

Abstract
Background
Glycosphingolipids are integral components of the cell membrane and have been shown to serve as messengers, transducing growth factor initiated phenotypes. Here we have examined whether inhibition of glycosphingolipid synthesis could ameliorate atherosclerosis and arterial stiffness in transgenic mice and rabbits.
Methods and Results
Apolipoprotein E−/− mice (12 weeks of age, n = 6) were fed regular chow or a western diet (1.25% cholesterol, 2% fat). Mice were fed 5mg/kg (mpk) or 10mpk of an inhibitor of glycosphingolipid synthesis, D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (D-PDMP), solubilized in vehicle (5% Tween-80 in PBS) and the placebo group received vehicle only. At 20 and 36 weeks of age, serial echocardiography was performed to measure aortic intima medial thickening (IMT). Aortic pulse wave velocity (PWV) measured vascular stiffness. Feeding mice a western diet markedly increased aortic PWV, IMT, oxidized LDL, Ca2+ deposits, and glucosyl- and lactosylceramide synthase activity. These were dose-dependently decreased by feeding D-PDMP. In liver, D-PDMP decreased cholesterol and triglyceride levels by raising the expression of SREBP2, LDL-r, HMGCo-A reductase, and cholesterol efflux genes (e.g., ABCG5, ABCG8). D-PDMP affected VLDL catabolism by increasing the gene expression for LPL and VLDLr. Rabbits fed a western diet for 90 days had extensive atherosclerosis accompanied by a 17.5-fold increase in total cholesterol levels and a 3-fold increase in lactosylceramide levels. This was completely prevented by feeding D-PDMP.
Conclusions
Inhibition of glycosphingolipid synthesis ameliorates atherosclerosis and arterial stiffness in ApoE−/− mice and rabbits. Thus, inhibition of glycosphingolipid synthesis may be a novel approach to ameliorate atherosclerosis and arterial stiffness.
Keywords: glycosphingolipids, Apolipoprotein E null mice, atherosclerosis, imaging
Introduction
Atherosclerosis contributes to nearly one half of the mortality in the western world and is growing in epidemic proportions in rapidly developing countries in Asia. High levels of blood cholesterol, high blood pressure, obesity, diabetes, stress, and lifestyles including smoking are among the risk factors contributing to this alarming increase in this disease. While several therapeutic modalities including cholesterol synthesis inhibitors (family of statins), cholesterol absorption inhibitor (ezetimibe), blood pressure lowering drugs, platelet adhesion inhibitors and many more are either prescribed alone and/or in combination, there is a need for novel approaches to mitigate the initiation and progression of atherosclerosis1. However, these therapeutic modalities do not decrease atherosclerotic plaque burden. A relationship between high blood levels of cholesterol and glycosphingolipids was suggested several decades ago2–7. Subsequently, a tighter correlation between the load of LDL cholesterol and GSL/lactosylceramide shedding in urinary proximal tubular cells in patients with the homozygous form of familial hypercholesterolemia was reported3. D-PDMP is an analog of glucosylceramide and was synthesized to inhibit glucosylceramide synthesis in patients with Gaucher’s disease. However, we observed that this compound could directly inhibit the activity of purified lactosylceramide synthase7. D-PDMP is a small molecular weight compound and is well tolerated by experimental animals e.g. mice, rats, rabbits up to 10 times the effective dose. In mice, the effective dose of D-PDMP is 10 mg per kg (mpk) body weight when given orally compared to the use of 100mpk of various adamanate derivatives used by other investigators8, 9. Since D-PDMP turnover time is a short ~52 min in mice10, it is rapidly detoxified and excreted with little or no side effects. Delivery of D-PDMP by oral gavage, or intraperitoneal injection, has no effect on appetite and overall wellbeing of the experimental animals. We report here that feeding a western diet to ApoE−/− mouse and normal rabbits leads to extensive atherosclerosis, vascular wall thickness and stiffness and increase in the arterial levels of glucosylceramide and lactosylceramide. Feeding these animals D-PDMP dose-dependently ameliorates atherosclerosis and vascular stiffness.
Materials and Methods
Animals and Treatments
Apolipoprotein E-deficient (ApoE−/−) male mice aged 11 weeks (Jackson Labs, Bar Harbor, Me) were purchased and baseline physiological parameters were measured prior to further experimentation. At the age of 12 weeks, the ApoE−/− mice were started on a high fat and high cholesterol diet (HFHC) of 4.5kcal/g, 2.0% fat, and 1.25% cholesterol (D12108C, Research Diet Inc., New Brunswick, NJ) for 20 to 36 weeks with and without treatment of D-PDMP (5mpk, 10 mpk) and compared to control mice fed only chow diet and placebo fed HFHC plus vehicle. Food was rationed once a week to estimate the weekly growth rate and food intake. Physiological studies were performed at around 12, 20 and 36 weeks. Tissues were harvested at 12, 20 and 36 weeks of age for molecular and histopathological studies.
D-PDMP was purchased from Matreya LLC (Pleasant Gap, PA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless mentioned otherwise. Animals were subject to anthropometric measurements (body weight, percent body fat) and physiological measurements [blood pressure; ultrasound to measure aortic intima-media thickening (AoIMT); pulse wave velocity (PWV), a measure of arterial stiffening and arteriosclerosis)]. A group of mice (n=5) were euthanized to obtain baseline values for aortic tissue and blood samples were collected.
The rest of the mice were divided into several groups. These were: Placebo (treated with vehicle only 5%Tween-80 in phosphate buffered saline), 5mpk of D-PDMP solubilized in vehicle, and 10mpk of D-PDMP solubilized in vehicle. Vehicle and D-PDMP was delivered daily by oral gavage. The physiological measurements were repeated at 20 and 36-week intervals and then the mice were euthanized. New Zealand white male rabbits (7lbs) were fed rabbit chow supplemented with 0.2% cholesterol and 14% coconut oil with and without 10mpk D-PDMP for 90 days. At 1, 2, 3 and 4 months, 5mL of blood was drawn from the ear vein. Serum was prepared and total cholesterol levels were measured at each time point. Lactosylceramide levels were measured at 3 months. All experimental protocols were approved by the Committee for Animal Care and Use at the Johns Hopkins University.
