

Abstract
Hydrophobic helical peptides interact with lipid bilayers in various modes, determined by the match between the length of the helix’s hydrophobic core and the thickness of the hydrocarbon region of the bilayer. For example, long helices may tilt with respect to the membrane normal to bury their hydrophobic cores in the membrane, and the lipid bilayer may stretch to match the helix length. Recent molecular dynamics simulations and potential of mean force calculations have shown that some TM helices whose lengths are equal to, or even shorter than, the bilayer thickness may also tilt. The tilt is driven by a gain in the helix precession entropy, which compensates for the free energy penalty resulting from membrane deformation. Using this free energy balance, we derived theoretically an equation of state, describing the dependence of the tilt on the helix length and membrane thickness. To this end, we conducted coarse-grained Monte Carlo simulations of the interaction of helices of various lengths with lipid bilayers of various thicknesses, reproducing and expanding the previous molecular dynamics simulations. Insight from the simulations facilitated the derivation of the theoretical model. The tilt angles calculated using the theoretical model agree well with our simulations and with previous calculations and measurements.
Introduction
Hydrophobic match or mismatch in transmembrane (TM) helices (or proteins) refers to the match or mismatch between the length of the hydrophobic core of the helix and the native thickness of the hydrocarbon region of the membrane (Figure 1).1−3 Positive mismatch refers to a situation in which the helix is longer than the membrane thickness (Figure 1A), and negative mismatch refers to a situation in which it is shorter (Figure 1C). Hydrophobic mismatch is a fascinating example of mutual protein–membrane interaction. In cell membranes, hydrophobic mismatch is one of the mechanisms driving the formation of microdomains (lipid rafts), in which membrane lipids and proteins of compatible length diffuse laterally and cluster together.4 Microdomains usually have important functional implications, for example in cell division and signal transduction.4 Moreover, hydrophobic mismatch is thought to be important in cellular processes such as the sorting of lipids and TM proteins into cellular compartments. This notion is supported by a recent survey that found differences in the lengths of TM helices from various cellular organelles, which were compatible with the differences in the thicknesses of the respective lipid bilayers.5
Figure 1.
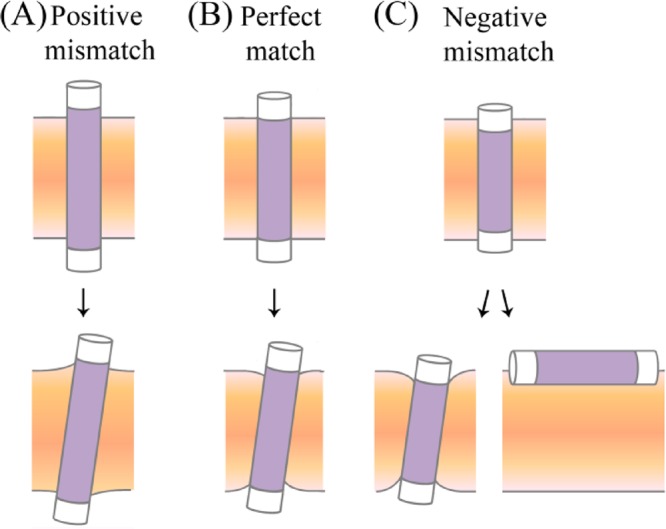
Helix-membrane configurations with (A) positive hydrophobic mismatch, (B) perfect match, and (C) negative hydrophobic mismatch. The helix is represented as a cylinder, with the hydrophobic core in purple and the hydrophilic termini in white. (A) At positive mismatch, the TM helix tilts and the membrane expands to match the helix hydrophobic core. (B) At perfect match, the helix tilts because of the favorable increase in precession entropy, and the membrane thins so that the polar helix termini can remain in the lipid headgroup region rather than partition into the hydrocarbon region of the membrane. (C) At slight negative mismatch (lower left panel), the TM helix tilts and the membrane thins locally as in perfect match. In cases of excessive mismatch, the helix adopts a surface orientation rather than forcing the membrane to thin beyond its elastic limit (lower right panel).
Both TM helices and lipids adapt to mismatch by minimizing the exposure of the polar side chains and backbone of the helix to the hydrophobic membrane environment while maximizing the favorable interaction between the hydrophobic amino acids and the lipid. Several means of system adaptation to both positive and negative mismatch have been observed in experiments.2,3 In positive mismatch, the helix tilts from the membrane normal and decreases its effective hydrophobic length (Figure 1A).2,3 Another adaptation to positive mismatch is kinking or flexing of the TM helix.3 Alternatively (or jointly), the acyl chains of the phospholipids surrounding the helix can stretch.1 The helix may also migrate to membrane regions with a better match to its hydrophobic length and/or interact with other TM helices/proteins.6 In cases of negative mismatch, the acyl chains have been shown to contract and reduce the bilayer thickness (Figure 1C).1 In extreme cases of negative mismatch, where TM orientation involves severe membrane deformation, the helix can be oriented in parallel to the membrane surface and reside at the water–membrane interface.7,8
Recent molecular dynamics (MD) simulations have demonstrated that tilting of TM helices occurs also at perfect match (Figure 1B) and negative mismatch (Figure 1C).9,10 Using potential of mean force calculations, Im and co-workers have attributed tilting under such conditions to the gain in precession entropy associated with the rigid-body translational-rotational motion of the tilted helix in the membrane.9,11,12 The authors demonstrated that, due to the precession entropy gain, TM helices tilt at least 10° from the membrane normal even under negative mismatch conditions.9 In addition, the authors showed that helix–lipid interactions may vary under different mismatch conditions. For instance, under negative mismatch conditions, helix–lipid interactions are energetically unfavorable and oppose the tilt.9 This might be related to the fact that under such conditions tilting involves a desolvation free energy penalty due to the transfer of the polar helix termini from the aqueous phase into the hydrocarbon region of the membrane. To avoid the associated free energy penalty, the membrane thins. The reduction in the entropy of the lipid chains due to membrane thinning can be balanced by the increase in precession entropy. Incorporating this free energy balance, we present below a simple theoretical model, an equation of state, for estimating the tilt angle according to the helix length and membrane thickness. The theoretical model integrates insights gained from Monte Carlo (MC) simulations using the method presented in refs (13−17) and below.
We use the equation of state to estimate the tilt angles of 17 synthetic peptides of the WALP/KALP/GWALP series of different lengths (Table 1) in six membrane types with various native thicknesses (Table 2). The peptides feature hydrophobic cores, composed of alanine and leucine amino acids, flanked by lysine (in KALP peptides) or tryptophan residues (in WALP and GWALP peptides). The results agree with experimental data, previous calculations, and our MC simulations.
