

Abstract
Background
Although the reference human genome sequence was declared finished in 2003, some regions of the genome remain incomplete due to their complex architecture. One such region, 1q21.1-q21.2, is of increasing interest due to its relevance to human disease and evolution. Elucidation of the exact variants behind these associations has been hampered by the repetitive nature of the region and its incomplete assembly. This region also contains 238 of the 270 human DUF1220 protein domains, which are implicated in human brain evolution and neurodevelopment. Additionally, examinations of this protein domain have been challenging due to the incomplete 1q21 build. To address these problems, a single-haplotype hydatidiform mole BAC library (CHORI-17) was used to produce the first complete sequence of the 1q21.1-q21.2 region.
Results
We found and addressed several inaccuracies in the GRCh37sequence of the 1q21 region on large and small scales, including genomic rearrangements and inversions, and incorrect gene copy number estimates and assemblies. The DUF1220-encoding NBPF genes required the most corrections, with 3 genes removed, 2 genes reassigned to the 1p11.2 region, 8 genes requiring assembly corrections for DUF1220 domains (~91 DUF1220 domains were misassigned), and multiple instances of nucleotide changes that reassigned the domain to a different DUF1220 subtype. These corrections resulted in an overall increase in DUF1220 copy number, yielding a haploid total of 289 copies. Approximately 20 of these new DUF1220 copies were the result of a segmental duplication from 1q21.2 to 1p11.2 that included two NBPF genes. Interestingly, this duplication may have been the catalyst for the evolutionarily important human lineage-specific chromosome 1 pericentric inversion.
Conclusions
Through the hydatidiform mole genome sequencing effort, the 1q21.1-q21.2 region is complete and misassemblies involving inter- and intra-region duplications have been resolved. The availability of this single haploid sequence path will aid in the investigation of many genetic diseases linked to 1q21, including several associated with DUF1220 copy number variations. Finally, the corrected sequence identified a recent segmental duplication that added 20 additional DUF1220 copies to the human genome, and may have facilitated the chromosome 1 pericentric inversion that is among the most notable human-specific genomic landmarks.
Keywords: 1q21, DUF1220 domain, Hydatidiform mole
Background
A major landmark in the modern era of medical genomics research is the sequence and assembly of the human genome. The current genome build, however, contains numerous gaps and areas of potential misassembly. Completion of an accurate assembly is a continuing challenge given the presence of multiple highly duplicated and complex regions that remain largely intractable to analysis with commonly used assembly techniques [1]. Nonetheless, finishing these regions has significant implications for identifying causative disease loci and in turn efficacious treatments for patients with genetic and genomic disorders [2]. The 1q21 region of chromosome 1 is a classic example, given its association with multiple clinical disorders and its complex architecture, with multiple regions of duplication that make complete assembly extremely difficult. The 2009 human genome assembly reflects this challenge, containing 14 gaps in the 7.7 Mb 1q21.1-2 region.
Closing these gaps in the current 1q21 build is a particularly pressing problem given that recurrent genetic and genomic variations in this region have been implicated in a multitude of disease phenotypes: neuropsychiatric diseases such as autism [3, 4] and schizophrenia [5, 6], microcephaly and macrocephaly [7, 8], cardiac conduction and structural defects [9, 10], multiple congenital anomalies [11–13], and ocular deficits [8]. Additionally, this region contains multiple Neuroblastoma Breakpoint Family (NBPF) genes encoding 238 of the known 270 copies of DUF1220, a protein domain that has undergone a striking copy number increase specifically in the human lineage [14, 15]. While this extreme DUF1220 copy number increase has been linked to the evolutionary expansion of the human brain [16, 17], the many interspersed and tandem DUF1220 paralogs in the 1q21 region are thought to be major contributors to 1q21 genomic instability leading to numerous disorders [16]. Ascertaining the exact involvement of DUF1220 and other 1q21 sequences in these diseases has been hindered by the incomplete nature of the 1q21 assembly and of the DUF1220-encoding gene family (NBPF) in particular. Without a complete, accurate assembly, genotype-phenotype associations are difficult to identify, and those that are found may not provide a complete picture of disease etiology and in some cases may even be incorrect and misleading.
To pursue the completion of the 1q21 genomic region, a haploid hydatidiform mole (CHM1) genome was utilized which reduces the challenges introduced by using a diploid, polymorphic genome [18]. Using bacterial artificial chromosomes (BACs) produced from the CHM1 genome the 14 gaps that remained in this region were closed and a single haploid genomic path generated that spans the 1q21.1-2 region. This new, completed assembly was used to more precisely analyze genomic structural variation in individuals with 1q21 CNVs and microcephaly or macrocephaly.
Results and discussion
Sequence finishing and assembly
A total of 48 BACs were sequenced generating a contig of 7,283,150 bp, covering the 1q21.1-q21.2 region of interest. This successfully closed all 14 gaps in the GRCh37/hg19 assembly, added 616,581 bp of sequence, and resulted in the addition of 12 new genes. In addition to these sequence additions, numerous differences between the previous 1q21 assembly and the CHM1 assembly were discovered, including changes in gene order, 2 inverted loci spanning multiple genes, and gene loss associated with multicopy genes (Figure 1, Table 1). While confirmation testing conducted on the assembly (see Confirmation of DUF1220 Copy Estimates section) indicates that the CHM1 library is representative of the general population, it cannot be ruled out that some of the changes may be true variants within the human population.