Glycosyltransferase Assays
Aortic tissue was homogenized in Tris buffer (pH 7.8) and centrifuged at 10,000 rpm for 10 minutes. The supernatant was used as a source for enzyme. [3H]UDP-Gal (American Radiolabeled Chemicals, St. Louis, MO) served as the nucleotide sugar donor in LacCer synthase assay and [3H] UDP-Glc served as the nucleotide sugar donor in GlcCer synthase assay. The details of these assays have been described previously11, 12. All assays were conducted in triplicate from 3 to 5 aortic samples from mice in each group.
Measurement of Atherosclerotic Lesions
Trans-thoracic echocardiography was performed in conscious mice using the 2100 Visualsonic ultrasound device (Toronto, Ontario, Canada), equipped with a 40MHz linear transducer respectively13, 14. The aorta was viewed in the two-dimensional (2D) mode and LV viewed along the parasternal long axis. The intima-media thickness (IMT) was measured from the ascending aortic wall and computed as the difference between external (Ao-ex) and internal (Ao-int) diameters.
Inner ascending aortic diameter was measured from the inner-to-inner edge and the external was measured from the external edge to the external edge of the ascending aortic wall. All measurements were performed according to the guidelines set by the American Society of Echocardiography. For each mouse, three to five values for each measurement were obtained and averaged for evaluation.
Blood pressure and Pulse wave velocity measurements
Systolic, diastolic and mean arterial blood pressure and heart rate were measured non-invasively in conscious mice using the CODA tail cuff blood pressure system (Kent Scientific Corporation USA). Data were recorded for later analysis.
Pulse wave velocity (PWV) measurement was performed non-invasively using the high frequency and high-resolution Doppler spectrum analyzer (DSPW). Mice were placed supine on a temperature and ECG controlled (Indus Instruments) plate and under anesthesia with 1.5% isoflurane. Core temperature was maintained at 37 °C. A 10-MHz Doppler probe was used to measure blood flow velocity signals at the thoracic and abdominal aorta sequentially. PWV was calculated by the thoracic-abdominal distance divided by the pulse transit time between flow pulses recorded at the thoracic and abdominal aortic sites. Pulse transit time was determined by the time delay between the foot of the proximal and distal aortic flow waves in reference to the R wave of the ECG. The sequential flow measurements in the aorta were taken a short time apart and there was no detectable difference in physiological parameters (e.g. heart rate) between the two measurements. Heart rate was maintained at a normal physiologic heart rate of approximately 500 (b/min)15–17. Estimated effect of mean pressure (MP (mmHg) on aortic PWV (PWVp, m/sec) was calculated using the following equation (Avolio A et al, unpublished observations):
This relationship was used to correct for changes in PWV due to mean pressure between the treated and control groups.
Histopathology
Masson trichrome staining was performed on 5μm thin slices of the ascending aorta from mice 36 weeks after treatment and HFHC diet. These samples were photographed using a Nikon 80I Eclipse equipped with Nikon DS-EI1 camera and the NIS-Elements software (Nikon, Japan) was used for image analysis.
Measurement of Triglyceride and Cholesterol Levels
The serum level of triglycerides, LDL cholesterol and HDL cholesterol were measured using commercially available kits from Wako Chemicals (Richmond, VA). Liver levels of these lipids were quantified by HPTLC followed by densitometric scanning of charred plates.
Measurement of oxidized LDL Levels
The serum level of oxidized LDL (oxLDL) was measured using an ELISA assay and monoclonal antibody against human oxLDL (Avanti Polar Lipids, Alabaster, AL). The ApoE−/− mouse serum was plated in a 96-well microtitration plate at a 1:100 dilution in PBS containing 0.27 mM EDTA and 20uM BHT overnight at 4 C. The oxLDL antibody was added after washing at a concentration of 5ug/mL in a solution of 0.27 mM EDTA, 0.02% sodium azide in PBS and incubated overnight at 4 C. The binding of the secondary antibody of goat anti-mouse IgM conjugated to alkaline phosphatase (Sigma-Aldrich, St. Louis, MO) was quantified using the chemiluminescent substrate pNPP (New England Biolabs, Ipswich, MA). Following a 4-hour incubation with the secondary antibody, the reaction was stopped with NaOH and the plate was read at 495 nm on a microplate reader (Bio-Rad Laboratories, Hercules, CA).
HPLC Analysis of Glycosphingolipid Levels in Liver Tissue in ApoE (−/−) Mice
Approximately 10 mg of liver tissue was homogenized in chloroform-methanol (2:1, v/v) and lipids were extracted according to the Bligh and Dyer method18. The total lipid extracts were dried in nitrogen and subject to deacylation using sphingosine ceramide N-deacylation19. Following deacylation, lipids underwent o-phthalaldehyde derivatization and quantification of the levels of glucosylceramide and lactosylceramide by reverse-phase high performance liquid chromatography (RP-HPLC). A C18 column was used with an isocratic organic mobile phase (methanol-water, 88:12, v/v) and calibrated with standard glycosphingolipids of known chemical structure and column affinity. All samples were analyzed in triplicate and a representative quantity (n = 3) of liver tissue samples was used for each treatment from control, placebo, 5mpk and 10mpk D-PDMP–treated ApoE−/− mice.
Analysis of gene expression by Quantitative Real-Time PCR
A 50-mg piece of liver tissue was homogenized from each subject and total RNA was isolated using TRIzol reagent according to the manufacturer’s instruction (Invitrogen, Camarillo, CA). Two micrograms of RNA were reverse-transcribed with SuperScript II using random primers. Real-time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) in an Applied Biosystems Step one Real time PCR system with the following thermal cycling conditions: 10 min at 95 °C, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min for denaturation, annealing and elongation. Relative mRNA levels were calculated by the method of 2−DDCt. Data were normalized to GAPDH mRNA levels. To determine the specificity of amplification, melting curve analysis was applied to all final PCR products. All samples were performed in triplicate. Primers used in the present study (Supplemental Table S1) were synthesized by Integrated DNA Technologies (Coralville, USA). Expression suite software (Applied Biosystems) was used to analyze the data.
