

Abstract
Accurate methods for predicting protein-ligand binding affinities are of central interest to computer-aided drug design for hit identification and lead optimization. Here, we used the mining minima (M2) method to predict cucurbit[7]uril binding affinities from the SAMPL4 blind prediction challenge. We tested two different energy models, an empirical classical force field, CHARMm with VCharge charges, and the Poisson-Boltzmann Surface Area (PBSA) solvation model; and a semiempirical quantum mechanical Hamiltonian, PM6-DH+, coupled with the COSMO solvation model and a surface area term for nonpolar solvation free energy. Binding affinities based on the classical force field correlated strongly with the experiments with a correlation coefficient (R2) of 0.74. On the other hand, binding affinities based on the quantum mechanical energy model correlated poorly with experiments (R2 = 0.24), due largely to two major outliers. As we used extensive conformational search methods, these results point to possible inaccuracies in the PM6-DH+ energy model or the COSMO solvation model. Furthermore, the different binding free energy components, solute energy, solvation free energy, and configurational entropy showed significant deviations between the classical M2 and quantum M2 calculations. Comparison of different classical M2 free energy components to experiments show that the change in the total energy, i.e. the solute energy plus the solvation free energy, is the key driving force for binding, with a reasonable correlation to experiment (R2 = 0.56); however, accounting for configurational entropy further improves the correlation.
Keywords: SAMPL4, supramolecular, binding affinity, host-guest, force field, semiempirical quantum
Introduction
Accurate methods for predicting protein-ligand binding affinities would greatly enhance the drug discovery process by accelerating progress and reducing costs [1,2]. While relatively simple approaches, such as docking and ligand-similarity [3], are routinely used to discover initial hits through virtual screening of compound libraries, more sophisticated free energy calculations should afford greater precision across subtle changes in ligand chemical structures, and thus promise to be particularly useful for lead optimization [4]. However, evidence for accurate prediction of binding affinities using free energy calculations is still largely anecdotal. Aspects of these calculations that merit attention include the accuracy of the energy models (force fields) and the adequacy of conformational sampling and convergence.
As methods evolve to address these issues, it is important that we critically assess our progress and identify problem areas that require further improvement. The Statistical Assessment of the Modeling of Proteins and Ligands (SAMPL) blind prediction challenge provides a valuable platform for comparing different methods to each other through validation against experimental data.[5-9] The prospective nature of the challenge reduces bias and provides for a realistic outlook on the performance of the different methods and the current state of the field.
Although it is necessary to test computational methods for biomolecular systems, as done in the SAMPL4 protein-ligand binding challenge [10], free energy calculations of protein-ligand systems can be problematic, due to their many degrees of freedom (often several thousand) and their conformational flexibility [11]. These make it difficult to assure numerical convergence, and hence to distinguish between convergence problems and force field errors when comparing computational results with experiment. In contrast, supramolecular host-guest systems are computationally quite tractable, as they typically comprise only a few hundred atoms and tend to be more rigid than proteins. As a consequence, convergence is relatively easy to achieve [12], so comparisons with experiment can report clearly on the accuracy of the energy models used [8], and the growing array of host-guest systems today provides many opportunities to test computational models of binding. In addition, host-guest systems are interesting in their own right, due to their practical applications, such as chemical sensing and drug delivery [13,14], and their convenience as model systems whose study can deepen our understanding of the physical chemistry of molecular recognition.
Here, we test the mining minima (M2) method in predictions of the binding affinities of cucurbit[7]uril (CB7) with guest molecules from the SAMPL4 challenge (Figure 1) [15]. The M2 technique combines either an empirical force field or a fast electronic structure method, with an implicit solvent model, and uses conformational search and the harmonic approximation (including, when possible, the mode-scanning correction for anharmonicity) to evaluate binding free energies that incorporate both conformational relaxation and the changes in configurational entropy of the host and guest. In the past, the M2 method using a classical empirical force field was applied successfully in both retrospective and prospective studies [16,17]. Nonetheless, our predictions for the previous SAMPL3 challenge using M2 deviated significantly from the experimental measurements [18], and we suspected that the errors likely originated from the underlying classical force field. We therefore developed a new approach, which combined the M2 method with the PM6-DH+ semiempirical quantum mechanical (QM) Hamiltonian and the COSMO solvation model [19]. The resulting QM/M2 method showed promise in initial tests for various CB7 systems [19]. Here, we report new results from M2 calculations using both the classical and semiempirical QM approaches.
Figure 1.

(A) Chemical structure of glycoluril, the repeating unit of CB7. (B) Crystallographic structure of CB7 shown in top and side views. (C) Chemical structures of SAMPL4 CB7 guest molecules. Protonation and tautomer states of the guest molecules are shown as used in the binding calculations.
Methods
Structure preparation
The structures of CB7 and the guest molecules were provided by SAMPL4 organizers, and are shown in Figure 1. Protonation and tautomer states of the guest molecules at pH 7.4 were determined using Marvin 6.04 software (ChemAxon). In order to address the uncertainty in the protonation of guest 10, we tested two different protonation states, with the central nitrogen atom carrying a formal charge of +1 or 0. The formal charges of the terminal nitrogen atoms were set to +1 in both cases. The protonation state with +2 net charge, i.e. with the central nitrogen atom deprotonated, exhibited a lower (more favorable) binding free energy using both classical and quantum M2 methods. Therefore, our predicted binding affinities for this guest are based on the +2 charge state.