Figure 1.

Comparison of GRCh37/hg19 assembly (left) with the WUSTL CHM1 assembly (right). NBPF genes are indicated in red, all other genes are in blue. Black boxes on the GRCh37 map denote gaps. The vertical bar to the right of the CHM1 map denotes the novel inversion spanning multiple genes discussed in the text.
Table 1.
Copy number differences in 1q21 between the GRCh37 build and the CHM1 assembly
| Gene name | GRCh37/hg19 | CHM1 assembly |
|---|---|---|
| FAM72D | 1 | 2 |
| FCGR1A | 1 | 2 |
| HYDIN | 0 | 1 |
| NBPF16 | 1 | 0 |
| NBPF24 | 1 | 0 |
| NBPF25 | 1 | 0 |
| NBPF26 | 0 | 1 |
| NOTCH2NL | 1 | 3 |
| PDE4DIP | 1 | 3 |
| PDZK1 | 3 | 2 |
| SEC22B | 1 | 3 |
| SRGAP2 | 1 | 2 |
| Total 1q21 DUF1220 | 242 | 238 |
| DUF1220 CON1 | 22 | 17 |
| DUF1220 CON2 | 13 | 11 |
| DUF1220 CON3 | 12 | 11 |
| DUF1220 HLS1 | 60 | 62 |
| DUF1220 HLS2 | 68 | 69 |
| DUF1220 HLS3 | 64 | 66 |
| DUF1220 Triplets | 51 | 59 |
Table 1 describes changes in copy number between the GRCh37 and new CHM1 assemblies. The majority of non-DUF1220 changes were gain in copy, with 8 genes previously under represented in GRCh37. Three NBPF genes are no longer present, although DUF1220 numbers remained close to the same with DUF1220 copies being mostly redistributed among the remaining NBPF genes. This may indicate that the gene loss is an artifact of misassembly rather than true gene copy number differences. Numbers do not include the additional DUF1220 domains and NBPF genes added to the 1p11.2 region as a result of the human-specific 1q21.2 segmental duplication described in the text.
The DUF1220-containing NBPF family of genes experienced by far the largest number of changes resulting from the CHM1 assembly (Figure 2, Table 2). While there was no significant change in DUF1220 number within the 1q21 region, the domains were redistributed among 11 predicted NBPF genes, rather than 13, resulting in sequence changes to 8 NBPF genes and the redesignation of many DUF1220 domains to different domain subtypes (hereafter referred to as clades). Finally, two NBPF genes were assigned to 1p11.2, resulting in the addition of 21 DUF1220 domains to the haploid genome. This gives a total haploid DUF1220 copy number of 289 domains in the GRCh38 genome build. It is important to note that this is likely the minimum number of copies in the human genome due to the tandemly duplicated nature of these domains and the resulting challenges to sequencing. Current sequencing technology often collapses tandemly duplicated reads into single copies or, less frequently, overestimates the number of copies within these regions. Therefore, the true number of copies in these duplications may be slightly different than that determined by sequencing.
Figure 2.

Organization of the DUF1220 domain and NBPF gene families in the 1q21.1-21.2 region in the GRCh37/hg19 assembly (black) and new CHM1 assembly (red). Three NBPF genes have been lost in the CHM1 assembly, and were likely artifacts of misassembly rather than true differences between the two. Six NBPF genes show different DUF1220 copy numbers between builds. The 6 different DUF1220 clades are denoted by colored boxes and DUF1220 triplets are underlined.
Table 2.
Description of NBPF genes in GRCh38 assembly
| Name | Location | No. of DUF1220 | No. of DUF1220 triplets |
|---|---|---|---|
| NBPF1 | 1p36.13 | 7 | 0 |
| NBPF2P | 1p36.12 | 3 | 0 |
| NBPF3 | 1p36.12 | 5 | 0 |
| NBPF4 | 1p13.3 | 4 | 0 |
| NBPF5P | 1p13.3 | 2 | 0 |
| NBPF6 | 1p13.3 | 4 | 0 |
| NBPF7 | 1p12 | 2 | 0 |
| NBPF8 | 1p11.2 | 8 | 1 |
| NBPF26 | 1p11.2 | 13 | 1 |
| NBPF23P | 1q21.1 | 0 | 0 |
| NBPF17P | 1q21.1 | 6 | 0 |
| NBPF15 | 1q21.1 | 6 | 1 |
| NBPF20 | 1q21.1 | 67 | 20 |
| NBPF25P | 1q21.1 | 6 | 1 |
| NBPF10 | 1q21.1 | 42 | 12 |
| NBPF12 | 1q21.1 | 11 | 2 |
| NBPF13P | 1q21.1 | 5 | 0 |
| NBPF11 | 1q21.2 | 7 | 0 |
| NBPF14 | 1q21.2 | 32 | 7 |
| NBPF9 | 1q21.2 | 9 | 1 |
| NBPF19 | 1q21.2 | 45 | 14 |
| NBPF18P | 1q21.3 | 0 | 0 |
| NBPF21P | 3p22.2 | 1 | 0 |
| NBPF22P | 5q14.3 | 2 | 0 |
Table 2 displays a summary of all annotated NBPF regions in the GRCh38 build. There are 23 NBPF-like regions, with 14 NBPF genes and 9 pseudogenes.