Western blot analysis
Approximately 90–100 mg of frozen liver tissue from each animal was homogenized in 1 mL of buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, pH 7.4) containing protease inhibitor cocktail (Roche). After protein quantitation using Bradford protein assay reagent (PIERCE), equal amount of homogenate proteins were resolved by SDS-PAGE. Respective proteins were detected by immunoblotting. Immunoreactive bands of predicted molecular mass were visualized using an ECL plus kit (GE Healthcare Life Sciences) and quantified with the KODAK Molecular Imaging Software (Kodak, New Haven, CT). GAPDH was used as a loading control. The data represent the mean ± SD of three independent experiments.
Statistical Analysis
All values are expressed as dot plots alongside mean± SEM. Comparison between groups was performed with Kruskal-Wallis test and Dunn’s multiple comparison post-test. For repeated measures in mice and rabbits, a two-way repeated measures ANOVA (RM-ANOVA) was performed with Bonferroni’s multiple comparison test for comparisons between groups. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.001. GraphPad PRISM and Excel statistical software were used.
Results
Aortic wall thickness, vascular stiffness and remodeling with and without treatment
Ultrasound studies revealed that, as compared to control mouse aorta (Fig 1A), feeding a western diet to mice from age 12 weeks to 20 weeks contributed to a marked increase in aortic wall thickening (indicated by arrows) (Fig 1B) in placebo mice. This was not observed in mice fed HFHC diet plus 10mpk of D-PDMP (Fig 1C). At 36 weeks of age the control mice show some Ca2+ deposit, perhaps due to aging (Fig 1D). However, extensive Ca2+ deposits were noted in placebo mice (Fig 1E) and this was prevented by treatment with 10mpk of D-PDMP (Fig 1F).
Figure 1.

Aortic wall thickening and pulse wave velocity in atherosclerotic mice is ameliorated by treatment with D-PDMP. Aortic ultrasound imaging: 2DB-mode ultrasound images of the aorta from ApoE−/− mice fed mice chow. Top panel (A, B, C) shows 20 week old mice. Bottom panel (D, E, F) shows 36 week old mice. Control (A) ApoE−/− mice fed mice chow. ApoE−/− mouse fed high fat, high cholesterol (HFHC) diet plus vehicle (B), HFHC plus 10mpk of D-PDMP (C). Thirty six week old control ApoE−/− mice fed mice chow (D); placebo fed HFHC chow (E) and HFHC fed mice treated with 10mpk D-PDMP (F). Note the arrows indicate marked increase in aortic wall thickening in HFHC plus vehicle fed; placebo (B mice) mice at 20 weeks of age. This was followed by a marked increase in Ca2+ deposits at 36 weeks of age (marked by asterisk) in this group of mice as compared to control mice. This was prevented by feeding D-PDMP (F). Aortic wall thickness and aortic wall stiffness in atherosclerotic mice is ameliorated by treatment with D-PDMP shown in graphs (G) and (H). Quantitation of intima-media thickness (AoIMT) (G) and pulse wave velocity (PWV) respectively (H), in ApoE−/− mouse on a mice chow diet (control 0.63 ± 0.04 mm/4.37±0.26 m/s), HFHC diet plus vehicle (Placebo 1.21 ± 0.06 mm/6.38±0.89 m/s) (H), HFHC diet plus treatment with 5 and 10mpk of D-DPMP (1.04 ± 0.04mm/6.07±0.5 m/s and 0.73 ± 0.04 mm/4.24±0.15 m/s). Note that both AoIMT and PWV increase continuously in placebo mice from 12 weeks to 36 weeks and this was ameliorated upon treatment with D-PDMP in a dose-dependent manner. A two-way RM ANOVA using the Bonferroni multiplo comparisons post-test was performed..* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; n = 3–5.
Further quantitative analysis revealed that aortic intima thickening progressed rapidly in placebo mice fed HFHC diet from 12 weeks to 36 weeks of age as compared to control mice. However, D-PDMP exerted a dose-dependent decrease in aortic intima thickening. In fact, the use of 10mpk of D-PDMP maintained the aortic intima-media thickening in HFHC fed mice comparable to control mice (Fig 1G). Likewise, pulse wave velocity measurement revealed a similar pattern, i.e., suggesting that the thickening of the aortic wall was associated with arterial stiffening. This was lowered to control levels upon treatment with 5 and 10mpk D-PDMP in a dose-dependent manner (Fig 1H). Since blood pressure did not change significantly with treatment, the increase in arterial stiffness is largely independent of blood pressure.
Body weight for baseline (29.4±0.93 g) the control (31.13±1.61 g) and the placebo (34.5±0.88 g) groups were not significantly different. At 36 weeks, the 5 and 10 mpk plus HFHC diet fed mice showed a dose -dependent increase in body weight (36.53 ±1.77 g and 41.03±0.55 g) compared to other groups. This was mainly because of an increase in bone mass density and muscle mass. These mice were also physically active and less aggressive than the placebo group on HFHC diet plus vehicle only. The latter instead showed an increase in percent body fat and were fragile. This suggests that HCHF may have an association with bone loss and calcified aortic wall shown in Fig 1E and no association with obesity.
Assessment of Atherosclerosis
Masson trichrome staining of 5μm thin slices of the aorta in control mice revealed normal aortic wall thickness with no fibrosis (Fig 2A). In contrast, 36-week-old placebo mice fed HFHC diet alone and daily dose of vehicle exhibited narrowed lumen volume, occupied largely by plaques loaded with cholesteryl esters (needle-like white crystals), amorphous Ca2+ deposits within the plaque and extensive fibrosis (Fig 2B).
Figure 2.