Mining Minima
The Mining Minima (M2) method is described in detail elsewhere [20-22], and so is only briefly summarized here. The host-guest binding free energy is determined from the standard chemical potential (μo) of the host, the guest, and their complex, as
Based on the predominant states approximation [21], the standard chemical potential of a molecule in solution is approximated as the sum over a collection of M local energy minima (or energy wells), i=1…M, according to the equation
where R, T, Co, E(X), and Zi are, respectively, the gas constant, the absolute temperature, the standard concentration, the energy as a function of the internal coordinates X, and the configuration integral over the internal coordinates X in energy well i. The energy E accounts for both the internal energy of the solute and the solvation free energy, both being functions of the internal coordinates. The M energy minima of a molecule or complex are identified through extensive conformational search. The configurational integral of each local energy minimum is calculated assuming either the quantized rigid-rotor harmonic oscillator (RRHO) approximation [23], for QM/M2, or the classical Harmonic Approximation with Mode Scanning (HA/MS) [22] for the classical M2. It is worth remarking, however, that the anharmonicity corrections calculated using mode scanning for host-guest systems are often so small as to be negligible [22]. See ref. [24] for a detailed description of RRHO approximation, and the associated equations.
Free energy decomposition
Decomposition of the binding free energy into energetic and entropic components provides additional insight into the driving forces for binding [25]. The ensemble average of an individual energy component is calculated as
where E now may be the solute energy, the polar solvation free energy, the nonpolar solvation free energy, or the sum of any of these quantities, for the local energy minimum associated with energy well i. The weighting factor Pi is the probability of finding the molecule in the ith local energy well, which in turn is given by the Boltzmann weight of the ith energy well. The total configurational entropy of a molecule or complex is calculated as,
where Siois the configurational entropy of the ith energy well, calculated with the RRHO or HA/MS approximation.
Classical M2
In the classical M2 method, the potential energy of the molecule is calculated with an empirical force-field energy model, while solvent effects are accounted for using a continuum solvation model. Here, the parameters for bond, angle, torsion and van der Waals parameters were assigned according the CHARMm force field [26], using Discovery Studio Visualizer (Accelrys, Inc.), and atomic partial charges were assigned using the VCharge software [27] (VeraChem, LLC). Initial structures of host-guest complexes were prepared by docking the guest molecules in the binding site of CB7 using the Autodock Vina program [28].
Binding free energy calculations were performed using the second-generation M2 software, available for download from http://pharmacy.ucsd.edu/labs/gilson/software1a.html. The protocol used for performing classical M2 calculations is identical to that reported in Moghaddam et al [17]. Briefly, each initial structure is subjected to energy minimization using a combination of conjugate gradient and truncated Newton methods, with an energy gradient tolerance of 0.001 kcal/mol/Å. Starting from the energy minimized structure, many local energy minima conformations were identified using the Tork conformational search algorithm [29]. Conformations within 10 kcal/mol of the lowest energy conformation were retained and filtered based on symmetry-corrected root-mean squared distance (RMSD) cutoff of 0.1 Å [30]. Local configuration integrals were computed using the harmonic approximation with mode scanning correction for the ten softest modes of vibration to account for possible anharmonicity.22 The Generalized-Born (GB) solvation model was used during energy minimization [31,32], conformational search, and the calculation of configurational integrals, due to its efficiency; and the final solvation free energies are corrected towards Poisson-Boltzmann/surface-area (PBSA) solvation model [33,34], computed for a single representative conformation for each local energy minima. Poisson-Boltzmann calculations were performed by setting the interior and exterior dielectric constants to 1 and 80, respectively, and the dielectric boundary is defined by the Richards molecular surface [35]. Successive iterations of conformational searching were carried out, where each iteration used the most stable conformations identified in the previous iteration as seeds for the conformational search algorithm. This iterative procedure was continued until the free energy converged to within 0.1 kcal/mol.
Quantum M2
The QM/M2 method is described in detail in a recent publication by our group [19]. In this method, we use the PM6-DH+ semiempirical QM Hamiltonian and the COSMO continuum solvation model as the energy model [36-39]. The initial structures of the host-guest complexes were prepared through manual docking, or by taking the lowest free-energy conformation identified in the classical M2 calculations. We submitted our predictions based on the starting structure that resulted in the lowest (i.e. most favorable) binding free energy. Many low-energy conformations of the host, guest, and complex molecules were generated using the OPLS-2005 force-field and the LMOD conformational search algorithm implemented in the program Macromodel (Schrodinger, LLC.) [40,41]. A maximum of 1000 steps of LMOD conformational search were performed, and conformations within 10 kcal/mol of the lowest energy conformation were retained. Duplicate conformations were filtered based on symmetry and a RMSD cutoff of 0.1 Å. The low-energy conformations generated using the classical OPLS energy model were then refined with the PM6-DH+/COSMO energy model implemented in program MOPAC 2009 using the eigenvector following method until the normalized gradient fell below 0.01 kcal/mol/Å [42]. The COSMO solvation parameters were set to that of water, i.e. a dielectric constant of 78.4 and solvent probe radius of 1.4 Å. The quantum optimized structures were further filtered to remove any duplicates. The configurational integral of each conformation at 300K, based on the RRHO approximation, was computed using MOPAC’s thermochemistry module, without the anharmonicity corrections. The polar solvation free energy determined using COSMO solvation model was supplemented with an additional nonpolar solvation free energy term that is proportional to surface area, to account for cavity formation and van der Waals interactions between the solute and the solvent.