Table 1. The WALP, KALP, and GWALP23 Peptides Used Herea.
| peptide | sequence | hydrophobic core (Å) |
|---|---|---|
| WALP17 | GWW(LA)5LWWA | 16.5 |
| WALP19 | GWW(LA)6LWWA | 19.5 |
| WALP21 | GWW(LA)7LWWA | 22.5 |
| WALP23 | GWW(LA)8LWWA | 25.5 |
| WALP25 | GWW(LA)9LWWA | 28.5 |
| WALP27 | GWW(LA)10LWWA | 31.5 |
| WALP29 | GWW(LA)11LWWA | 34.5 |
| WALP31 | GWW(LA)12LWWA | 37.5 |
| KALP17 | GKK(LA)5LKKA | 16.5 |
| KALP19 | GKK(LA)6LKKA | 19.5 |
| KALP21 | GKK(LA)7LKKA | 22.5 |
| KALP23 | GKK(LA)8LKKA | 25.5 |
| KALP25 | GKK(LA)9LKKA | 28.5 |
| KALP27 | GKK(LA)10LKKA | 31.5 |
| KALP29 | GKK(LA)11LKKA | 34.5 |
| KALP31 | GKK(LA)12LKKA | 37.5 |
| GWALP23 | GGALW(LA)6LWLAGA | 19.5 |
The lengths of their hydrophobic cores were estimated assuming a translation of 1.5 Å per residue along the helix axis, as in a perfect α-helix.
Table 2. Phospholipid Types Used Herea.
| phospholipid | width of the hydrocarbon region (Å) |
|---|---|
| 1,2-didecanoyl-sn-glycero-3-PC | 16.6 |
| 1,2-dilauroyl-sn-glycero-3-PC (DLPC) | 21 |
| 1,2-dimyristoyl-sn-glycero-3-PC (DMPC) | 25.4 |
| 1,2-dipalmitoyl-sn-glycero-3-PC (DPPC) | 29.8 |
| 1,2-distearoyl-sn-glycero-3-PC | 34.3 |
| 1,2-diarachidoyl-sn-glycero-3- PC | 38.7 |
The width of the hydrophobic core is calculated as described in ref (22). PC, phosphatidylcholine.
Methods
Monte Carlo Simulations
The peptide was described in a reduced way; each amino acid was represented by two interaction sites, corresponding to the α-carbon and side chain.17 The interaction sites and sequential α-carbons were connected by virtual bonds. The membrane hydrophobicity was represented as a smooth profile, corresponding to the native thickness of the hydrocarbon region.17
The total free energy difference between a peptide in the aqueous phase and in the membrane (ΔGtot) was calculated as18,19
| 1 |
where ΔGcon is the free energy change due to membrane-induced conformational changes in the peptide. At constant (absolute) temperature T, it can be calculated as ΔGcon = ΔE – TΔS, where ΔE is the internal energy difference between the peptide in water and in the membrane. The internal energy is a statistical potential derived from available three-dimensional (3D) protein structures.20 The energy function assigns a score (energy) to each peptide conformation according to the conformation’s abundance in the Protein Data Bank. Common conformations are assigned high scores (low energy), while rare conformations are assigned lower scores (higher energy). ΔS refers to the entropy difference between the water and membrane-bound states, while the entropy (S) in each state is determined by the distribution of the virtual bond rotations in the reduced peptide representation.
ΔGdef is the free energy penalty associated with fluctuations of the membrane thickness around its native (resting) value, calculated following the estimation of Fattal and Ben-Shaul.21 Their calculations were based on a statistical-thermodynamic molecular model of the lipid chains. Their model fits a harmonic potential of the form ΔGdef = ΩΔL2, where ΔL is the difference between the native and actual thickness of the membrane and Ω is a harmonic-force constant related to the membrane elasticity and is equal to Ω = 0.22 kT/Å2,21 where k is the Boltzmann constant. In the model, the membrane is allowed to deform within its elastic range, that is, ±20% of its native thickness.22
ΔGCoul stands for the Coulombic interactions between titratable residues of the peptide and the (negative) surface charge of the membrane. It is calculated using the Gouy–Chapman theory that describes how the electrostatic potential depends on the distance from the membrane surface in an electrolyte solution.13 We used electrostatically neutral membranes, corresponding to the zwitterionic lipid phosphatidylcholine, so ΔGCoul = 0.
ΔGsol is the free energy of transfer of the peptide from the aqueous phase to the membrane. It takes into account electrostatic contributions resulting from changes in solvent polarity, as well as nonpolar effects, both resulting from differences in the van der Waals interactions of the peptide with the membrane and aqueous phases, and from solvent structure effects. ΔGimm is the free energy penalty resulting from the confinement of the external translational and rotational motion of the peptide inside the membrane. ΔGlip is the free energy penalty resulting from the interference of the peptide with the conformational freedom of the aliphatic chains of the lipids in the bilayer while the membrane retains its native thickness. The latter three terms, i.e., ΔGsol, ΔGimm, and ΔGlip, are calculated using the Kessel and Ben-Tal hydrophobicity scale.18 The scale accounts for the free energy of transfer of the amino acids, located in the center of a polyalanine α-helix, from the aqueous phase into the membrane midplane. In order to avoid the excessive penalty associated with the transfer of charged residues into the bilayer, in the model the titratable residues are neutralized gradually upon insertion into the membrane, so that a nearly neutral form is desolvated into the hydrophobic core.
To calculate the free energy change in eq 1, we conducted MC simulations of the peptide in water and in membrane environments. In water, the peptide is subjected solely to internal conformational modifications. In one MC cycle, the number of internal modifications attempted is equal to the number of residues in the peptide. In the membrane, each MC cycle includes additional external rigid body rotational and translational motions to allow the peptide to change its location in the membrane and its orientation with respect to the membrane normal. A helical peptide in a membrane typically adheres to one of the two following configurations: TM orientation with the helices’ principal axis roughly along the membrane normal or surface orientation with the axis approximately in parallel to the membrane surface. The transition between the two configurations is associated with a high free energy barrier. Thus, for simulations in the membrane environment, each of the two configurations is used as the initial orientation for three independent simulations of 500 000 MC cycles. Simulations in water (i.e., without the membrane) are also carried out in three independent runs of 500 000 MC cycles each.
Results
MC Simulations
We conducted MC simulations with 16 peptides of the WALP and KALP series interacting with six membrane types of varying thicknesses. The results for WALPs, which are more commonly used in experiments and MD simulations, are presented below (Figure 2), and the results for KALPs are presented in the Supporting Information (Figure S1). Throughout the simulations, the peptides were, in essence, helical in both the aqueous and membrane environments (Figure S2), which is anticipated for these peptides, composed mostly of Ala and Leu, two amino acids with high helix propensity.20
Figure 2.