The two NBPF genes newly placed at 1p11.2 are the result of a human lineage specific (HLS) segmental duplication (SD) from the 1q21.2 region to the 1p11.2 region, discovered through sequencing efforts for the CHM1 1q21 region (Figure 3). This SD is not present in other primates and is particularly interesting as both it and its paralog in the 1q21.2 region were identified in Szamalek et al. [19] as the breakpoint regions for a HLS pericentric inversion event. This event is hypothesized to be the catalyst for the expansion of the 1q12 C-band and the hyper-amplification of the HLS DUF1220 triplet in the 1q21 region [20]. The discovery of this SD is an important finding as it helps narrow the time frame in human evolution when the HLS chromosome 1 pericentric inversion took place.
Figure 3.
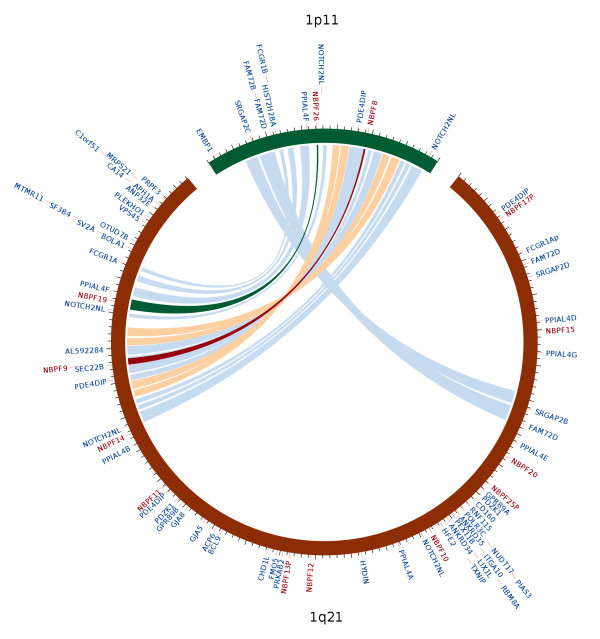
Comparison of 1q21 to 1p11.2 showing two separate duplication events between the two regions: 1) a segmental duplication between 1q21.2 and 1p11.2 containing 11 genes, including 2 NBPF genes and 2) a smaller duplication from 1q21.1 to 1p11.2[21].
Confirmation of DUF1220 Copy Number Estimates
Digital droplet PCR
DUF1220 clade (subtype) copy number estimates were compared within each NBPF gene between the new and old assemblies (Figure 2). DUF1220 domains are subdivided into six clades based on sequence similarity, referred to as conserved (CON) clades 1 through 3 and human lineage specific (HLS) clades 1 through 3. CON1 and HLS1 were analyzed for DUF1220 copy number validation. The CON1 copy number determined by ddPCR of CHM1 DNA was comparable to that seen across multiple control samples, suggesting that the assembly of this clade within NBPF genes accurately reflects the general population. The HLS1 copy number, meanwhile, was slightly lower than that seen in the majority of control samples, suggesting that the CHM1 genome may have fewer HLS copies than would be found in healthy individuals. It should be noted that HLS1 DUF1220 domains are highly polymorphic within the population and as such these results do not necessarily suggest that either version of the 1q21 assembly will be more useful for future analysis of copy number-phenotype correlations for this locus. Copy number measurements of PDE4DIP as measured by ddPCR reflected those predicted by the molar assembly and mirrored those seen in healthy controls. In addition, a primer set mapping uniquely to NBPF11 and NBPF24 (NBPF genes differing by only 3 nucleotides) from the GRCh37 assembly was used to determine if the loss of one of these regions was unique to the CHM1 cell line or representative of healthy individuals as well. ddPCR results demonstrate a single copy, confirming loss of NBPF24 and retention of the one copy of NBPF11. Overall, results indicate that the CHM1 assembly generally is representative of healthy individuals and can be used in place of the current human genome build.
Irys
Single DNA molecules (>150 kb) fluorescently labeled at BspQI sites were used to examine genome segment length within NBPF genes in BACs used to produce the new assembly. Segment lengths between BspQI labels observed in consensus maps de novo assembled from BAC molecules harboring NBPF10, NBPF12 and NBPF15 (NBPF12 shown in Figure 4) were consistent to those observed in in silico maps of the CHM1 assembly.
Figure 4.