Empirical corrections to continuum solvation free energies
In a previous study [19], we found that the solvation free energy calculated using a continuum solvation model showed correlation to the errors in calculated binding free energies for CB7 host-guest systems. To correct for this systematic error, we developed CB7-specific empirical corrections for the COSMO and PBSA solvation models; the corrected solvation free energy was computed as
where ⟨W⟩ is the polar solvation free energy calculated using COSMO or PB, Δ⟨SASA⟩ is the change in the average surface area of host and guest upon binding, and α, γ and δ are fitted parameters. The standard values of α, γ and δ, i.e. without these CB7-specific corrections, are 1.0 (no scaling), 0.006 Å−2, and 0.0 kcal/mol (no shift). The value of 0.006 kcal/mol/Å−2 for γ was derived from fitting the experimental hydration free energies of linear alkanes [43]. In addition to using these standard parameter values, we also tried a set of parameters previously optimized specifically for CB7, through fitting of calculated binding affinities to experimental affinities of previously published host-guest complexes [19]. The fitted values of (α, γ, δ) are (0.9628, 0.0086 kcal/mol/Å−2, −5.83 kcal/mol), and (1.005, 0.0055 kcal/mol/Å−2, 3.37 kcal/mol), for the COSMO and PBSA solvation models, respectively. Note that these parameters were determined prior to the SAMPL4 challenge, so the fitting did not include the present SAMPL4 experimental data. Also, these empirical parameters are likely to be dependent on the associated solute energy model, PM6-DH+ or CHARMm/VCharge, and may not be transferable to other solute energy models.
Results
Binding free energy predictions
We computed the binding free energies of CB7-guest complexes using classical and quantum M2 methods, both with and without CB7-specific empirical corrections to the continuum solvation free energy. Figure 2 shows a comparison of the calculated binding affinities to the experimental measurements. Binding free energies computed using the classical M2 method correlate well with the experimental free energies, with a correlation coefficient (R2) of 0.74, and a root mean square error (RMSE) of 6.8 kcal/mol.
Figure 2.

Comparison of M2 predictions with experimental binding free energies. Left panes show the results of classical M2 with (top) and without (bottom) the empirical correction of solvation free energy. Right panes show analogous results for quantum M2.
Despite the good correlation between computed and experimental binding affinities, the slope of the linear regression fit is 2.0, a considerable deviation from the ideal value of 1.0. A similarly large slope was observed in a previous study of CB7 systems with the classical M2 method [16], but not with all force field parameters, and also not for other host-guest systems [12]. Thus, the large slope observed here is not a uniform attribute of the classical M2 method itself. The high RMSE results in part from a constant shift of the binding free energies, as reflected by the mean signed error (MSE) of −5.9 kcal/mol; i.e. the binding free energies are consistently predicted to be too favorable compared to experiments. Applying the CB7-specific empirical correction to the PBSA solvation model improved the RMSE from 6.8 to 3.9 kcal/mol, but the correlation remained the same at R2 = 0.74. To assess the error in relative binding affinities, we calculated RMSE after subtracting the mean unsigned error, to yield the RMSE_o statistic. This measure of error is 3.5 kcal/mol for the classical M2 method, only slightly lower than the RMSE of the absolute binding affinities.
Although the linear regression slope between the quantum method and experiment is better than that obtained using the classical M2 method, 0.7 vs 2.0, the quantum calculations yield a poorer correlation to experiment, with a correlation coefficient of 0.27 and RMSE of 6.7 kcal/mol. Applying the CB7-specific empirical corrections to the COSMO solvation model resulted in only slight improvement of the RMSE from 6.7 to 5.8 kcal/mol, and no improvement in the correlation. The quantum M2 binding free energies without solvation corrections were consistently less favorable than experiment, by 5.4 kcal/mol on average; with the solvation correction, the binding free energies are consistently more favorable than experiment, by 4.9 kcal/mol on average. The poor experimental correlation of the quantum M2 results is largely attributable to two outlying guest molecules, 3 and 14, whose computed binding free energies deviate from experiment by more than 10 kcal/mol (with solvation corrections), a large error relative to the other guest molecules. Excluding these two outliers, the correlation coefficient was 0.61 and 0.68 and the RMS error was 4.2 kcal/mol and 7.1 kcal/mol, with and without the solvation corrections, respectively. The lack of improvement in correlation with empirical solvation corrections is not totally surprising, as we found in a previous study that these corrections only slightly improve the correlation, but significantly improve the absolute binding free energies [19]. The RMSE_o of the quantum M2 predictions is 3.2 kcal/mol, somewhat better than that of the classical M2 predictions, owing to the better slope of quantum M2 method.
Analysis of guests 3 and 14
No guest molecules emerged as major outliers in the classical M2 calculations, but guests 3 and 14 showed large deviations in the quantum M2 calculations, with deviations in binding free energy of 11.6 and 11.0 kcal/mol, respectively (Table 1), after applying the CB7-specific empirical solvation corrections. Guest 3 is, essentially, n-hexane with one terminal hydroxyl and one terminal primary amine group, and guest 14 is a disubstituted adamantane compound. A larger error for guest 14 was somewhat expected, as we previously observed significant deviation between calculation and experiment for a disubstituted adamantane compound with two primary amine groups [19]. As is the case here, the binding free energy was calculated to be significantly more favorable than experiment. Our earlier study also included several mono-substituted adamantane guests, and the calculated binding affinities for these guests show only small deviations from the experimental values. Interestingly, the error for guest 14 in the classical M2 calculations was also relatively high compared to the other guests (Table 1).