MC simulations and comparison to the theoretical model. The results were obtained from MC simulations of eight WALP peptides (Table 1) interacting with membranes of six different types (Table 2). The standard errors are marked; in many cases, the error bars are smaller than the symbols. (A) The dependence of the tilt angle on the hydrophobic mismatch. A tilt angle of 0° corresponds to a helix with its principal axis perpendicular to the membrane plane; a tilt angle of 90° corresponds to a helix with its principal axis parallel to the membrane plane. The inset shows the theoretical dependence of the tilt angle of WALP21 on the hydrophobic mismatch (solid curve) in comparison to the values obtained from the MC simulations (triangles). (B) Membrane adaptation vs hydrophobic mismatch. The inset demonstrates the results for WALP21. (C) Location of the flanking residues in the membrane vs hydrophobic mismatch. For clarity, the data for only three peptides are shown. ΔZ is the shortest distance between the average position of the α-carbon of the flanking residue and the boundary of the hydrocarbon region of the membrane. The dotted lines were added to guide the eye. (D) Correlation between the theoretically predicted tilt angles of WALPs and the values estimated from the MC simulations; Theoretical_tilt = 0.97 × MC_tilt + 0.6, R2 = 0.99. The dashed line represents the ideal fit, i.e., Theoretical_tilt = MC_tilt. The theoretical tilts were calculated using eq 6. The values based on the MC simulations were reproduced from A.
Tilting
Figure 2A shows the dependence of the helix tilt angle on the hydrophobic mismatch. The results are similar to those of previous MD studies.10,23 As anticipated, helices whose hydrophobic cores were longer than the membrane thickness were in TM orientation with their principal axes tilted with respect to the membrane normal. In cases of negative mismatch, the helices assumed a tilted TM orientation as well, provided that the difference between the length of the helical peptide and the thickness of the bilayer did not exceed approximately 10 Å. To accommodate helical peptides of greater negative mismatch in TM orientation, the membrane would be forced to deform beyond its elastic limit, which was not allowed in the simulations. Such peptides could not span the membrane and adsorbed at the membrane–water interface as helices with their principal axes approximately parallel to the membrane surface (e.g., Figure S3), in line with previous experimental studies.7,24,25 Within the boundaries of the elastic region of the membrane, the tilt angle decreased as the mismatch decreased, with a minimal value of ∼10° at a mismatch of −10 Å.
We compared the tilt angles calculated in the MC simulations to the available data, obtained using various experimental techniques and MD simulations. Results for WALPs are shown in Table 3; Table S1 shows results for KALPs. Good agreement was observed in all cases, but it is noteworthy that in some cases the range of tilt values obtained in previous studies is rather large. In particular, the range of tilt values obtained in previous studies of WALP23 in DMPC and DLPC membranes exceeds 20° (Table 3).
Table 3. Comparison of α (degrees), Calculated Using the Theoretical Model, to the MC Simulations and Previous Dataa.
| peptide | membrane | theoretical model | MC | previous data |
|---|---|---|---|---|
| WALP19 | DLPC | 19 | 17.1 ± 7.5 | 11 (NMR)41 |
| 13.5 ± 7.2 (MD)33 | ||||
| DMPC | 14 | 4 (NMR)41 | ||
| 13.3 ± 6.6 | 12.1 (MD)9 | |||
| 13 (MD)38 | ||||
| WALP23 | DLPC | 28 | 24.4 ± 9.7 | 17.5 ± 7.6 (MD)33 |
| 23.7 ± 8.8 (MD)35 | ||||
| 29 ± 5 (NMR)37 | ||||
| 36 (NMR)41 | ||||
| DMPC | 19 | 17.3 ± 8.4 | 14 ± 5 (NMR)37 | |
| 20.8 ± 1.4 (NMR)34 | ||||
| 22 (NMR)41 | ||||
| 26.9 ± 6.7 (MD)40 | ||||
| 28 (MD)38 | ||||
| 28.1 (MD)9 | ||||
| 33.5 (MD)36 | ||||
| 36 ± 19 (MD)28 | ||||
| DPPC | 13 | 13.1 ± 7.1 | 12.3 ± 6.5 (MD)35 | |
| ∼15 (MD)39 | ||||
| WALP27 | DMPC | 21 | 29.2 ± 9.7 | 43.3 (MD)9 |
The method used is listed in parentheses. Where possible, the values are shown as average ± standard deviation. NMR, nuclear magnetic resonance; MD, molecular dynamics.
Membrane Adaptation
Besides helix tilting, an additional possible mechanism of adaptation to hydrophobic mismatch is stretching and contraction of the acyl chains of the lipids surrounding the peptides. This phenomenon has been observed in studies using experimental26,27 and computational10,28 techniques. We estimated the membrane adaptation as the average deviation of the thickness of the hydrocarbon core during the simulation from its initial value, deduced from X-ray studies of pure (peptide-free) lipid bilayers.22 Figure 2B shows the dependence of membrane adaptation on the hydrophobic mismatch. For helical peptides that were too short to span the membrane and resided on the surface, the membrane thickness fluctuated around the initial native value, as it should. Longer peptides assumed TM orientation, causing membrane contraction. Interestingly, membrane thinning of up to 1 Å was detected also when the helix’s hydrophobic core was up to 5 Å longer than the thickness of the hydrocarbon region of the membrane (Figure 2B). Finally, helices with hydrophobic TM cores that were more than 5 Å longer than the membrane thickness caused the membrane to expand slightly to improve the fit to their long TM cores (Figure 2B). Clearly, the changes in the membrane thickness upon incorporation of a TM helix are small and might appear to be negligible in view of the implicit and crude representation of the membrane in the model. However, the same pattern was repeatedly observed in simulations of various peptide-membrane systems, consolidating the observation.
Membrane Interaction of the Flanking Trp (Lys) Residues
We inspected the membrane location of the Trp (Lys) residues at the edges of the hydrophobic core of each WALP (KALP) peptide, namely, the residues in the third positions from the N- and from the C-termini of the peptides. Figures 2C and S1C show the average deviation of the α-carbons of these residues from the nearest membrane boundary as a function of the hydrophobic mismatch. When the hydrophobic mismatch was strongly negative, the Cα of these residues remained at the membrane boundary. However, as the mismatch became less negative, the Cα position extended farther away from the boundary into the polar headgroup region. At a positive mismatch of around 5 Å, the Cα positions of Trp saturated at their maximal values of ∼3 Å (Figure 2C) from the membrane boundary, and Lys saturated at a value of ∼4 Å (Figure S1C). Using Figures 2C and S1C, we estimated Peff, i.e., the length of the helix interacting with lipid chains, to be used in the theoretical model (Table S3).