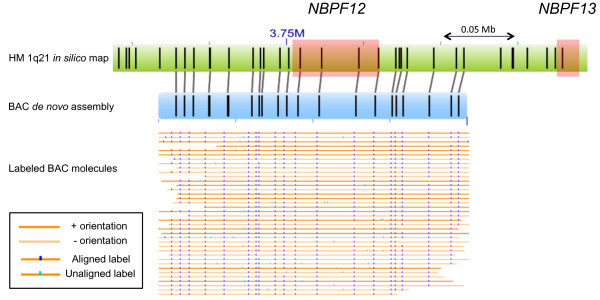
Single-molecule genome maps (orange) from three hydatidiform mole BACs were assembled de novo into consensus genome maps (blue). One of the assembled consensus genome maps is shown here (BAC CH17-112A12, blue) and is aligned to an in silico map based on the 1q21 sequence assembly described in this paper (green). Locations of the NBPF12 and NBPF13 genes on the 1q21 sequence assembly are marked in red. Segment lengths between labels in the NBPF12 gene are consistent across in silico and de novo maps.
Evolutionary Analysis
All complete HLS DUF1220 triplet sequences were used to create a phylogenetic tree (Figure 5). The phylogeny reveals first and foremost a confirmation that only NBPF genes within the 1q21 region have undergone triplet hyper-amplification. While two new NBPFs found to reside in 1p11.2 each contain a HLS DUF1220 triplet, they remain in the unexpanded form and cluster with the first triplets of other NBPF genes within the phylogeny. Second, as the 1p11.2 NBPF genes are unexpanded when compared to their 1q21.2 counterparts, the most parsimonious conclusion is that the SD from 1q21.2 to 1p11.2 occurred prior to the genomic changes that led to the HLS DUF1220 triplet hyper-amplification. This, combined with the previous discussion of the SD being found to be the breakpoints for the HLS pericentric inversion event, lends more support for the hypothesis put forth recently [20] that the 1q21.1-q21.2 region within the pericentric inversion is a unique genomic environment that allowed the HLS DUF1220 triplet to hyper-amplify.
Figure 5.
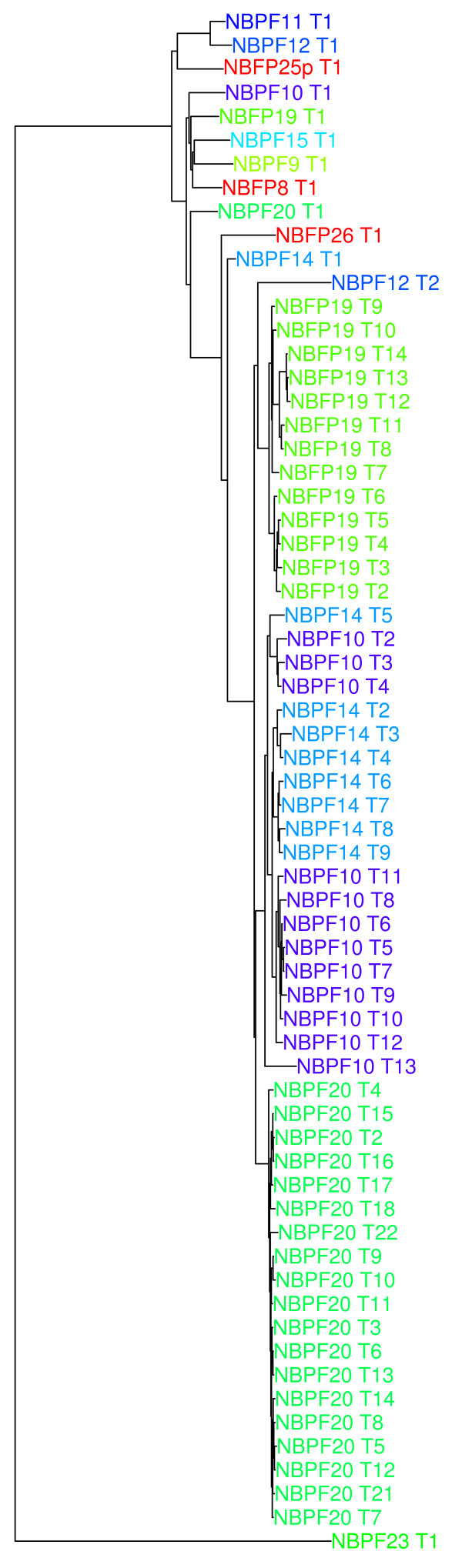
Phylogeny of DUF1220 triplets in the CHM1 assembly.
Mapping disease samples
ArrayCGH results of patient samples from the Baylor College of Medicine with known 1q21 CNVs [17] were mapped to the 2009 assembly and the new WUSTL CHM1 assembly. Figure 6 shows a comparison of these data plotted on the 2009 assembly (upper image) and the CHM1 assembly (lower image). Regions affected by these copy changes overlapped, but were not identical, between assemblies. In addition, there are genes that do not appear to be affected by deletion/duplication in the 2009 assembly (or are not present in that assembly) that fall in affected regions in the CHM1 1q21 assembly (not shown).Of note, one segment of the 2009 assembly that contains multiple genes is inverted in the new assembly (Figure 1). By mapping patient data to this new assembly, it was discovered that this inversion, while representative of the CHM1 genome, may be polymorphic in the human population (or may be inverted uniquely in the CHM1 genome). This conclusion is based on the fact that in the new CHM1 assembly, samples with Type I deletions (a class of deletions found in the distal 1q21 region) have a contiguous gene deletion region, and an additional region deleted proximally that is not contiguous, suggesting two deletion events (Figure 6). In the 2009 assembly, these two deleted regions are contiguous, suggesting one deletion event rather than two. As a single-deletion event is more likely than multiple deletions events, it is likely that this inversion occurred in the CHM1 genome but may not be the more common allelic form in the human population.