Table 1.
Summary of computed binding free energy components, including solute energy, solvation free energy, and entropy, from the quantum M2 and classical M2 methodsa
| PM6-DH+/C0SM0 |
CHARMm/V charge/PBSA |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Guest | ΔGexp | Δ⟨U⟩ | -TS° | Δ⟨Wp⟩ | Δ⟨Wnp⟩ | Δ⟨U⟩ | -TS° | Δ⟨Wp⟩ | Δ⟨Wnp⟩ | ||
| 1 | −10.0 | −15.5 | −187.9 | 8.9 | 171.8 | −2.5 | −10.3 | −185.6 | 13.6 | 160.8 | −2.4 |
| 2 | −9.7 | −12.5 | −99.9 | 6.6 | 88.3 | −1.7 | −7.6 | −96.1 | 9.8 | 77.3 | −2.0 |
| 3 | −6.7 | −18.3 | −98.5 | 10.5 | 77.9 | −2.3 | −7.8 | −105.3 | 15.3 | 81.2 | −2.3 |
| 4 | −8.5 | −9.7 | −178.7 | 12.5 | 164.8 | −2.5 | −7.8 | −184.2 | 16.6 | 158.9 | −2.5 |
| 5 | −8.6 | −13.3 | −189.9 | 11.6 | 172.5 | −1.6 | −7.6 | −168.2 | 13.1 | 146.1 | −1.9 |
| 6 | −8.0 | −13.9 | −101.8 | 6.9 | 88.9 | −2.2 | −11.4 | −100.0 | 10.4 | 76.9 | −2.1 |
| 7 | −10.2 | −12.6 | −99.3 | 10.0 | 84.6 | −2.1 | −11.0 | −98.4 | 10.9 | 75.1 | −2.0 |
| 8 | −12.0 | −16.1 | −102.4 | 9.5 | 84.8 | −2.2 | −15.5 | −102.7 | 12.9 | 73.0 | −2.1 |
| 9 | −12.8 | −14.0 | −103.4 | 8.2 | 89.3 | −2.4 | −16.3 | −107.1 | 12.9 | 76.9 | −2.3 |
| 10 | −8.0 | −11.4 | −184.3 | 11.6 | 169.6 | −2.5 | −2.6 | −177.8 | 11.5 | 162.6 | −2.3 |
| 11b | −11.2 | −16.0 | −102.6 | 7.0 | 87.5 | −2.1 | −13.2 | −101.0 | 10.1 | 76.4 | −2.1 |
| 12 | −13.4 | −15.9 | −110.2 | 12.5 | 90.1 | −2.5 | −18.8 | −108.8 | 13.9 | 75.1 | −2.4 |
| 13 | −14.3 | −21.4 | −107.6 | 5.6 | 88.7 | −2.3 | −19.5 | −107.1 | 11.2 | 75.3 | −2.2 |
| 14 | −11.7 | −22.7 | −110.3 | 9.5 | 86.4 | −2.5 | −20.9 | −110.9 | 13.6 | 75.3 | −2.4 |
The polar solvation energy term Δ⟨Wp⟩ and the non-polar solvation energy term Δ⟨Wnp⟩ shown for both M2 methods are empirically corrected. See Methods section.
The calculated binding free energy is that of the racemic mixture.
Our prediction for guest 3 using the classical M2 method was significantly better than that using the quantum M2 method. Therefore, we compared the binding modes of guest 3 in the most stable conformations of the complexes generated by the classical and quantum M2 methods (Figure 3a). In the most stable quantum M2 conformation, the guest molecule adopts a parallel orientation within the host's binding site, with the skeleton of the hexane curled up in the horizontal plane of symmetry of CB7 and both the terminal hydrogen donating groups form hydrogen bonds with the carbonyl groups of the CB7 (Figure 3a, left). On the other hand, in the most stable classical M2 conformation, guest 3 adopts a more perpendicular orientation, with the terminal hydroxyl and amine groups forming hydrogen bonds to opposite carbonyl portals of the host (Figure 3a, right). On the quantum mechanical energy landscape, this most stable classical M2 conformation is less stable than the most stable quantum M2 conformation by 4.1 kcal/mol. We also examined the binding modes of guest 4, an n-hexane derivative with two terminal primary amine groups, which is similar to guest 3 in the sense that it also has two terminal hydrogen bond donors and a hydrophobic center. Both classical and quantum M2 predictions for guest 4 were significantly better compared to guest 3. Binding modes of guest 4 observed in the most stable conformation from classical and quantum M2 methods are somewhat similar (Figure 3b); interestingly, this guest molecule adopts an extended conformation in both the classical and quantum M2 calculations. Despite the similarity in structure, the free energy of the most stable classical M2 conformation is less stable than the most stable quantum M2 conformation by 5.6 kcal/mol, when it is reevaluated on the quantum mechanical energy landscape. These results suggest that the large error in computed binding affinity of guest 3 resulted from the inaccuracy of the energy model, rather than from an inadequate conformational search.