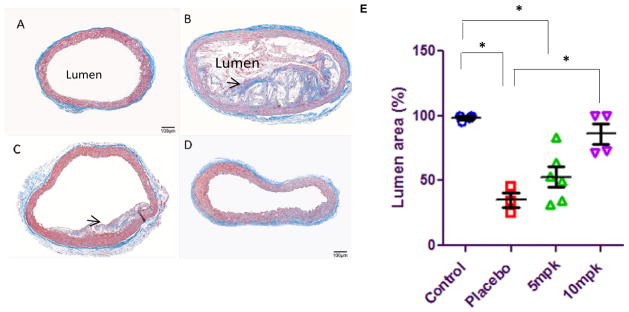
D-PDMP treatment ameliorates atherosclerotic plaque buildup and lumen volume in ApoE−/− mice fed a western diet. Masson Trichrome stained ascending aortic rings of ApoE−/− mouse: Control mice fed regular mice chow (A), mice fed high fat, high cholesterol (HFHC) diet consisting of 2% fat and 1.25 cholesterol plus vehicle (Placebo) (B), HFHC plus 5mpk, D-PDMP(C), and HFHC plus 10mpk, D-PDMP (D). Bar = 50 μm. Elastin fibers (E) decreased following treatment with D-PDMP. Quantification of lumen area (F) reveals a decrease in lumen volume in the mice fed HFHC. Lumen area is significantly reduced due to increased plaque accumulation in placebo mice aorta (Black arrow, B). Treatment with D-PDMP significantly reduced and/or delayed medial thickening, elastin fibers, and plaque accumulation fragmentation in a dose-dependent manner. A nonparametric one-way ANOVA using the Kruskal-Wallis test and Dunn’s multiple comparison post-test were performed. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; n = 3–5.
Treatment with 5mpk of D-PDMP significantly improved lumen volume; cholesteryl esters were expunged from the plaque (Fig 2C) compared to placebo (Fig 2B), reduced fibrosis, and elastin fragmentation. The most unexpected result was the observation that treatment with 10mpk of D-PDMP completely prevented atherosclerosis in these mice at age 36 weeks (Fig 2D). Notably, coronary artery disease, the principle cause of morbidity and mortality due to atherosclerosis, was drastically prevented by the treatment. A clear lumen was observed at 36 weeks for control mice, while a narrowed lumen volume was observed for placebo mice fed HFHC diet (Fig 2E). This was mitigated by treatment with 5mpk and 10mpk D-PDMP (Fig 2E). Histopathological analysis of the left coronary artery and quantification of lumen area revealed that D-DPMP reduces obstruction and plaque deposition in apoE−/− mice (Supp. Fig. 1).
Treatment with D-PDMP dose-dependently decreases the activity of glycosyltransferases in the aorta in ApoE−/− mice
High activity of glucosylceramide synthase and lactosylceramide synthase in the aorta in ApoE−/− mice was observed in 20-week-old mice fed HCHF diet (Fig 3A, 3B). When these mice were given a daily supply of D-PDMP by oral gavage during this period (at age 20 weeks) it dose -dependently decreased the activity of these enzymes. We noted a 10-fold increase in the activity of LacCer synthase in 36-week-old mice ApoE−/− mice fed the western diet (Fig 3C). However, up to the age of 36 weeks did not exhibit any further increase in the activity of GlcCer synthase compared to 20-week-old mice (Fig 3D). Notwithstanding these observations, some of this increase may be attributed to effects of aging. D-PDMP dose-dependently decreased the activity of both GlcCer synthase and LacCer synthase in 36 week old mice, as indicated by the mass of LacCer (Fig 3E) and GlcCer (Fig 3F).
Figure 3.

Glycosphingolipid glycosyltransferase activity in ApoE−/− mice fed high fat and high cholesterol diet with and without D-PDMP. Freshly harvested aorta tissue was stored in Tris HCl buffer supplemented with a protease inhibitor cocktail, homogenized and the activity of glucosylceramide synthase and lactosylceramide synthase were measured. Lactosylceramide synthase activity significantly decreased at 20 weeks (A) and 36 weeks (C) following treatment with D-PDMP. A significant change in glucosylceramide synthase activity was also observed at 20 weeks (B) and 36 weeks (D). In (E) and (F), treatment with D-PDMP decreases the mass of glycosphingolipids in the liver of ApoE−/− mice fed a western diet. Glycosphingolipids were extracted from 10 mg of harvested liver tissue and homogenized in CHCl3-MeOH, 2:1). Following treatment with the enzyme SCDase and derivatization by OPA, mass was calculated via liquid chromatography and tandem mass spectrometry against glucosylceramide and lactosylceramide standards. A significant decrease from the placebo in the mass of glucosylceramide was seen in the 5mpk treated and 10mpk treated mice fed the HFHC diet. A nonparametric one-way ANOVA using the Kruskal-Wallis test and Dunn’s multiple comparison post-test were performed. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; n = 3.
Treatment with D-PDMP ameliorates hyperlipidemia in ApoE−/− mice
Feeding a high fat and cholesterol diet markedly increased the serum level of oxidized LDL measured using an ELISA assay. In contrast, feeding the glycosyltransferase inhibitor dose-dependently decreased the serum levels of ox-LDL level to below baseline levels in the ApoE−/− mice serum (Fig 4A). Similarly, the serum level of LDLc (Fig 4B) and triglycerides (Fig 4C) were increased in western diet fed mice serum. This was also decreased to baseline values in mice fed D-PDMP. Interesting, although feeding the western diet decreased the serum levels of HDLc (Fig 4D), treatment with D-PDMP raised HDLc level in the serum. Thus D-PDMP may be cardioprotective.
Figure 4.

Plasma levels of oxidized LDL, cholesterol, triglycerides, and HDL-c in ApoE−/− mice fed a high fat and high cholesterol diet with and without D-PDMP. Serum extracted from ApoE−/− mice was analyzed for the presence of oxLDL (A), LDLc (B), triglycerides (C), and HDLc (D) on microtiter plates. Serum concentrations of oxLDL were determined using an immunohistochemical ELISA assay with an antibody against oxLDL. Concentrations of LDLc triglycerides and HDLc concentrations were taken from microtiter readings following Wako kit assays. Values are means ± SEM. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001; n = 3.