Theoretical Model
Here, we develop a simple theoretical model as a closed-form expression to estimate the dependence of α, the angle at which the principal axis of the TM helix tilts from the membrane normal, on the length of the hydrophobic core of the helix (P), on the effective length of the helix, i.e., the length of the helix portion that interacts with the lipid chains (Peff, Peff < P), and on the native (peptide-free) thickness of the hydrocarbon region of the membrane (L). The model parameters were derived from the experimental data and MC simulations.
First, we deal with the two limiting cases: helices that are substantially longer, or shorter, than the native thickness of the lipid bilayer. For (P – L) ≥ 5 Å, i.e., positive mismatch of 5 Å or more, the hydrophobic effect dominates, and α is determined mostly by the tendency of the hydrophobic core of the helix to be buried in the hydrocarbon region of the membrane (Figure 1A). Because of the low free energy penalty of membrane expansion, the membrane may slightly expand;21 indeed, our MC simulations showed that it expands by approximately 1 Å (Figure 2B). In addition, the ends of the hydrophobic core of the helix extend out of the hydrocarbon region of the membrane such that only Peff interacts with the lipids (Figure 2C). Thus, in this limit cos α = (L + 1)/Peff.
The other limit addresses the case of large negative mismatch, i.e., when the hydrophobic core of the helix is substantially shorter than the hydrocarbon region of the membrane (Figure 1C, lower right panel). In this case, the membrane would need to deform beyond its elastic range (±20% of its native thickness22) to accommodate the helix in TM orientation, which is energetically unfavorable. Thus, when Peff is less than 80% of the membrane’s native thickness, the helix resides in surface orientation, and α = 90°, i.e., when Peff < 0.8L, cos α = 0.
Now we turn to the intermediate region, i.e., when P – L < 5 Å and Peff ≥ 0.8L. According to the MC simulations and previous calculations,9,10 TM helices tilt even at negative mismatch (Figure 1C, lower left panel), in spite of the free energy penalty due to membrane deformation (ΔGdef). The driving force for this is the free energy gain from the increase in precession entropy (TΔS).9,11,12 Here, we exploit the balance between these opposing contributions to derive an expression for α.
We make several assumptions. First, in accordance with the MC simulations, we assume that the peptide adopts, in essence, the same (helical) conformation regardless of the tilt angle, and that its internal energy (ΔE) is independent of the tilt. Additionally, we assume that all residues are preserved in the same local environment, i.e., the hydrocarbon or headgroup region of the lipid bilayer or the aqueous phase. We also limit the possible changes in the thickness of the hydrophobic region of the membrane to up to 20% of its native value.22 Finally, we assume that helix librations in the membrane (maximum amplitude denoted as β) are independent of the tilt α (Figure 3). Under these assumptions, eq 1 reduces to the free energy balance:
| 2 |
Figure 3.
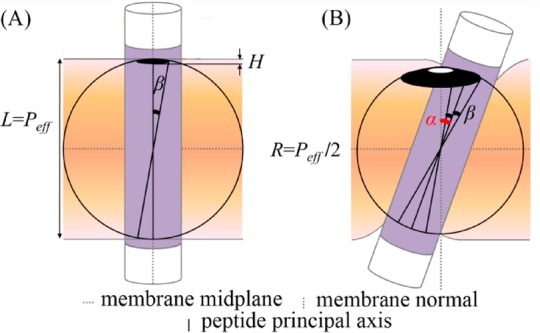
The precession entropy gain associated with TM helix tilting in the membrane. (A) Schematic illustration of the spherical surface area corresponding to the precession entropy of a hypothetical helix that spans the membrane vertically. Helix librations around the membrane normal, with a maximum amplitude of β, generate rotational entropy that is proportional to the dark cap-like surface area. (B) The precession entropy of a helix, which is tilted at an angle α from the normal, is larger (larger area). Assuming that β is independent of α, the rotational entropy of the tilted helix corresponds to the dark belt-like area. The helix is represented as a cylinder with the hydrophobic core in purple and hydrophilic termini in white. The helix’s principal axis is marked by the solid line. L is the native (peptide-free) width of the hydrocarbon region of the membrane. Peff is the length of the portion of the helix’s hydrophobic core that spans the hydrocarbon region of the membrane. R is the radius of the helix rotational sphere. H is the height of the dark sector of the sphere.
Figure 3 illustrates the precession entropy (S) of the helix in vertical vs tilted configurations; in each case, the entropy is proportional to the logarithm of the shaded area in the corresponding panel of the figure. This estimation of the precession entropy is somewhat different from the derivation of Im and Lee.12 In particular, it includes the contribution of helix librations also in the vertical orientation. In the vertical configuration, the precession entropy is proportional to the surface area of a small sector of a sphere calculated as 2πRH, where R = 1/2Peff is the sphere’s radius and H is the height of the small sector (Figure 3A). The surface area of the sphere sector is 1/2πPeff2(1 – cos β). For a helix tilted by α, the entropy corresponds to a larger belt-like section of the same sphere (Figure 3B). The surface area of the belt-like section is the difference between the areas of two sphere sectors, denoted, respectively, by 2πR2(1 – cos(α + β)) and 2πR2(1 – cos(α – β)). Using R = 1/2Peff and one of the trigonometric identities, the area of the belt-like section can be written as πPeff2 × sin α × sin β.
For negative mismatch, one has to compare between two alternative states: In the first state, the (short) TM helix is in vertical orientation, and the membrane thins to match its hydrophobic core (Figure 4A, i). The precession entropy of this state corresponds to the area of the small sphere sector of Figure 3A. In the alternative state, the helix tilts away from the normal, and the membrane thins further (Figure 4A, ii). Here, the precession entropy is higher and corresponds to the area of the larger belt-like region in Figure 3B. Substitution in eq 2 gives
| 3 |
where the left-hand side is associated with the precession entropy and the right-hand side with the membrane deformation. In eq 3, ω is a harmonic-force constant reflecting the membrane elasticity.17,21 For a cylinder of radius 5 Å, approximating the helix, ω = 0.075 kT/Å2.19,21 The derivation of eq 3 with respect to α gives a simpler expression:
| 4 |
Figure 4.