Figure 3.

The most stable conformations of CB7-guest 3 complexes (a) and CB7-guest 4 complexes (b) generated by the quantum M2 and the classical M2 method, shown in two perspectives. Hydrogen bonds are shown as green dashed lines.
Comparison of classical and quantum M2 free energy components
For a better understanding of the varying performances of the classical and quantum M2 method, both based on the same idea of mining minima and differing only in the energy model, we decomposed the binding affinities of SAMPL4 host-guest complexes computed by both methods into contributions from the solute energy Δ⟨U⟩, the entropy term -TS°, the polar solvation term, Δ⟨Wp⟩ and the non-polar solvation term, Δ⟨Wnp⟨. The binding free energies and the respective decomposition are summarized in Table 1. Overall the quantum and classical M2 method show similar ranges of values for each individual term, and the favorable contribution to the binding free energy is found always to result from the solute energy and a small contribution from the non-polar solvation energy. The polar solvation free energy and the entropy consistently oppose binding.
The binding free energies computed using classical and quantum M2 methods correlate with each other to some extent, R2 = 0.53 and 0.58, with and without CB7-specific empirical solvation free energy corrections, respectively (Figure 4a). We further compared the individual energy components to directly assess the differences in the classical and quantum M2 calculations. For clarity, we present our results based on the calculations with the CB7-specific empirical corrections for solvation free energies, but similar results were obtained even without the solvation free energy corrections. Binding contribution of solute energies, polar solvation energies and nonpolar solvation energies calculated by quantum and classical M2 methods correlate strongly with each other (R2 = 0.97, 0.97 and 0.82, respectively), however, this strong correlation is a result of having two well-separated groups of data points (Figure 4b-d). The total energy change, i.e. the solute energy change plus the solvation free energy change, showed some correlation between the two methods (R2 = 0.48; Figure 4e), with an RMS deviation of 10.2 kcal/mol. The RMS deviations of the solute energy change and the polar solvation free energy change, were 6.7 and 12.6 kcal/mol, respectively, suggesting significant differences between the solute Hamiltonians and the solvation models used in the classical and quantum M2 methods, although it is important to keep in mind that the conformations from the two methods are not identical, so the energy comparison is not rigorous. The RMS deviation in the nonpolar solvation free energy was relatively small, 0.1 kcal/mol, which is expected since both the methods use a simple surface area model. The entropic contributions to binding from the classical and quantum M2 calculations were somewhat correlated (R2 = 0.48), with an RMS deviation of 3.6 kcal/mol (Figure 4f). Overall, these results are consistent with our previous study comparing classical and quantum M2 method using ten CB7 host-guest complexes [19].
Figure 4.

Comparison of free energy components, computed by classical and quantum M2 methods.
Driving forces for binding
The decomposition of binding free energy into energetic and entropic components can provide insights into the underlying driving forces for binding [12,25]. Given the much better performance of the classical M2 method in predicting the binding affinities, we analyze the decomposition of the classical M2 binding free energies (with CB7-specific empirical corrections to solvation free energy) to assess the driving forces of CB7 binding. (Note that the solvation free energy implicitly includes the entropy change of the solvent upon guest binding. As a consequence, the decomposition provided here cannot be directly compared to experiments, for example, isothermal titration calorimetry measurements.) The average change in binding energy (solute energy plus solvation free energy) ranged from −17.5 to −38.0 kcal/mol. Although, solute energy and solvation free energy individually are considerably more negative and positive respectively, the compensation between these two quantities results in relative small and favorable energy changes. The nonpolar solvation free energy contributes a small and nearly constant value of −2.2 ± 0.2 kcal/mol, towards the binding free energy of the current host-guest systems. The loss in configurational entropy is unfavorable and adds significant free energy cost to the guest binding; the entropy contributions for the present host-guest complexes ranged from 9.8 to 16.6 kcal/mol.
Interestingly, none of the individual binding free energy components correlated well with the experimental binding affinities (R2 < 0.23). On the other hand, the total energy change, i.e. the sum of changes in solute energy and solvation free energy, showed decent correlation (R2 = 0.56) to the experimental binding affinities (Figure 5).
Figure 5.
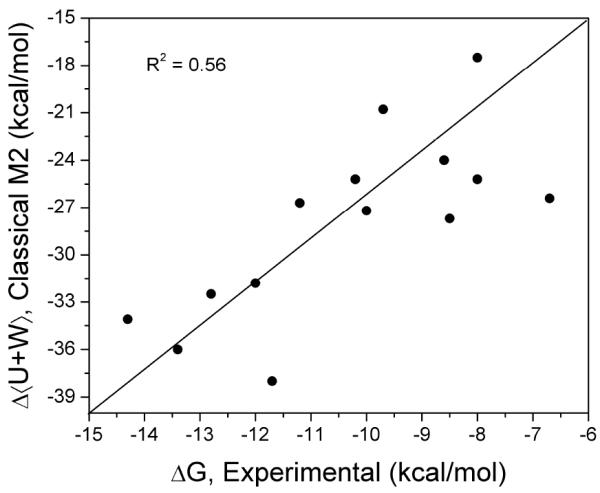
Comparison of change in total energy (solute energy plus solvation free energy) to experimental binding free energy.