Effect of D-PDMP on the expression of genes involved in LDL metabolism
As shown in Fig 5A, treatment with 10mpk D-PDMP increased the mRNA levels of 3-hydroxy-3-methylglutaryl-Coenzyme A reductase and sterol regulatory element binding transcription factor 2 up to approximate 5.2-fold (P < 0.0001) and 3.6-fold (P < 0.0001), respectively. The mRNA level of Low density lipoprotein receptor was also elevated by D-PDMP respectively.
Figure 5.

Effect of D-PDMP on the expression of hepatic genes, which play roles in cholesterol and lipid metabolism. Expression of hepatic genes involved in LDL metabolism; 3-hydroxy-3-methylglutaryl-Coenzyme A reductase (Hmgcr), Low density lipoprotein receptor (Ldlr), Sterol regulatory element binding transcription factor 1(Srebf1), Sterol regulatory element binding transcription factor 2(Srebf2) (A) HDL metabolism; apolipoprotein A-I(Apoa1), scavenger receptor class B (SRB-1), CD36 antigen (B) VLDL and Triglyceride metabolism; lipoprotein lipase (Lpl), very low density lipoprotein receptor (Vldlr) (C) and Cholesterol efflux and uptake; ATP-binding cassette sub-family ABCA1(Abca1), cholesterol 7 alpha-hydroxylase(Cyp7A1) (D) as assessed by quantitative real-time PCR. Expressions of these genes were significantly up regulated in mice treated with D-PDMP in a dose dependent manner. Values are means ± SEM. A nonparametric one-way ANOVA using the Kruskal-Wallis test and Dunn’s multiple comparison post-test were performed. * p ≤ 0.05, ** p ≤ 0.01, **** p ≤ 0.0001; n = 3.
Effect of D-PDMP on the expression of transporter genes involved in cholesterol efflux, bile synthesis and triglyceride metabolism
Real time PCR showed that the expression level of CD36, SRB-1 and ABCA1 genes were significantly upregulated by D-PDMP treatment (Fig 5B). These transporters play a major role in cholesterol efflux from peripheral tissues. As shown in Fig 5C, treatment with D-PDMP dose dependently increased the mRNA levels of lipoprotein lipase and very low-density lipoprotein receptor to approximate 4.8-fold (P < 0.0001) and 2.4-fold (P < 0.0001) in liver samples of drug treated mice. The level of Cyp7A1 was also significantly up regulated by the drug treatment (Fig 5D), indicating cholesterol catabolism and bile acid synthesis upon drug treatment.
Effect of D-PDMP on the expression of proteins involved in LDL metabolism
Western immune-blot assays revealed that placebo mice liver tissue had significantly lower expression of SREBP2 protein as well as LDL receptor protein mass (Fig 6A,B). In contrast, treatment with D-PDMP markedly increased the expression of these proteins. These findings are in tandem with the gene expression data above, suggesting that drug treatment prevented atherosclerosis by increasing cholesterol metabolism via the LDL receptor pathway.
Figure 6.


Western blot analysis of liver Srebp-2, LDLR, ABCG5 and ABCG8 proteins. Western blots illustrate D-PDMP induces the expression of Srebp-2 (A), LDL receptor (B), ABCG5 (C) and ABCG8 (D) proteins in liver tissues. Values are means ± SEM. A nonparametric one-way ANOVA using the Kruskal-Wallis test and Dunn’s multiple comparison post-test were performed. * p ≤ 0.05, **** p ≤ 0.0001; n = 3.
Treatment with D-PDMP ameliorates atherosclerosis in normal rabbits fed a western diet
Treatment with the glycosphingolipid glycosyltransferase inhibitor D-PDMP prevented atherosclerosis in New Zealand white rabbits. Rabbits fed HFHC diet showed a marked increase in aortic lactosylceramide levels (Fig 7A) accompanied by extensive atherosclerosis and a 17-fold increase in serum cholesterol (Fig 7B). In contrast to rabbits fed HFHC diet alone, rabbits treated with D-PDMP showed a prevention of atherosclerosis as measured by lactosylceramide and cholesterol levels which were similar to the control (Fig 7).
Figure 7.
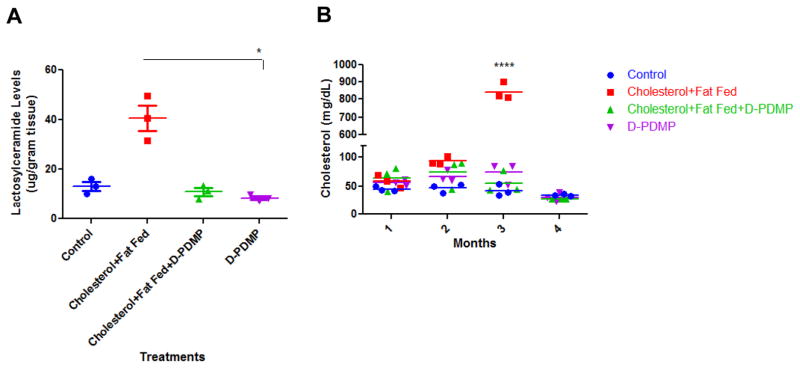
Inhibiting glycosphingolipid synthesis inhibits atherosclerosis in rabbits. New Zealand white rabbits (n=3) were fed a diet with and without cholesterol (2%) and fat (14%) and with and without D-PDMP (10mpk) for three months. Marked increase in lactosylceramide levels in the aorta tissue was observed (A) accompanied by extensive atherosclerosis and marked increase (17-fold) in serum cholesterol levels (B) were found in rabbits fed the hyperlipidemic diet in comparison to control and drug-treated rabbits at 3 months. This was prevented by treatment with D-PDMP. Statistical analysis: In 7A, a nonparametric one-way ANOVA using the Kruskal-Wallis test and Dunn’s multiple comparison post-test were performed. In 7B, a two-way repeated measures ANOVA was performed with Bonferroni’s multiple comparisons post-test. * p ≤ 0.05, **** p ≤ 0.0001; n = 3.