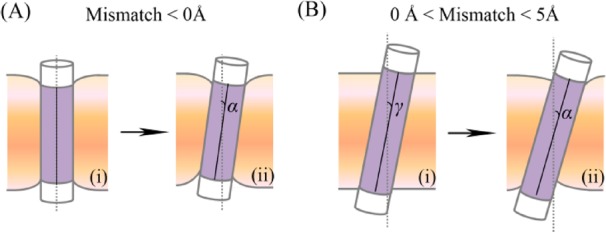
Derivation of the theoretical model. (A) Two hypothetical configurations of the system at (small) negative mismatch: (i) The helix is in vertical orientation, and the membrane thins to match the helix’s hydrophobic length. (ii) Driven by the precession entropy gain, the helix tilts by α from the normal, and the membrane thins further. (B) Two alternative configurations of the system for (small) positive mismatch in the range of 0–5 Å: (i) The helix is tilted by an angle γ from the membrane normal to match the native width of the hydrocarbon region of the membrane. (ii) Driven by the precession entropy contribution, the tilt angle increases to α (α > γ), and the membrane slightly thins. The helix is represented by a cylinder, with the hydrophobic core in purple and the polar termini in white. The membrane normal is marked by the vertical dashed line; the helix’s principal axis is marked by the solid line.
The MC simulations indicated that the tilt is driven by the precession entropy also in cases of positive mismatch of up to about 5 Å. In this respect, a small positive mismatch is similar to a negative mismatch. This notion is based on the trends of membrane adaptation (Figure 2B) and the location of the flanking hydrophilic residues relative to the membrane’s hydrophobic core (Figure 2C). In both cases, different patterns were observed for different degrees of mismatch, and the border between them was at a positive hydrophobic mismatch of approximately 5 Å rather than at a perfect match. Figure 4B shows two hypothetical helix-membrane configurations for a small positive hydrophobic mismatch in the range between 0 and 5 Å. In the first, the membrane retains its native thickness, and the helix tilts by an angle γ from the membrane normal to match the width of the hydrocarbon region of the membrane (Figure 4B, i). In the alternative configuration, the membrane thins, and the tilt angle increases to its final value of α (α > γ) to facilitate the favorable increase in helix precession entropy (Figure 4B, ii). Substitution in eq 2 gives
| 5 |
Conveniently, the derivation of eq 5 with respect to α leads to eq 4. To summarize:
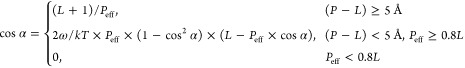 |
6 |
Equation 6 can be viewed as an equation of state of the helix in the lipid bilayer. To understand it better, we plotted α as a function of ω, L, and Peff in the physiologically relevant region of parameter space (Figure 5). The tilt angle α decreases with an increase in ω; i.e., the membrane rigidity limits the tilt, as anticipated. However, the dependence is marginally weak (Figure 5A). Additionally, the tilt angle α increases with increases in Peff and with decreases in L, as it should (Figure 5A). To explore these relations further, we plotted α as a function of Peff/L at constant ω (Figure 5B). This revealed a linear relation between α and the Peff-to-L ratio at constant Peff – L, with an increase in α when the helix length decreases (and the membrane thins). The increased tilt for shorter helices (in thinner membranes) is due to the decrease in the perturbation to the lipid; the lipid chains are shorter. The theoretical tilt angles agree well with previous calculations and measurements (Tables 3 and S1). To examine the equation of state further, we compared the results to MC simulations of various peptides within lipid bilayers over a broad range of hydrophobic mismatch scenarios.
Figure 5.
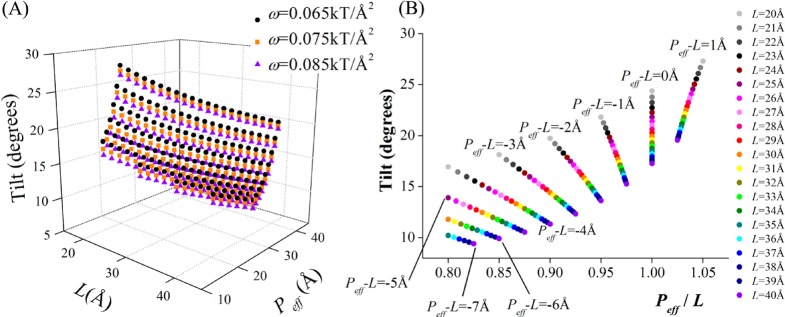
Equation of state of the helix in the membrane. (A) The dependence of α on Peff and L, for three different ω values. The tilt angle (α) was calculated using the second line of eq 6 in the range 15 Å < Peff < 50 Å and 20 Å < L < 40 Å, using the appropriate limitations on L and Peff (i.e., (P – L) < 5 Å and Peff ≥ 0.8L). Clearly, the dependence of α on ω is marginally weak. As expected, α decreases with L and increases with Peff. (B) The dependence of α on Peff/L for ω = 0.075 kT/Å2. The tilt angle α increases with an increase in Peff at the same L (symbols with the same color). Interestingly, there is a linear relation between α and the Peff-to-L ratio at constant (Peff – L).
The Theoretical Model vs MC Simulations
Figure 2A (inset) shows the good correlation between the theoretical model and MC simulations for the WALP21 peptide in bilayers of various thicknesses, and Figure 2D shows that the agreement extends throughout the WALP series (correlation coefficient of 0.99, slope of about 1, and small intercept). Similar agreement was obtained also for the KALP series (Figures S1A and S1D). Interestingly, in both cases, the minimal tilt angle was approximately 10°, in agreement with previous calculations.9,10
In addition, we also studied GWALP23, a representative peptide from the newly introduced series of GWALP peptides, which feature a single Trp residue at their termini29−33 (Table 1). The tilt values obtained via the theoretical model correlated well with the MC simulations and available data (Figure S4, Table S2).
Discussion
Implications and Limitations of the Study
We presented a theoretical derivation of an equation of state relating the tilt of a TM helix to the helix length and bilayer thickness. The equation of state (eq 6) was utilized to investigate 17 peptides of various lengths interacting with membranes of six different thicknesses, covering hydrophobic mismatch in the range of approximately ±25 Å, much broader than ranges used in previous studies.9,11,28,33−41 The tilt angles calculated using the theoretical model correlated well with our MC simulations and with data from previous experiments and calculations (Tables 3 and S1). In this respect, it is important to note that the first nuclear magnetic resonance (NMR) studies in WALP and KALP peptides reported very small tilt angles.42−44 However, more recent publications have shown that this is because the NMR experiments were interpreted using an overly simplified model of helix motion; interpretation of the same data using several dynamic models revealed larger tilt angles36,37,41,45 that are closer to those obtained in computational studies, including our theoretical model and MC simulations. The proper model for interpretation of the NMR data is debatable, but clearly the external helix motion in the membrane should be considered.46,47
The equation of state captures the thermodynamic determinants of tilting of all hydrophobic α-helical peptides, regardless of their sequences. It provides a back-of-the-envelope estimate of the tilt angle of any arbitrary peptide, given the peptide’s (effective hydrophobic) length and the membrane thickness. It could be useful for the design and interpretation of experiments, as well as for preparation of initial peptide-membrane conformations to be used, for instance, in MC or MD simulations. This way, the initial configuration of the system should be close to its energetic minimum, which should facilitate rapid convergence.