Discussion
We used classical and quantum mining minima (M2) methods to compute the binding affinities of 14 CB7 host-guest complexes in the SAMPL4 challenge. While the classical M2 binding affinities correlated strongly with experiment, the quantum M2 binding affinities were poorly correlated and suffered from two major outliers. It is unlikely that the errors in the quantum M2 calculations resulted from not finding the most stable conformation in their associated energy landscapes, given that we used aggressive conformational algorithms to enumerate the local energy minima. Moreover, finding even more stable conformations for these complexes would only shift the calculated binding free energies further from the experimental value.
One of the key approximations made by the M2 method, both classical and quantum, is their use of continuum solvation models. It is not clear how well the conformational distributions of the host, guest, and complex obtained using a continuum solvation model match what would be obtained from an explicit treatment of water molecules [44]. Indeed, it is known that the conformational distributions of proteins are markedly different between explicit-water and implicit water simulations [45,46]. Nevertheless, our classical M2 calculations showed good correlation with experiment. It is possible that the differences between the classical and quantum M2 methods lie within the different continuum solvation models used, PB and COSMO. Alternatively, the errors in quantum M2 might have resulted from the approximations in the semiempirical QM Hamiltonian, PM6-DH+. However, it would seem surprising for this energy model to underperform the relatively simplistic force field used in the classical M2 calculations. It is worth noting that another SAMPL4 participant, using a free energy method similar in structure to the quantum M2 method, obtained more accurate results for the CB7 systems in the SAMPL4 host-guest challenge [15]. They used a QM Hamiltonian based on density functional theory (DFT) with a three-body dispersion correction, a continuum solvation model, and a modified RRHO approximation. Further studies are underway to compare PM6-DH+ to DFT methods for computing host-guest binding free energies.
Our predictions for the previous SAMPL3 challenge using the classical M2 method were less than satisfactory [18]. While CB7 was also included in the SAMPL3 challenge, there were only two guests for this host, so the overall performance of our calculations was dominated by the results for a different host molecule, which posed extra challenges due to its uncertain protonation states and more flexible structure [8]. To avoid the protonation and flexibility issues faced in the SAMPL3 challenge, the experimentalists chose the CB7 host as one of the hosts for SAMPL4. The performance of classical M2 method in the current challenge is more in line with our expectations based on previous studies of CB7 binding, both retrospective and prospective [17,16].
The binding free energies predicted with both the classical and quantum M2 methods with CB7-specific empirical solvation free energy corrections were consistently more favorable than the measured binding free energies. We speculate that this systematic bias in the predictions might result from an incomplete treatment of the salt effects on binding affinity. Cations in solution bind to the carbonyl portals of cucurbiturils and tend to compete with the binding of guest molecules, thus lowering the affinities of the guests for CB7 [47]. As a consequence, a higher salt concentration will tend to lower these host-guest binding affinities. Our CB7-specific empirical corrections to the solvation free energy were based on previous binding studies of CB7 host-guest complexes measured in a 50 mM sodium acetate (deuterated; CD3COONa) buffer or deionized water, whereas the present experimental measurements were obtained in 100 mM sodium phosphate (Na3PO4) buffer [19,15]. Thus, the empirical fit may have underestimated the affinity lowering effects of the higher salt concentrations in the SAMPL4 experiments. Indeed, CB7 binding affinities of two of the present guests (1 and 4) were previously measured in a 50 mM sodium acetate buffer [48], and the low-salt binding affinities differ from the current measurements by 2.8 and 2.5 kcal/mol, respectively. Thus, the high salt concentration in the present experimental measurements led to higher (less favorable) binding free energies for these guests. This is somewhat sobering, since empirical scoring functions are often parameterized using data from several different sources, which may differ in solvent conditions in a way that could potentially influence the measured binding affinity.
The M2 method provides a detailed breakdown of the binding free energy into solute energy, polar solvation, nonpolar solvation free energy, and configurational entropy. A comparison of each of these components to the experimental binding affinities indicated that none of these free energy components is an independent predictor of binding affinity. However, the solute energy change plus the solvation free energy change showed some correlation to the experimental binding affinity, and so one should minimally account for these factors in developing approximate computational models of binding. Also, the change in the solute energy plus solvation free energy was found to correlate reasonably well with experiment, substantiating the use of methods such as docking, which are based largely on interaction models which focus on these contributions, to enrich the yield of ligands by initial coarse screens of compound libraries. Nevertheless, accounting for the loss of configurational entropy helps improve the correlation between calculation and experiment.