Discussion
The following major findings emerged from the present study. D-PDMP, an inhibitor of glucosylceramide synthase and Lactosylceramide synthase dramatically and dose-dependently ameliorated atherosclerosis in ApoE−/− mice and normal rabbits fed a western diet. In mice this was accompanied with complete reversal of aortic intima-media thickening, and pulse wave velocity, an index of vascular stiffness. Our mechanistic studies revealed that D-PDMP decreased serum levels of cholesterol by way of regulating the expression of genes implicated in the biosynthesis, egress, and conversion of cholesterol to bile acids. The triglyceride levels were also dramatically reduced in the D-PDMP -treated mice by an increase in VLDL receptor and lipoprotein lipase gene expression.
A close association between glycosphingolipids and atherosclerosis was suggested by us3, 7 and others2, 20, 21. However, few attempts have been made to examine the effects of inhibiting/depleting the level of GSLs in atherosclerosis in experimental animal models. Recently, Bietrix et al. used an iminosugar, N-5--adamatane -1 ′-ul-methoxy)-pentyl1-deoxynojirimycin (AMP-DNM), an inhibitor of glucosylceramide synthase, which markedly reduced plasma cholesterol and inhibited atherosclerosis development in ApoE*3 Leiden and LDL receptor −/− mice. Additional studies have made use of this inhibitor in enhancing insulin responsiveness in a rat model of diabetes22 and in liver steatosis23. Another inhibitor of GlcCer synthase, Zavesca (1,5- butylimino) 1,5-dideoxy-D- glucitol and Genz-112638/miglustat is already in use for substrate inhibition therapy24 in patients with Gaucher’s disease. D-PDMP inhibits both GlcCer synthase and LacCer synthase activity in cultured mammalian cells and can directly inhibit the activity of pure LacCer synthase7. Our in vivo studies have shown its efficacy in mitigating VEGF/FGF-induced angiogenesis, restenosis in rabbits following balloon angioplasty25 and lowering renal tumor volume by 50%26. Since arteriosclerosis is a multi-factorial disease, we hypothesized that a compound such as D-PDMP could be efficacious in lowering the GSL load and the phenotypes above.
An anticipated observation in this study was the remarkable filling of the lumen with atherosclerotic plaques in ApoE −/− mice fed high fat and cholesterol diet which progressively worsened from 20 weeks to 36 weeks of age. This was accompanied with extensive arterial wall thickening and an associated increase in the arterial wall stiffness measured by ultrasound. Consequently, pulse wave velocity also increased. This was ameliorated upon treatment with D-PDMP. An unexpected observation was that from week 12 to week 20 there were no visible Ca 2+ deposits in the arterial wall. However, at 36 weeks of age, even mice fed mice chow alone had a few Ca2+ deposits. Whereas, mice fed HFHC diet had extensive Ca2+ deposits, and this was prevented by treatment with D-PDMP. These observations were made using 5 and 10mpk of D-PDMP in a dose- dependent manner. Not only are these drug concentrations 10 -fold lower than a previous report8, but also these mice did not accumulate subcutaneous fat. There was also a higher bone density27 as compared to the placebo over the 36-week study, and no overt obesity was observed. As expected, D-PDMP treatment not only decreased the activity of GlcCer synthase but also LacCer synthase in the aorta in these mice. This was accompanied by a decrease in the level of GlcCer, LacCer as well as ceramide in the liver tissue. D-PDMP also decreased the level of serum cholesterol and triglycerides via recruiting multiple genes/pathways in lipid metabolism. For example, we noted that D-PDMP increased LDL receptor and SREBP2 gene expression and decreased HMG-CoA reductase gene expression. These findings are suggestive of inhibition of cholesterol biosynthesis and increased LDL uptake. It also increased the expression of genes responsible for cholesterol efflux by way of increasing the expression of ABCA1 and increased expression of ABCG 5, ABCG 8 proteins responsible for pumping cholesterol out from liver and intestine for excretion. In a previous study it was shown that GSL accumulation in particular lactosylceramide in cultured cells can inhibit the efflux of cholesterol via the ABCA1/Apolipoprotien A-1 pathway. In contrast, D-PDMP was found to serve as a cholesterol efflux accelerator by inducing the expression of these genes. Our findings are in agreement and expand these observations to an animal model of atherosclerosis. A 7-hydroxylase enzyme, coded by the gene Cyp7A1, can convert cholesterol to bile acid. The expression of this gene was increased in mice treated with D-PDMP. This gene plays a pivotal role in the expression of an enzyme required for the conversion of cholesterol to bile acids. In a previous study the iminosugar N sugar AMP- DNM was also shown to increase the expression of this gene in liver tissue in ApoE*3 Leiden mice and facilitate the lowering of cholesterol load by conversion to bile acids and their subsequent excretion8. Moreover, in our study HDL cholesterol was increased about 2-fold in the serum of D-PDMP treated mice. Thus a combination of genes implicated in the lipid and lipoprotein metabolic pathways contributed to the complete reversal of a marked increase in cholesterol level and atherosclerosis in the ApoE−/− mice fed HFHC diet. Previous studies have shown that an enzyme lipoprotein lipase plays a crucial role in linking up VLDL, the major triglyceride carrying lipoprotein to VLDL receptors28. We observed that the liver tissue in D-PDMP treated mice had increased expression of gene required for the expression of lipoprotein lipase1 as well as VLDL receptors. Such observations could explain why treatment with D-PDMP could reduce the markedly increased serum levels of triglycerides in ApoE−/− mice a HFHC diet for 36 weeks (Fig 4C).