Although the theoretical tilt estimation agreed with the available data, one should keep in mind that the crude estimation has inherent limitations. The equation of state is based on the balance between the precession entropy and ΔGdef, but other terms in eq 1 may also contribute. For instance, the helix may tilt even further, inserting the polar termini deeper into the membrane core, and the precession entropy may compensate for the associated desolvation penalty. In addition, the assumptions made to derive the theory clearly simplified the mechanisms affecting helix tilt. For instance, the model assumes that the internal energy (ΔE) is independent of the tilt. In fact, at larger tilt angles, the side chains of the helix become more restrained, thus causing entropy reduction. This is not considered in the theoretical calculation. Similarly, the assumption of the same helix librations in the vertical and tilted configurations is questionable. Moreover, the equation of state does not include possible specific peptide–lipid interaction. Finally, the theoretical model assumes that the helix can be approximated by a perfectly symmetrical cylinder with no preference to any rotational angle around the principal axis.
Comparison with Other Studies
The results of the theoretical model and MC simulations are compatible with previous systematic studies of hydrophobic mismatch, performed using explicit MD simulations and potential of mean force calculations. In particular, our simulations fully agree with previous calculations showing that the helix tilt from the membrane normal is at least 10°, regardless of the extent of the hydrophobic mismatch.9,10 Regarding membrane adaptation, there are some discrepancies between the studies. Im and Kim reported membrane thinning up to 7 Å and membrane thickening up to 5 Å.9 Kandasamy and Larson, simulating KALP peptides, demonstrated membrane thinning of up to 6 Å and thickening of up to 3 Å,10 values closer to our estimations (Figures 2B and S1B), as well as to previous experimental assessments.27 Regardless of the exact values, the three studies agree that the magnitude of membrane-thinning is larger than that of thickening, as anticipated.
Additionally, Kandasamy and Larson demonstrated that in KALPs the lysine side chains extend further into the lipid polar headgroup region as the hydrophobic mismatch increases. We observed a similar effect in the flanking Trp residues in WALPs (Figure 2C) and in the flanking Lys residues in KALPs (Figure S1C). Kandasamy and Larson attributed this phenomenon to specific interactions between the lipids’ phosphate oxygen atoms and ammonium groups of the flanking Lys side chains. Im and colleagues also emphasized the role of specific lipid–peptide interactions in the balance between precession entropy gain and free energy penalty driving the TM helix tilt; they did not demonstrate the exact nature of these interactions.9,11 In contrast, our theoretical model is based on the balance between the precession entropy and nonspecific helix–lipid interactions. It should be noted that specific lipid–peptide interactions are also not taken into account in our MC simulations, and yet the results of both the theoretical model and simulations agree well with experimental data. This suggests that specific lipid–peptide interactions play only a secondary role, at least for the WALP/KALP/GWALP peptides.
TM vs Surface Configuration
The theoretical model, derived from the MC simulations, showed that if the effective length of the helix is shorter than the minimal thickness of the bilayer, the helix resides at the water–membrane interface.1 Clearly, the situation is slightly more complicated than this.48 A surface configuration of a hydrophobic helix such as a WALP or KALP is energetically favorable irrespective of the hydrophobic mismatch. In contrast, the free energy of membrane association of a helix in TM configuration depends on the hydrophobic mismatch (Figure S3). Therefore, regardless of the hydrophobic mismatch, the peptide partitions between the two configurations, and the partition ratio depends on the free energy of the two states. A similar trend was demonstrated for polyleucine peptides, either Ln or GGPG-Ln-GPGG, where n is the number of Leu residues,49−51 as well as for peptides with the sequence of the form KK-(LA)n-KK.8
In both WALPs and KALPs, the helices’ surface and TM configurations differ from each other in their helical content. The TM configuration is almost a perfect helix (Figure S2). In contrast, the surface configuration has lower helical content with decreased helicity in the helix core (Figure S2). This is because formation of a perfect α-helix in the surface configuration involves insertion of the polar Trp or Lys side chains and helix termini into the membrane core, which is energetically unfavorable. This is in agreement with the all-atom molecular dynamics simulations of Ulmschneider and colleagues.51
KALPs vs WALPs
It has been suggested that WALPs tilt to a larger degree compared with KALPs of similar length, since Trp residues partition deeper into the headgroup region than do Lys residues.7,52,53 This proposition is guided by the difference in the free energy penalty of transfer of Trp and Lys from the aqueous phase into the membrane (1.3 versus 7.4 kcal/mol in the hydrophobicity scale used here).18 The transfer free energy difference is not taken into account explicitly in the theoretical model, but it is considered implicitly since it affects the effective hydrophobic length of the helix Peff (Table S3). Application of a paired t-test to the MC simulations showed that the tilt angles of the WALPs were larger than those of the KALPs at a confidence level of 0.95. However, the average difference was only 2.8°, which is probably below the resolution of the MC model because of the use of a reduced representation for the peptide.
Conclusions
Following previous works on tilting under hydrophobic mismatch conditions,9,11,12 we demonstrated that precession entropy can contribute to the tilting of TM helices under conditions of perfect match and negative mismatch, despite the unavoidable membrane deformation. We utilized the energy balance between the precession entropy ΔS and free energy of membrane deformation ΔGdef to derive an equation of state describing the dependence of the tilt on the helix length and membrane thickness. The theoretical tilt values are similar to measurements, previous MD simulations, and our MC simulations. Thus, the equation of state can be used for a quick estimation of the helix tilt.
Notably, our simple theoretical model managed to reproduce the tilt angles observed for 17 different peptides in membranes of various thicknesses. This supports the model’s underlying assumption, namely, that the tilt is determined by the free energy balance between the helix precession entropy and lipid perturbation. However, one should take into account that the 17 peptides are synthetic and very similar to each other. It may well be that in reality more free energy contributions should be taken into account.
Acknowledgments
We thank Avinoam Ben-Shaul, Haim Diamant, Michael Levitt, Yasmine Meroz, and Kim Sharp for helpful discussions. This work was supported by the ISF-VATAT Converging Technologies Program, by the European Commission under the sixth Framework Program through the Marie-Curie Action:BIOCONTROL, contract number MCRTN - 33439, and by NATO traveling grant no. CBP.MD.CLG 984340. T.H. acknowledges the support of the Turkish Academy of Sciences and Sate Planning Organization (DPT 2009K120520).