Figure 6.
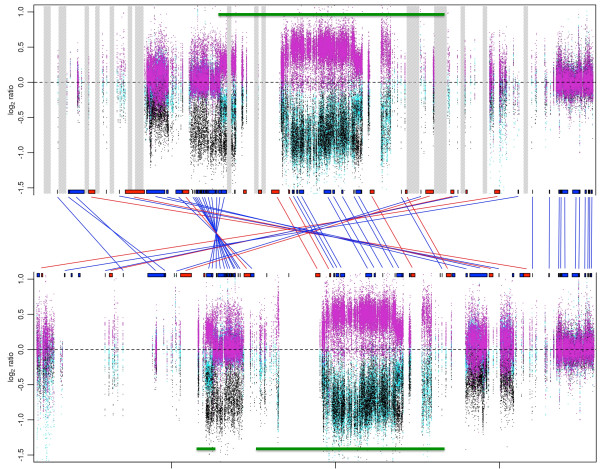
Comparison of arrayCGH profiles of patients with 1q21 deletions and duplications between the GRCh37/hg19 assembly and the CHM1 assembly. Samples with known duplications are represented in pink, Type I deletions in blue, and Type II deletions, which are larger than Type I deletions and include the thrombocytopenia-absent radius (TAR) region, in black. Gray vertical regions in the GRCh37/hg19 assembly represent gaps that were eliminated in the CHM1 assembly. Green bars above the GRCh37map and below the CHM1 map indicate the approximate location of the Type I deletion in each assembly. Note that the inverted gene segment in the CHM1 assembly requires a two-deletion event rather than single-deletion event to explain the Type I deletion mapping pattern. Tick marks at the bottom of the figure are separated by 2 Mb; the GRCh37 assembly starts at 142,000,000 and the CHM1 assembly starts at 0.
By remapping array results for 40 patients with 1q21 CNVs to the new assembly, regions affected by deletions and duplications were identified with better certainty and precision. In the future, the more accurate and complete 1q21 assembly will also allow for better mapping of breakpoints of these copy number changes, which will in turn aid in localizing disease-causing genes and regulatory loci, and in the development of a morbidity map of the region.
Conclusions
This investigation demonstrated that resources developed from a haploid hydatidiform mole genome could effectively be used to complete assembly of chromosome 1q21, one of the most complex and evolutionarily dynamic regions of the human genome. Completing this region also has significant implications for studying human disease given the numerous disorders associated with CNVs, mutations and chromosomal aberrations in the 1q21 region. Additionally, the complete 1q21 assembly will play an integral role in studies of human evolution, as 1q21 contains the majority of the 289 DUF1220 protein domain copies, 160 of which were added specifically to the human genome since the Homo/Pan divergence [15][20]. The new 1q21 assembly has already led to the discovery of a novel copy of SRGAP2, a gene in the 1q21 region that may be important for the elaboration of neuronal processes in the human brain [21]. Efforts to localize disease-causing genes and regulatory regions that have previously been hindered by the incomplete nature of the 1q21 assembly and inaccuracies in the region may now move forward with a complete and reliable map to identify causative sequence variations.
Methods
Hydatidiform mole
Hydatidiform moles are human conception abnormalities that most often arise from the fertilization of an anucleate ovum by a single X-bearing sperm. Subsequent diploidization results in a 46 XX karyotype in which all allelic variation has been eliminated allowing the unambiguous delineation of duplicated DNA as well as haplotype characterization. The hydatidiform mole (CHM1) BAC library (CHORI-17) was previously created by the Children’s Hospital Oakland Research Institute BACPAC Resource by Mikhail Nefedov in Pieter de Jong’s laboratory. The library was prepared from a well-characterized haploid cell line (CHM1htert) from Dr. Urvashi Surti, Director of the Pittsburgh Cytogenetics, laboratory, using the cloning approach described in Osoegawa et al. [22]. This library was used for subsequent analysis.
Sequence finishing and assembly
A minimum tiling path of single haplotype clones was selected based off of a fingerprint map and alignment of existing BAC end sequences (BES) to span the 1q21.1-q21.2 region of interest. Sequences were generated to cover each BAC insert as described below. The clones were pooled prior to sequencing in groups of 25, in equal molar ratio, and a single 454 fragment library and a single 3730 plasmid library were produced. This approach leveraged the high throughput, unbiased 454 data with the 3730 data, which provides long-range linkage, long reads for assembly, and template availability. The 454 pools were sequenced to greater than 25× coverage, and 3730 libraries to a coverage of 4×. In addition, BACs difficult to resolve due to multiple paralog content had individual 3730 libraries created and sequenced.