We have previously shown that oxidized LDL can dose- dependently increase the activity of LacCer synthase in cultured human arterial smooth muscle cells by phosphorylating the serine, threonine and tyrosine residues in this enzyme. This was mitigated by pre- treatment of cells with D-PDMP29. The activation of this enzyme generated LacCer that, in turn, activated a signaling cascade involving ROS generation, p44MAPK activation, nuclear transcription factor c-Fos and cyclin A to induce cell proliferation29. In the present study we show that feeding a western diet significantly increases the level of Ox-LDL in the serum of ApoE−/− mice. This may have contributed to the activation of LacCer synthase in the arterial wall, thus contributing to an increase in the levels of LacCer. In contrast, in mice fed western diet plus D-PDMP the level of Ox-LDL was decreased to basal levels (Fig 4A). This reduction in LacCer level might have contributed to the resumption of cholesterol efflux at the normal level (Fig 8). An important observation made in our present study is that fibrosis contributes greatly towards arterial wall thickness in our western diet fed model of atherosclerosis and this too was prevented in mice fed D-PDMP.
Figure 8.

D-PDMP inhibits ox-LDL induced LCS activity and atherosclerosis development. Oxidized LDL (ox-LDL) activates LacCer synthase (LCS) to produce large quantities of LacCer, which stimulates the activity of NADPH oxidase. Concomitantly, there is an increase in the generation of superoxide radicals (ROS). ROS mediates p44MAPK phosphorylation thereby stimulating c-fos expression, promoting cell proliferation of vascular-smooth-muscle cells further to angiogenesis and generation of atherosclerosis. D-PDMP, a potent inhibitor of LCS, impaired ox-LDL mediated induction of LCS activity thereby inhibiting the above pathway.
In sum, we conclude that there is a tight relationship between glycosphingolipid metabolism, and lipoprotein metabolism, which heavily impacts upon aortic intima media thickening via fibrosis and lipid deposition. Such multigeneic changes bring about arteriosclerosis and its pathological sequelae. Herein, we demonstrate that several genes responsible for the biosynthesis of cholesterol, its efflux, absorption, bile acid production, and excretion were improved upon treatment with a glycosphingolipid glycosyltransferase inhibitor. Additionally, genes responsible for triglyceride metabolism were also improved due to treatment resulting in decreased triglyceride levels in apoE−/− mice. An important observation was the increased expression of genes responsible for apoA1, the major protein in HDL, thus contributing to an increase in the plasma levels of HDL in treated mice.
Limitations
The glycosphingolipid inhibitor used in this study inhibits the synthesis of glucosylceramide synthase as well as lactosylceramide synthase and therefore is not specific for a particular enzyme in the biosynthetic pathway. However, this compound is not toxic to animals when given for the duration of this study -6 months. Clearly, further studies are required using relatively more specific inhibitors in the near future. PWV depends on arterial pressure, but in these experiments PWV was measured using external Doppler transducers so it was not possible to obtain PWV-pressure relationships by altering pressure with vasoactive agents. However, the changes in pressure between the groups was not large as assessed by tail-cuff measurements, and when there was a change the PWV values were adjusted for pressure differences using relationships obtained in mice from other studies. Results showed that pressure adjustments did not produce any major alterations, hence this is not considered to be a significant limitation in these studies.
Supplementary Material
Acknowledgments
We thank Sara Kimiko Suzuki, Maya Hernandez, Fraulein Li, Bryan Brensinger and Jennifer Hou for assistance during various phases of this research.
Funding Sources: This study was funded by NIH grant’s P01HL107153 and 3PO1HL107153-03S1 (SC).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Kwiterovich P. The johns hopkins textbook of dyslipidemia. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2010. [Google Scholar]
- 2.Dawson G, Kruski AW, Scanu AM. Distribution of glycosphingolipids in the serum lipoproteins of normal human subjects and patients with hypo- and hyperlipidemias. J Lipid Res. 1976;17:125–131. [PubMed] [Google Scholar]
- 3.Chatterjee S, Kwiterovich PO. Glycosphingolipids of human plasma lipoproteins. Lipids. 1976;11:462–466. doi: 10.1007/BF02532836. [DOI] [PubMed] [Google Scholar]
- 4.Bhunia AK, Arai T, Bulkley G, Chatterjee S. Lactosylceramide mediates tumor necrosis factor-alpha-induced intercellular adhesion molecule-1 (icam-1) expression and the adhesion of neutrophil in human umbilical vein endothelial cells. J Biol Chem. 1998;273:34349–34357. doi: 10.1074/jbc.273.51.34349. [DOI] [PubMed] [Google Scholar]
- 5.Gong N, Wei H, Chowdhury SH, Chatterjee S. Lactosylceramide recruits pkcα/ε and phospholipase a2 to stimulate pecam-1 expression in human monocytes and adhesion to endothelial cells. Proc Natl Acad Sci U S A. 2004;101:6490–6495. doi: 10.1073/pnas.0308684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glaros EN, Kim WS, Quinn CM, Wong J, Gelissen I, Jessup W, Garner B. Glycosphingolipid accumulation inhibits cholesterol efflux via the abca1/apolipoprotein a-i pathway: 1-phenyl-2-decanoylamino-3-morpholino-1-propanol is a novel cholesterol efflux accelerator. The J Biol Chem. 2005;280:24515–24523. doi: 10.1074/jbc.M413862200. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee S, Ghosh N. Oxidized low density lipoprotein stimulates aortic smooth muscle cell proliferation. Glycobiology. 1996;6:303–311. doi: 10.1093/glycob/6.3.303. [DOI] [PubMed] [Google Scholar]