Supporting Information Available
Figure S1 shows the results for KALPs (equivalent to Figure 2 for WALPs). Figure S2 shows the helical content of WALP23 in the aqueous phase and in a DMPC membrane. Figure S3 shows the free energy of WALP19 and WALP23 association with membranes. Figure S4 shows the theoretical and simulated tilt values of GWALP23 in various membrane types. Table S1 compares the theoretical and simulated tilt values of the KALP peptides with previous data (equivalent to Table 3). Table S2 compares the theoretical and simulated tilt values of the GWALP23 peptide with previous data. Table S3 reports the effective lengths of WALPs and KALPs in various membrane types. This material is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare no competing financial interest.
Supplementary Material
References
- Killian J. A. Synthetic peptides as models for intrinsic membrane proteins. FEBS Lett. 2003, 555, 134–138. [DOI] [PubMed] [Google Scholar]
- Killian J. A.; Nyholm T. K. Peptides in lipid bilayers: the power of simple models. Curr. Opin. Struct. Biol. 2006, 16, 473–479. [DOI] [PubMed] [Google Scholar]
- Holt A.; Killian J. A. Orientation and dynamics of transmembrane peptides: the power of simple models. Eur. Biophys. J. 2010, 39, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Maxfield F. R. Membrane domains. Annu. Rev. Cell. Dev. Biol. 2004, 20, 839–866. [DOI] [PubMed] [Google Scholar]
- Sharpe H. J.; Stevens T. J.; Munro S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 2010, 142, 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel A.; Ben-Tal N.. Protein-membrane interactions. In Introduction to Proteins: Structure, Function and Motion; CRC Press: Boca Raton, FL, 2010; pp 450–463. [Google Scholar]
- de Planque M. R.; Bonev B. B.; Demmers J. A.; Greathouse D. V.; Koeppe R. E. 2nd; Separovic F.; Watts A.; Killian J. A. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry 2003, 42, 5341–5348. [DOI] [PubMed] [Google Scholar]
- Harzer U.; Bechinger B. Alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: a CD, 15N and 31P solid-state NMR spectroscopy investigation. Biochemistry 2000, 39, 13106–13114. [DOI] [PubMed] [Google Scholar]
- Kim T.; Im W. Revisiting hydrophobic mismatch with free energy simulation studies of transmembrane helix tilt and rotation. Biophys. J. 2010, 99, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy S. K.; Larson R. G. Molecular dynamics simulations of model trans-membrane peptides in lipid bilayers: a systematic investigation of hydrophobic mismatch. Biophys. J. 2006, 90, 2326–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Im W. Transmembrane helix tilting: insights from calculating the potential of mean force. Phys. Rev. Lett. 2008, 100, 018103. [DOI] [PubMed] [Google Scholar]
- Lee J.; Im W. Restraint potential and free energy decomposition formalism for helical tilting. Chem. Phys. Lett. 2007, 441, 132–135. [Google Scholar]
- Shental-Bechor D.; Haliloglu T.; Ben-Tal N. Interactions of cationic-hydrophobic peptides with lipid bilayers: a Monte Carlo simulation method. Biophys. J. 2007, 93, 1858–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon-Grossman M.; Gofman Y.; Zimmermann H.; Frydman V.; Shai Y.; Ben-Tal N.; Goldfarb D. A combined pulse EPR and Monte Carlo simulation study provides molecular insight on peptide-membrane interactions. J. Phys. Chem. B 2009, 113, 12687–12695. [DOI] [PubMed] [Google Scholar]
- Gofman Y.; Linser S.; Rzeszutek A.; Shental-Bechor D.; Funari S. S.; Ben-Tal N.; Willumeit R. Interaction of an antimicrobial peptide with membranes: experiments and simulations with NKCS. J. Phys. Chem. B 2010, 114, 4230–4237. [DOI] [PubMed] [Google Scholar]
- Dorosz J.; Gofman Y.; Kolusheva S.; Otzen D.; Ben-Tal N.; Nielsen N. C.; Jelinek R. Membrane interactions of novicidin, a novel antimicrobial peptide: phosphatidylglycerol promotes bilayer insertion. J. Phys. Chem. B 2010, 114, 11053–11060. [DOI] [PubMed] [Google Scholar]
- Kessel A.; Shental-Bechor D.; Haliloglu T.; Ben-Tal N. Interactions of hydrophobic peptides with lipid bilayers: Monte Carlo simulations with M2delta. Biophys. J. 2003, 85, 3431–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel A.; Ben-Tal N.. Free energy determinants of peptide association with lipid bilayers. In Current Topics in Membranes; Simon S. A., McInotosh T. J., Eds.; Academic Press: San Diego, CA, 2002; Vol. 52, pp 205–253. [Google Scholar]
- Ben-Tal N.; Ben-Shaul A.; Nicholls A.; Honig B. Free-energy determinants of alpha-helix insertion into lipid bilayers. Biophys. J. 1996, 70, 1803–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I.; Kaplan M.; Jernigan R. L. Short-range conformational energies, secondary structure propensities, and recognition of correct sequence-structure matches. Proteins 1997, 29, 292–308. [DOI] [PubMed] [Google Scholar]
- Fattal D. R.; Ben-Shaul A. A molecular model for lipid-protein interaction in membranes: the role of hydrophobic mismatch. Biophys. J. 1993, 65, 1795–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D. Energetics of hydrophobic matching in lipid-protein interactions. Biophys. J. 2008, 94, 3996–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli L.; Kandasamy S. K.; Periole X.; Larson R. G.; Tieleman D. P.; Marrink S.-J. The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [DOI] [PubMed] [Google Scholar]
- Bechinger B. Solid-state NMR investigations of interaction contributions that determine the alignment of helical polypeptides in biological membranes. FEBS Lett. 2001, 504, 161–165. [DOI] [PubMed] [Google Scholar]
- Webb R. J.; East J. M.; Sharma R. P.; Lee A. G. Hydrophobic mismatch and the incorporation of peptides into lipid bilayers: a possible mechanism for retention in the Golgi. Biochemistry 1998, 37, 673–679. [DOI] [PubMed] [Google Scholar]
- Nezil F. A.; Bloom M. Combined influence of cholesterol and synthetic amphiphillic peptides upon bilayer thickness in model membranes. Biophys. J. 1992, 61, 1176–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Planque M. R.; Greathouse D. V.; Koeppe R. E. 2nd; Schafer H.; Marsh D.; Killian J. A. Influence of lipid/peptide hydrophobic mismatch on the thickness of diacylphosphatidylcholine bilayers. A 2H NMR and ESR study using designed transmembrane alpha-helical peptides and gramicidin A.. Biochemistry 1998, 37, 9333–9345. [DOI] [PubMed] [Google Scholar]