The data was assembled using a de novo assembly approach using both pcap [23] and newbler (Roche 454 software package) assembly algorithms to assemble the data. These assemblies were then compared to one another as well as to the human reference sequence, to further guide the assembly and resolve any sequencing ambiguities. This approach has been applied extensively to whole genome bacterial projects in the size range of 5 Mb, with great success, as well as clone pools in human structural variation fosmid projects [21]. An automated improvement process called prefinishing was performed to choose directed work for low quality regions and gaps, and then a manual process of finishing the regions to a level of less than 1 error per 10,000 bp was employed. At the end of this process, a high quality product suitable for identification of sequence differences between the reference sequence and the single haplotype was achieved. The new assembly can be found at accession number JH636052.4.
NBPF gene annotation
All DUF1220/NBPF homologous regions were evaluated using the criteria published in O’Bleness et al. [20]. Through a collaboration between the Sikela laboratory, RefSeq at NCBI, and the HUGO Gene Nomenclature Committee, a consensus gene nomenclature was decided upon. This nomenclature is used in the GRCh38 release of the human genome.
Analyses of segmental duplications and DUF1220 HLS triplet expansion events
Evaluation of the 1q21 to 1p11 duplication events was generated by aligning the CHM1 1q21 region to the CHM1 1p11 region using the Exonerate alignment tool with the genome to genome option [24] and visualized in GBrowse for manual annotation and confirmation. The largest and highest scoring alignments were plotted using the Circos visualization tool (Figure 3) [25]. Evaluation of the relationship between the DUF1220 HLS triplet (hls1-hls2-hls3) sequences in each NBPF gene was performed by aligning each DUF1220 HLS triplet using the PRANK multiple sequence aligner [26] and generating a phylogenetic tree using the APE package in R (Figure 5) [27].
Mapping of disease samples to the new 1q21 assembly
40 patient samples from the Baylor College of Medicine with 1q21 microduplications or microdeletions were identified by array comparative genomic hybridization (arrayCGH) using probes specific to the 2009 1q21 assembly. Arrays were previously constructed using Agilent custom array capabilities, designed and processed as described in Dumas et al. [17]. Probes from the initial run based on the old assembly were re-mapped to the new assembly in order to identify loci affected by deletion/duplication status that vary by assembly map.
Droplet digital PCR confirmation of copy number estimates in the new 1q21 assembly
Droplet digital PCR (ddPCR), a third-generation PCR technique [28], was used to check the copy number estimates of multiple loci in the new assembly. DNA was extracted from CHM1htert cell pellets, provided by Dr. Urvashi Surti, using a Qiagen DNA extraction kit following manufacturer’s protocols. Extracted DNA was digested with the restriction enzyme DDE1. Digested DNA was then added to a PCR mix according to the manufacturer’s protocol including fluorescently tagged probes specific to the region of interest (separate reactions for conserved clade 1 (CON1) (Left ‘AATGTGCCATCACTTGTTCAAATAG’ , Right – ‘GACTTTGTCTTCCTCAAATGTGATTTT’ , Hyb – ‘CATGGCCCTTATGACTCCAACCAGCC’), human lineage specific clade 1 (HLS1) (Left – ‘GCTGTTCAAGACAACTGGAAGGA’, Right - ‘GGGAGCTGCTGGAGGTAGT’ , Hyb – ‘AGAGCCTGAAGTCTTGCAGGACTCAC’), PDE4DIP (Left – ‘GCCTTATTAGCATCCCAAGACAA’ , Right – ‘CCCTGAACAGCCTTTCCTTCT’ , Hyb – ‘CATGCTGTGAAGAAGTCGGTCTACCCCAC’), and a unique region mapping to NBPF11 (Left - ‘GGAAAGTCGGGTTTGTGAGA’, Right – ‘TGGCACAACATCCTGGAATA’ , Hyb – ‘ACAACAGAGGAGAGCGGAGA’) and to a reference sequence of known copy number, RPP30 (Left – ‘GATTTGGACCTGCGAGCG’, Right – ‘GCGGCTGTCTCCACAAGT’ , Hyb – ‘TTCTGACCTGAAGGCTCTGCGC’). Oil droplets containing this mixture were produced using a BioRad droplet generator, resulting in over 14,000 droplets per well. Droplets were then subject to a thermocycling protocol with an annealing temperature of 56°C and read single-file on a droplet reader to compare fluorescence of the target and reference in each droplet. Results were merged to produce a final copy number estimate and this estimate was compared to that provided by the new assembly and to ddPCR results examining the same loci in healthy controls from the Coriell dataset.
Confirmation of NBPF data using Irys technology
The Irys platform automates high-resolution genome mapping by imaging labeled single DNA molecules in nanochannels. Three hydatidiform mole BACs (CH17-112A12, CH17-353B19 and CH17-382H24) containing NBPF12, NBPF10 and NBPF15, respectively, were selected for genome mapping to validate the sequence assembly of the chr1q21 region. BAC DNA was extracted with Large Construct kit (Qiagen, Valencia, CA) and 300 ng of purified BAC DNA were used for nicking and labeling according to the irysPrep protocol (BioNano Genomics, San Diego, CA). In brief, 300 ng of purified BAC DNA were incubated in a 10 μL nicking reaction at 37°C with 7 U Nt.BspQI (NEB, Ipswich, MA) for two hours followed by heat inactivation of the nicking enzyme at 80°C for 20 minutes. Five microliters of labeling master mix, consisting of 1.5× labeling buffer, 1.5× labeling mix (BioNano Genomics) and 1 U Taq polymerase (NEB), was added to the heat-inactivated nicking reaction mixture and this labeling reaction mixture was incubated at 72°C for an hour. The nicked and labeled DNA was repaired by PreCR® repair mix that contained 10 mM dNTP mix (NEB). One microliter of stop solution (BioNano Genomics) was added to stop the repair reaction. Lastly, the nicked, labeled and repaired DNA was stained overnight with DNA stain (BioNano Genomics). The nicked, labeled, repaired and stained BAC DNA samples (20 μL each) were combined before they were loaded on the IrysChip for genome mapping on the Irys system (BioNano Genomics).