Acknowledgements
This study was made possible in part by grant GM61300 from the NIGMS. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Principal Investigator MKG is a founder of and has an equity interest in VeraChem LLC. Although grant GM61300 has been identified for conflict of interest management based on the overall scope of the project and its potential benefit to VeraChem LLC, the research findings included in this particular publication may not necessarily relate to the interests of VeraChem LLC. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
REFERENCES
- 1.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303(5665):1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 2.MK Gilson, Zhou HX. Calculation of protein-ligand binding affinities. Annu Rev Bioph Biom. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 3.PCD Hawkins, AG Skillman, Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J Med Chem. 2007;50(1):74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 4.Jorgensen WL. Efficient Drug Lead Discovery and Optimization. Accounts Chem Res. 2009;42(6):724–733. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guthrie JP. A Blind Challenge for Computational Solvation Free Energies: Introduction and Overview. J Phys Chem B. 2009;113(14):4501–4507. doi: 10.1021/jp806724u. [DOI] [PubMed] [Google Scholar]
- 6.MT Geballe, AG Skillman, A Nicholls, JP Guthrie, Taylor PJ. The SAMPL2 blind prediction challenge: introduction and overview. J Comput Aid Mol Des. 2010;24(4):259–279. doi: 10.1007/s10822-010-9350-8. [DOI] [PubMed] [Google Scholar]
- 7.MT Geballe, Guthrie JP. The SAMPL3 blind prediction challenge: transfer energy overview. J Comput Aid Mol Des. 2012;26(5):489–496. doi: 10.1007/s10822-012-9568-8. [DOI] [PubMed] [Google Scholar]
- 8.HS Muddana, CD Varnado, CW Bielawski, AR Urbach, L Isaacs, MT Geballe, Gilson MK. Blind prediction of host-guest binding affinities: a new SAMPL3 challenge. J Comput Aid Mol Des. 2012;26(5):475–487. doi: 10.1007/s10822-012-9554-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skillman AG. SAMPL3: blinded prediction of host-guest binding affinities, hydration free energies, and trypsin inhibitors. J Comput Aid Mol Des. 2012;26(5):473–474. doi: 10.1007/s10822-012-9580-z. [DOI] [PubMed] [Google Scholar]
- 10.DL Mobley, S Liu, NM Lim, N Deng, K Branson, AL Perryman, S Forli, RM Levy, E Gallicchio, Olson AS. Blind prediction of HIV integrase binding from the SAMPL4 challenge. J Comput Aid Mol Des. 2014 doi: 10.1007/s10822-014-9723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.CD Snow, EJ Sorin, YM Rhee, Pande VS. How well can simulation predict protein folding kinetics and thermodynamics? Annu Rev Bioph Biom. 2005;34:43–69. doi: 10.1146/annurev.biophys.34.040204.144447. [DOI] [PubMed] [Google Scholar]
- 12.W Chen, CE Chang, Gilson MK. Calculation of cyclodextrin binding affinities: Energy, entropy, and implications for drug design. Biophys J. 2004;87(5):3035–3049. doi: 10.1529/biophysj.104.049494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macartney DH. Encapsulation of Drug Molecules by Cucurbiturils: Effects on their Chemical Properties in Aqueous Solution. Isr J Chem. 2011;51(5-6):600–615. [Google Scholar]
- 14.AL Koner, Nau WM. Cucurbituril encapsulation of fluorescent dyes. Supramol Chem. 2007;19(1-2):55–66. [Google Scholar]
- 15.HS Muddana, AT Fenley, DL Mobley, Gilson MK. Blind prediction of the host-guest binding affinities from the SAMPL4 challenge. J Comput Aid Mol Des. 2014 doi: 10.1007/s10822-012-9554-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.S Moghaddam, C Yang, M Rekharsky, YH Ko, K Kim, Y Inoue, Gilson MK. New Ultrahigh Affinity Host-Guest Complexes of Cucurbit[7]uril with Bicyclo[2.2.2]octane and Adamantane Guests: Thermodynamic Analysis and Evaluation of M2 Affinity Calculations. J Am Chem Soc. 2011;133(10):3570–3581. doi: 10.1021/ja109904u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.S Moghaddam, Y Inoue, Gilson MK. Host-Guest Complexes with Protein-Ligand-like Affinities: Computational Analysis and Design. J Am Chem Soc. 2009;131(11):4012–4021. doi: 10.1021/ja808175m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.HS Muddana, Gilson MK. Prediction of SAMPL3 host-guest binding affinities: evaluating the accuracy of generalized force-fields. J Comput Aid Mol Des. 2012;26(5):517–525. doi: 10.1007/s10822-012-9544-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.HS Muddana, Gilson MK. Calculation of Host-Guest Binding Affinities Using a Quantum-Mechanical Energy Model. J Chem Theory Comput. 2012;8(6):2023–2033. doi: 10.1021/ct3002738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.CE Chang, Gilson MK. Free energy, entropy, and induced fit in host-guest recognition: Calculations with the second-generation mining minima algorithm. J Am Chem Soc. 2004;126(40):13156–13164. doi: 10.1021/ja047115d. [DOI] [PubMed] [Google Scholar]
- 21.MS Head, JA Given, Gilson MK. "Mining minima": Direct computation of conformational free energy. J Phys Chem A. 1997;101(8):1609–1618. [Google Scholar]
- 22.CE Chang, MJ Potter, Gilson MK. Calculation of molecular configuration integrals. J Phys Chem B. 2003;107(4):1048–1055. [Google Scholar]
- 23.Hill TL. Dover Publications; New York: 1986. An introduction to statistical thermodynamics. [Google Scholar]