- Esteban-Martin S.; Gimenez D.; Fuertes G.; Salgado J. Orientational landscapes of peptides in membranes: prediction of (2)H NMR couplings in a dynamic context. Biochemistry 2009, 48, 11441–11448. [DOI] [PubMed] [Google Scholar]
- Vostrikov V. V.; Daily A. E.; Greathouse D. V.; Koeppe R. E. 2nd. Charged or aromatic anchor residue dependence of transmembrane peptide tilt. J. Biol. Chem. 2010, 285, 31723–31730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrikov V. V.; Grant C. V.; Daily A. E.; Opella S. J.; Koeppe R. E. 2nd. Comparison of “Polarization inversion with spin exchange at magic angle” and “geometric analysis of labeled alanines” methods for transmembrane helix alignment. J. Am. Chem. Soc. 2008, 130, 12584–12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrikov V. V.; Hall B. A.; Greathouse D. V.; Koeppe R. E. 2nd; Sansom M. S. Changes in transmembrane helix alignment by arginine residues revealed by solid-state NMR experiments and coarse-grained MD simulations. J. Am. Chem. Soc. 2010, 132, 5803–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrikov V. V.; Koeppe R. E. 2nd. Response of GWALP transmembrane peptides to changes in the tryptophan anchor positions. Biochemistry 2011, 50, 7522–7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C.-K.; Han W.; Wu Y.-D. Parameterization of PACE force field for membrane environment and simulation of helical peptides and helix–helix association. J. Chem. Theory Comput. 2012, 8, 300–313. [DOI] [PubMed] [Google Scholar]
- Holt A.; Rougier L.; Reat V.; Jolibois F.; Saurel O.; Czaplicki J.; Killian J. A.; Milon A. Order parameters of a transmembrane helix in a fluid bilayer: case study of a WALP peptide. Biophys. J. 2010, 98, 1864–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli L.; Tieleman D. P.; Fuchs P. F. Interpretation of 2H-NMR experiments on the orientation of the transmembrane helix WALP23 by computer simulations. Biophys. J. 2010, 99, 1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdirekcan S.; Etchebest C.; Killian J. A.; Fuchs P. F. On the orientation of a designed transmembrane peptide: toward the right tilt angle?. J. Am. Chem. Soc. 2007, 129, 15174–15181. [DOI] [PubMed] [Google Scholar]
- Strandberg E.; Esteban-Martin S.; Salgado J.; Ulrich A. S. Orientation and dynamics of peptides in membranes calculated from 2H-NMR data. Biophys. J. 2009, 96, 3223–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im W.; Lee J.; Kim T.; Rui H. Novel free energy calculations to explore mechanisms and energetics of membrane protein structure and function. J. Comput. Chem. 2009, 30, 1622–1633. [DOI] [PubMed] [Google Scholar]
- Bond P. J.; Wee C. L.; Sansom M. S. Coarse-grained molecular dynamics simulations of the energetics of helix insertion into a lipid bilayer. Biochemistry 2008, 47, 11321–11331. [DOI] [PubMed] [Google Scholar]
- Kim T.; Jo S.; Im W. Solid-state NMR ensemble dynamics as a mediator between experiment and simulation. Biophys. J. 2011, 100, 2922–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandberg E.; Esteban-Martin S.; Ulrich A. S.; Salgado J. Hydrophobic mismatch of mobile transmembrane helices: Merging theory and experiments. Biochim. Biophys. Acta 2012, 1818, 1242–1249. [DOI] [PubMed] [Google Scholar]
- Ozdirekcan S.; Rijkers D. T.; Liskamp R. M.; Killian J. A. Influence of flanking residues on tilt and rotation angles of transmembrane peptides in lipid bilayers. A solid-state 2H NMR study. Biochemistry 2005, 44, 1004–1012. [DOI] [PubMed] [Google Scholar]
- Strandberg E.; Ozdirekcan S.; Rijkers D. T.; van der Wel P. C.; Koeppe R. E. 2nd; Liskamp R. M.; Killian J. A. Tilt angles of transmembrane model peptides in oriented and non-oriented lipid bilayers as determined by 2H solid-state NMR. Biophys. J. 2004, 86, 3709–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel P. C.; Strandberg E.; Killian J. A.; Koeppe R. E. 2nd. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane alpha-helix determined by (2)H NMR. Biophys. J. 2002, 83, 1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grage S. L.; Strandberg E.; Wadhwani P.; Esteban-Martin S.; Salgado J.; Ulrich A. S.. Comparative analysis of the orientation of transmembrane peptides using solid-state (2)H- and (15)N-NMR: mobility matters. Eur. Biophys. J. 2012. [DOI] [PubMed] [Google Scholar]
- Fuchs P. F. Molecular dynamics of membrane peptides and proteins: principles and comparison to experimental data. Methods Mol. Biol. 2010, 654, 403–421. [DOI] [PubMed] [Google Scholar]
- Bechinger B.; Resende J. M.; Aisenbrey C. The structural and topological analysis of membrane-associated polypeptides by oriented solid-state NMR spectroscopy: established concepts and novel developments. Biophys. Chem. 2011, 153, 115–125. [DOI] [PubMed] [Google Scholar]
- de Planque M. R.; Killian J. A. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 2003, 20, 271–284. [DOI] [PubMed] [Google Scholar]
- Ulmschneider J. P.; Smith J. C.; White S. H.; Ulmschneider M. B. In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J. Am. Chem. Soc. 2011, 133, 15487–15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider J. P.; Andersson M.; Ulmschneider M. B. Determining peptide partitioning properties via computer simulation. J. Membr. Biol. 2011, 239, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider M. B.; Doux J. P.; Killian J. A.; Smith J. C.; Ulmschneider J. P. Mechanism and kinetics of peptide partitioning into membranes from all-atom simulations of thermostable peptides. J. Am. Chem. Soc. 2010, 132, 3452–3460. [DOI] [PubMed] [Google Scholar]
- Killian J. A.; von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 2000, 25, 429–434. [DOI] [PubMed] [Google Scholar]
- de Planque M. R.; Kruijtzer J. A.; Liskamp R. M.; Marsh D.; Greathouse D. V.; Koeppe R. E. 2nd; de Kruijff B.; Killian J. A. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane alpha-helical peptides. J. Biol. Chem. 1999, 274, 20839–20846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.