Image detection, genome map alignment, and assembly were performed using software tools developed in-house and packaged into IrysView at BioNano Genomics. Briefly, the DNA backbone and fluorescent labels were detected, integrated, and converted into single-molecule genome maps. De novo assembly of genome maps was performed using a graph-based assembler. Consensus genome maps were then aligned to an in silico map based on the 1q21 sequence assembly.
Acknowledgements
This work was supported by grants 1R01 MH081203-01A2 (JMS), 3R01 MH081203-02S1 (JMS) and R01 HG005946 (PYK).
The authors would like to acknowledge Drs. S.W. Cheung, S.S.C. Nagamani and A. Erez from the Baylor College of Medicine for providing 1q21 deletion and duplication samples, and Dr. Ernest T. Lam at BioNano Genomics for help with data analysis and editing.
Abbreviations
- ArrayCGH
Array comparative genomic hybridization
- DUF1220
Domain of unknown function 1220
- NBPF
Neuroblastoma breakpoint family
- Hg19
Human genome version 19
- GRCh37
Genome Reference Consortium Human Build 37
- BAC
Bacterial artificial chromosome
- HM
Hydatidiform mole
- HLS
Human lineage-specific
- SD
Segmental duplication
- ddPCR
Droplet digital PCR
- CON1
Conserved clade 1
- PDE4DIP
Phosphodiesterase 4D-interacting protein
- HLS1/2/3
Human lineage specific clades 1/2/3
- CHORI-17
Children’s Hospital Oakland Research Institute-17 (name of Hydatidiform Mole BAC Library)
- BES
BAC end sequences
- WUSTL
Washington University St. Louis.
Footnotes
Competing interests
JMS is founder and primary shareholder of GATC Science, LLC.
Authors’ contributions
MO is co-first author. She oversaw the annotation and genomic and evolutionary analyses of the DUF1220 domains and NBPF genes and co-wrote the final manuscript. VBS is co-first author. She performed all droplet digital PCR protocols, contributed to analyzing genomic and evolutionary features of the new assembly and co-wrote the final manuscript. CMD provided bioinformatics support for analysis of segmental duplications, gene annotations and DUF1220 clade classifications. DA provided bioinformatic support and analysis of the 1p11 segmental duplication DA worked on BAC mapping, assembling BAC clones in a TPF and submission of scaffold data. ACYM worked on map construction and data analysis. YYYL worked on map construction and data analysis. CL grew BACs and prepared DNA samples for Irys processing CYMC performed all Irys labeling and nanochannel experiments for the project TG managed the CHM1 BAC library sequencing effort. P-YK provided funding and overall direction in Irys study design. RW is the director of the DNA sequencing lab at WUSTL. JMS oversaw the project from design to completion. All authors read and approved the final manuscript.
Contributor Information
Majesta O’Bleness, Email: Majesta.OBleness@ucdenver.edu.
Veronica B Searles, Email: Veronica.Searles@ucdenver.edu.
C Michael Dickens, Email: cmdckens@memphis.edu.
David Astling, Email: David.Astling@ucdenver.edu.
Derek Albracht, Email: dalbrach@genome.wustl.edu.
Angel C Y Mak, Email: maka@humgen.ucsf.edu.
Yvonne Y Y Lai, Email: LaiY@derm.ucsf.edu.
Chin Lin, Email: Chin.Lin@ucsf.edu.
Catherine Chu, Email: ChuCatherine@derm.ucsf.edu.
Tina Graves, Email: tgraves@genome.wustl.edu.
Pui-Yan Kwok, Email: pui.kwok@ucsf.edu.
Richard K Wilson, Email: rwilson@wustl.edu.
James M Sikela, Email: James.Sikela@ucdenver.edu.