- 24.HX Zhou, Gilson MK. Theory of Free Energy and Entropy in Noncovalent Binding. Chem Rev. 2009;109(9):4092–4107. doi: 10.1021/cr800551w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.AT Fenley, HS Muddana, Gilson MK. Entropy-enthalpy transduction caused by conformational shifts can obscure the forces driving protein-ligand binding. P Natl Acad Sci USA. 2012;109(49):20006–20011. doi: 10.1073/pnas.1213180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.FA Momany, Rone R. Validation of the General-Purpose Quanta(R)3.2/Charmm(R) Force-Field. J Comput Chem. 1992;13(7):888–900. [Google Scholar]
- 27.MK Gilson, HSR Gilson, Potter MJ. Fast assignment of accurate partial atomic charges: An electronegativity equalization method that accounts for alternate resonance forms. Journal of chemical information and computer sciences. 2003;43(6):1982–1997. doi: 10.1021/ci034148o. [DOI] [PubMed] [Google Scholar]
- 28.O Trott, Olson AJ. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.CE Chang, Gilson MK. Tork: Conformational analysis method for molecules and complexes. J Comput Chem. 2003;24(16):1987–1998. doi: 10.1002/jcc.10325. [DOI] [PubMed] [Google Scholar]
- 30.W Chen, J Huang, Gilson MK. Identification of symmetries in molecules and complexes. Journal of chemical information and computer sciences. 2004;44(4):1301–1313. doi: 10.1021/ci049966a. [DOI] [PubMed] [Google Scholar]
- 31.D Qiu, PS Shenkin, FP Hollinger, Still WC. The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate Born radii. J Phys Chem A. 1997;101(16):3005–3014. [Google Scholar]
- 32.WC Still, A Tempczyk, RC Hawley, Hendrickson T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J Am Chem Soc. 1990;112(16):6127–6129. [Google Scholar]
- 33.MK Gilson, Honig B. Calculation of the Total Electrostatic Energy of a Macromolecular System - Solvation Energies, Binding-Energies, and Conformational-Analysis. Proteins-Structure Function and Genetics. 1988;4(1):7–18. doi: 10.1002/prot.340040104. [DOI] [PubMed] [Google Scholar]
- 34.JD Madura, JM Briggs, RC Wade, ME Davis, BA Luty, A Ilin, J Antosiewicz, MK Gilson, B Bagheri, LR Scott, Mccammon JA. Electrostatics and Diffusion of Molecules in Solution - Simulations with the University-of-Houston Brownian Dynamics Program. Comput Phys Commun. 1995;91(1-3):57–95. [Google Scholar]
- 35.B Lee, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55(3):379–IN374. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 36.J Rezac, J Fanfrlik, D Salahub, Hobza P. Semiempirical Quantum Chemical PM6 Method Augmented by Dispersion and H-Bonding Correction Terms Reliably Describes Various Types of Noncovalent Complexes. J Chem Theory Comput. 2009;5(7):1749–1760. doi: 10.1021/ct9000922. [DOI] [PubMed] [Google Scholar]
- 37.M Korth, M Pitonak, J Rezac, Hobza P. A Transferable H-Bonding Correction for Semiempirical Quantum-Chemical Methods. J Chem Theory Comput. 2010;6(1):344–352. doi: 10.1021/ct900541n. [DOI] [PubMed] [Google Scholar]
- 38.Korth M. Third-Generation Hydrogen-Bonding Corrections for Semiempirical QM Methods and Force Fields. J Chem Theory Comput. 2010;6(12):3808–3816. [Google Scholar]
- 39.A Klamt, Schuurmann G. Cosmo - a New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J Chem Soc Perk T. 1993;2(5):799–805. [Google Scholar]
- 40.F Mohamadi, NGJ Richards, WC Guida, R Liskamp, M Lipton, C Caufield, G Chang, T Hendrickson, Still WC. Macromodel - an Integrated Software System for Modeling Organic and Bioorganic Molecules Using Molecular Mechanics. J Comput Chem. 1990;11(4):440–467. [Google Scholar]
- 41.I Kolossvary, WC Guida. Low mode search. An efficient, automated computational method for conformational analysis: Application to cyclic and acyclic alkanes and cyclic peptides. J Am Chem Soc. 1996;118(21):5011–5019. [Google Scholar]
- 42.Stewart JJP. Mopac - a Semiempirical Molecular-Orbital Program. J Comput Aid Mol Des. 1990;4(1):1–45. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 43.RA Friedman, Honig B. A free energy analysis of nucleic acid base stacking in aqueous solution. Biophys J. 1995;69(4):1528–1535. doi: 10.1016/S0006-3495(95)80023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.HS Muddana, NV Sapra, AT Fenley, MK Gilson. The electrostatic response of water to neutral polar solutes: Implications for continuum solvent modeling. J Chem Phys. 2013;138(22) doi: 10.1063/1.4808376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou RH. Free energy landscape of protein folding in water: Explicit vs. implicit solvent. Proteins-Structure Function and Genetics. 2003;53(2):148–161. doi: 10.1002/prot.10483. [DOI] [PubMed] [Google Scholar]
- 46.H Nymeyer, Garcia AE. Simulation of the folding equilibrium of alpha-helical peptides: A comparison of the generalized born approximation with explicit solvent. P Natl Acad Sci USA. 2003;100(24):13934–13939. doi: 10.1073/pnas.2232868100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.W Ong, Kaifer AE. Salt effects on the apparent stability of the cucurbit[7]uril-methyl viologen inclusion complex. J Org Chem. 2004;69(4):1383–1385. doi: 10.1021/jo035030+. [DOI] [PubMed] [Google Scholar]
- 48.SM Liu, C Ruspic, P Mukhopadhyay, S Chakrabarti, PY Zavalij, Isaacs L. The cucurbit[n]uril family: Prime components for self-sorting systems. J Am Chem Soc. 2005;127(45):15959–15967. doi: 10.1021/ja055013x. [DOI] [PubMed] [Google Scholar]