase and phosphatidylinositol 3-kinase cascades and requires de novo protein synthesis in basolateral nucleus of amygdala. Neuroscience. 2005;134:247–260. doi: 10.1016/j.neuroscience.2005.04.003. [DOI] [PubMed] [Google Scholar]