References
- 1.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011;13:34–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis JM, Searles VB, Anderson N, Keeney J, Dumas L, Sikela JM. DUF1220 dosage is linearly associated with increasing severity of the three primary symptoms of autism. PLoS Genet. 2014;10(3):e1004241. doi: 10.1371/journal.pgen.1004241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:178–179. doi: 10.1038/455178a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, Zhang N, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Kendler KS, Freedman R, Dudbridge F, Pe’er I, Hakonarson H, Bergen SE, Fanous AH, Holmans PA, Gejman PV. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, Shen J, Kang SH, Pursley A, Lotze T, Kennedy G, Lansky-Shafer S, Weaver C, Roeder ER, Grebe TA, Arnold GL, Hutchison T, Reimschisel T, Amato S, Geragthy MT, Innis JW, Obersztyn E, Nowakowska B, Rosengren SS, Bader PI, Grange DK, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EM, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christiansen J, Dyck JD, Elyas BG, Lilley M, Bamforth JS, Hicks M, Sprysak KA, Tomaszewski R, Haase SM, Vicen-Wyhony LM, Somerville MJ. Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ Res. 2004;94:1401–1402. doi: 10.1161/01.RES.0000130528.72330.5c. [DOI] [PubMed] [Google Scholar]
- 10.Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, Gorham JM, Gabriel S, Altshuler DM, Quintanilla-Dieck Mde L, Artunduaga MA, Eavey RD, Plenge RM, Shadick NA, Weinblatt ME, De Jager PL, Hafler DA, Breitbart RE, Seidman JG, Seidman CE. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klopocki E, Schulze H, Strauss G, Ott CE, Hall J, Trotier F, Fleischhauer S, Greenhalgh L, Newbury-Ecob RA, Neumann LM, Habenicht R, König R, Seemanova E, Megarbane A, Ropers HH, Ullmann R, Horn D, Mundlos S. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet. 2007;80:232–240. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ledig S, Schippert C, Strick R, Beckmann MW, Oppelt PG, Wieacker P. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Küster-Hauser syndrome. Fertil Steril. 2011;95:1589–1594. doi: 10.1016/j.fertnstert.2010.07.1062. [DOI] [PubMed] [Google Scholar]
- 13.Weber S, Landwehr C, Renkert M, Hoischen A, Wühl E, Denecke J, Radlwimmer B, Haffner D, Schaefer F, Weber RG. Mapping candidate regions and genes for congenital anomalies of the kidneys and urinary tract (CAKUT) by array-based comparative genomic hybridization. Nephrol Dial Transplant. 2011;26:136–143. doi: 10.1093/ndt/gfq400. [DOI] [PubMed] [Google Scholar]
- 14.Fortna A, Kim Y, MacLaren E, Marshall K, Hahn G, Meltesen L, Brenton M, Hink R, Burgers S, Hernandez-Boussard T, Karimpour-Fard A, Glueck D, McGavran L, Berry R, Pollack J, Sikela JM. Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol. 2004;2:E207. doi: 10.1371/journal.pbio.0020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Popesco MC, Maclaren EJ, Hopkins J, Dumas L, Cox M, Meltesen L, McGavran L, Wyckoff GJ, Sikela JM. Human lineage-specific amplification, selection, and neuronal expression of DUF1220 domains. Science. 2006;313:1304–1307. doi: 10.1126/science.1127980. [DOI] [PubMed] [Google Scholar]
- 16.Dumas L, Sikela JM. DUF1220 domains, cognitive disease, and human brain evolution. Cold Spring Harb Symp Quant Biol. 2009;74:375–382. doi: 10.1101/sqb.2009.74.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dumas LJ, O’Bleness MS, Davis JM, Dickens CM, Anderson N, Keeney JG, Jackson J, Sikela M, Raznahan A, Giedd J, Rapoport J, Nagamani SS, Erez A, Brunetti-Pierri N, Sugalski R, Lupski JR, Fingerlin T, Cheung SW, Sikela JM. DUF1220-domain copy number implicated in human brain-size pathology and evolution. Am J Hum Genet. 2012;91:444–454. doi: 10.1016/j.ajhg.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eichler EE. Proposal for Construction a Human Haploid BAC library from Hydatidiform Mole Source Material. 2002. [Google Scholar]
- 19.Szamalek J, Goidts V, Cooper D, Hameister H, Kherer-Sawatzki H. Characterization of the human lineage-specific pericentric inversion that distinguishes human chromosome 1 from the homologous chromosomes of the great apes. Hum Genet. 2006;120:126–138. doi: 10.1007/s00439-006-0209-y. [DOI] [PubMed] [Google Scholar]
- 20.O’Bleness MS, Dickens CM, Dumas LJ, Kehrer-Sawatzki H, Wyckoff GJ, Sikela JM. Evolutionary history and genome organization of DUF1220 protein domains. G3 (Bethesda) 2012;2:977–986. doi: 10.1534/g3.112.003061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dennis MY, Nuttle X, Sudmant PH, Antonacci F, Graves TA, Nefedov M, Rosenfeld JA, Sajjadian S, Malig M, Kotkiewicz H, Curry CJ, Shafer S, Shaffer LG, de Jong PJ, Wilson RK, Eichler EE. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell. 2012;149(4):912–922. doi: 10.1016/j.cell.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osoegawa K, Woon PY, Zhao B, Frengen E, Tateno M, Catanese JJ, de Jong PJ. An improved approach for construction of bacterial artificial chromosome libraries. Genomics. 1998;52(1):1–8. doi: 10.1006/geno.1998.5423. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Wang J, Aluru S, Yang SP, Hillier L. PCAP: a whole-genome assembly program. Genome Res. 2003;13(9):2164–2170. doi: 10.1101/gr.1390403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slater G, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinforma. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Löytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci. 2005;102(30):10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 28.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddeson AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittman DK, Erndt NG, Cauley TH, Koehler RT, So AP, Dube S, Rose KA, Montesclaros L, Wang S, Stumbo DP, Hodges SP, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]