- 8.Bietrix F, Lombardo E, van Roomen CP, Ottenhoff R, Vos M, Rensen PC, Verhoeven AJ, Aerts JM, Groen AK. Inhibition of glycosphingolipid synthesis induces a profound reduction of plasma cholesterol and inhibits atherosclerosis development in apoe* 3 leiden and low-density lipoprotein receptor−/− mice. Arterioscler Thromb Vasc Biol. 2010;30:931–937. doi: 10.1161/ATVBAHA.109.201673. [DOI] [PubMed] [Google Scholar]
- 9.Kamani M, Mylvaganam M, Tian R, Rigat B, Binnington B, Lingwood C. Adamantyl glycosphingolipids provide a new approach to the selective regulation of cellular glycosphingolipid metabolism. J Biol Chem. 2011;286:21413–21426. doi: 10.1074/jbc.M110.207670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Radin NS, Shayman J, Inokuchi J-I. Metabolic effects of inhibiting glucosylceramide synthesis with pdmp and other substances. Adv Lipid Res. 1993;26:183. [PubMed] [Google Scholar]
- 11.Chatterjee S. Assay of lactosylceramide synthase and comments on its potential role in signal transduction. Meth Enzymol. 2000;311:73–81. doi: 10.1016/s0076-6879(00)11068-7. [DOI] [PubMed] [Google Scholar]
- 12.Basu M, De T, Das KK, Kyle JW, Chon HC, Schaeper RJ, Basu S. Glycolipids. Meth Enzymol. 1987;138:575–607. doi: 10.1016/0076-6879(87)38053-x. [DOI] [PubMed] [Google Scholar]
- 13.Habashi JP, Holm TM, Doyle JJ, Aziz H, Bedja D, Dietz HC. At2 signaling is a positive prognostic and therapeutic modifier of marfan syndrome: Lessons on the inequality of acei and arbs. Circulation. 2009;120:S963–S963. [Google Scholar]
- 14.Olson LE, Bedja D, Alvey SJ, Cardounel A, Gabrielson KL, Reeves RH. Protection from doxorubicin-induced cardiac toxicity in mice with a null allele of carbonyl reductase 1. Cancer Res. 2003;63:6602–6606. [PubMed] [Google Scholar]
- 15.Jun J, Reinke C, Bedja D, Berkowitz D, Bevans-Fonti S, Li J, Barouch LA, Gabrielson K, Polotsky VY. Effect of intermittent hypoxia on atherosclerosis in apolipoprotein e-deficient mice. Atherosclerosis. 2010;209:381–386. doi: 10.1016/j.atherosclerosis.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, Polotsky VY, Biswal S. Disruption of nrf2, a key inducer of antioxidant defenses, attenuates apoe-mediated atherosclerosis in mice. PLoS One. 2008:3. doi: 10.1371/journal.pone.0003791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sussan TE, Rangasamy T, Blake DJ, Malhotra D, El-Haddad H, Bedja D, Yates MS, Kombairaju P, Yamamoto M, Liby KT, Sporn MB, Gabrielson KL, Champion HC, Tuder RM, Kensler TW, Biswal S. Targeting nrf2 with the triterpenoid cddo-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci U S A. 2009;106:250–255. doi: 10.1073/pnas.0804333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 19.Zama K, Hayashi Y, Ito S, Hirabayashi Y, Inoue T, Ohno K, Okino N, Ito M. Simultaneous quantification of glucosylceramide and galactosylceramide by normal-phase hplc using o-phtalaldehyde derivatives prepared with sphingolipid ceramide n-deacylase. Glycobiology. 2009;19:767–775. doi: 10.1093/glycob/cwp047. [DOI] [PubMed] [Google Scholar]
- 20.Garner B, Priestman DA, Stocker R, Harvey DJ, Butters TD, Platt FM. Increased glycosphingolipid levels in serum and aortae of apolipoprotein e gene knockout mice. J Lipid Res. 2002;43:205–214. [PubMed] [Google Scholar]
- 21.Glaros EN, Kim WS, Quinn CM, Jessup W, Rye K-A, Garner B. Myriocin slows the progression of established atherosclerotic lesions in apolipoprotein e gene knockout mice. J Lipid Res. 2008;49:324–331. doi: 10.1194/jlr.M700261-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M, Dubbelhuis PF, Aten J, Kuipers F, Serlie MJ, Wennekes T. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes. 2007;56:1341–1349. doi: 10.2337/db06-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bijl N, Sokolović M, Vrins C, Langeveld M, Moerland PD, Ottenhoff R, van Roomen CP, Claessen N, Boot RG, Aten J. Modulation of glycosphingolipid metabolism significantly improves hepatic insulin sensitivity and reverses hepatic steatosis in mice. Hepatology. 2009;50:1431–1441. doi: 10.1002/hep.23175. [DOI] [PubMed] [Google Scholar]
- 24.McEachern KA, Fung J, Komarnitsky S, Siegel CS, Chuang W-L, Hutto E, Shayman JA, Grabowski GA, Aerts JM, Cheng SH. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of gaucher disease. Molec Genet Metab. 2007;91:259–267. doi: 10.1016/j.ymgme.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Chatterjee S. Methods for treatment of angiogenesis. US Patent application #20090202439. Issued Aug 13, 2009.
- 26.Chatterjee S, Alsaeedi N, Hou J, Bandaru VVR, Wu L, Halushka MK, Pili R, Ndikuyeze G, Haughey NJ. Use of a glycolipid inhibitor to ameliorate renal cancer in a mouse model. PLoS One. 2013;8:e63726. doi: 10.1371/journal.pone.0063726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hyder JA, Allison MA, Barrett-Connor E, Detrano R, Wong ND, Sirlin C, Gapstur SM, Ouyang P, Carr JJ, Criqui MH. Bone mineral density and atherosclerosis: The multi-ethnic study of atherosclerosis, abdominal aortic calcium study. Atherosclerosis. 2010;209:283–289. doi: 10.1016/j.atherosclerosis.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takahashi S, Suzuki J, Kohno M, Oida K, Tamai T, Miyabo S, Yamamoto T, Nakai T. Enhancement of the binding of triglyceride-rich lipoproteins to the very low density lipoprotein receptor by apolipoprotein e and lipoprotein lipase. J Biol Chem. 1995;270:15747–15754. doi: 10.1074/jbc.270.26.15747. [DOI] [PubMed] [Google Scholar]
- 29.Chatterjee S, Bhunia AK, Snowden A, Han H. Oxidized low density lipoproteins stimulate galactosyltransferase activity, ras activation, p44 mitogen activated protein kinase and c-fos expression in aortic smooth muscle cells. Glycobiology. 1997;7:703–710. doi: 10.1093/glycob/7.5.703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.