

Abstract
Background
The giant Galápagos tortoise, Chelonoidis nigra, is a large-sized terrestrial chelonian of high patrimonial interest. The species recently colonized a small continental archipelago, the Galápagos Islands, where it has been facing novel environmental conditions and limited resource availability. To explore the genomic consequences of this ecological shift, we analyze the transcriptomic variability of five individuals of C. nigra, and compare it to similar data obtained from several continental species of turtles.
Results
Having clarified the timing of divergence in the Chelonoidis genus, we report in C. nigra a very low level of genetic polymorphism, signatures of a weakened efficacy of purifying selection, and an elevated mutation load in coding and regulatory sequences. These results are consistent with the hypothesis of an extremely low long-term effective population size in this insular species. Functional evolutionary analyses reveal a reduced diversity of immunity genes in C. nigra, in line with the hypothesis of attenuated pathogen diversity in islands, and an increased selective pressure on genes involved in response to stress, potentially related to the climatic instability of its environment and its elongated lifespan. Finally, we detect no population structure or homozygosity excess in our five-individual sample.
Conclusions
These results enlighten the molecular evolution of an endangered taxon in a stressful environment and point to island endemic species as a promising model for the study of the deleterious effects on genome evolution of a reduced long-term population size.
Background
Evolution on islands is a fascinating topic. A number of plant and animal species are known to be endemic from small islands or archipelagos, having evolved in isolation from their continental relatives during long periods of time. Such systems are typically seen as natural laboratories for the study of adaptation [1]. Invading an island means entering a new biotic environment, that is, a new community of competitors, predators, preys and parasites, and a reduced total amount of available food. This sudden ecological challenge must be faced by a supposedly small number of migrants, in a context of reduced or null gene flow from the mainland. The successful colonization of an island by a new species is therefore likely to be driven by rapid adaptive evolution. Consistently, evolution on islands is often associated with rapid morphological changes [2], the observation of which has been of major importance in Darwin’s thoughts and conceptions. In the genomic era, the search for the molecular targets of such adaptive processes appears as a promising quest.
A second reason why island endemic species are of specific interest to evolutionary biologists is their supposedly reduced population size. The effective population size (Ne) is a central parameter of the population genetic theory, which determines the strength of genetic drift, the random fluctuation of allele frequencies generation after generation. The theory makes a number of important predictions regarding the influence of Ne on patterns of molecular diversity. First, small populations are expected to be genetically less diverse than large populations because of the reduced sojourn time of neutral mutations in the former. The existing data seem in broad agreement with this prediction at a wide phylogenetic scale [3,4]. In studies of more restricted taxonomic groups, a relationship between population size predictors and genetic diversity has been reported in fish [5], but not in mammals [6] or birds [7], despite abundant genetic data in the latter two taxa.
Importantly, genetic drift is also expected to decrease the efficiency of natural selection, as it pushes the frequency of an allele up and down irrespective of its contribution to fitness. Consequently, natural selection in favor of slightly advantageous mutations and in disfavor of slightly deleterious mutations is supposed to be less efficient in small than in large populations [8]. It was convincingly argued that the Ne effect is the major explanation for the difference in genome architecture between prokaryotes and large organisms [9]. Besides this contrast, it would appear important to determine whether the Ne effect on selection efficiency is detectable at a more recent phylogenetic scale, that is, between closely-related species. In particular, whether species affected by a recent drop in population size are ‘genetically endangered’ (that is, suffer from an increased load of deleterious mutations) is still debated [6,10].
To date, empirical evidence regarding the influence of Ne on the efficiency of natural selection is not so abundant. The most convincing contribution was the report in mammals of a positive correlation between body mass and the ratio of non-synonymous to synonymous substitutions (dN/dS) [11]. An increased dN/dS ratio is expected when the efficiency of purifying selection against deleterious non-synonymous changes is weakened. The mammalian pattern was therefore interpreted as a Ne effect, plausibly assuming that populations of large animals tend to be smaller than populations of small animals, on average. Still in mammals, the evolutionary rate of non-coding sequences upstream and downstream of genes was reported to be faster in primates than in rodents, which was interpreted in terms of a reduced Ne in primates [12].
One problem for testing the population size effect on patterns of molecular variation is that we typically have no direct measurement of Ne, which in the above-cited studies was indirectly approached through various surrogates (for example, body mass, conservation status, marine vs. terrestrial habitat). The long-term average Ne, which is the relevant parameter in molecular evolution, may be badly predicted by current species abundance [6,7]. Islands provide a good opportunity to cope with these problems: island endemic species are likely to have evolved in small populations during long periods of time, owing to the overall limitation in space and resource availability. Consistent with this prediction, an increased dN/dS ratio for mitochondrial genes was reported in island endemic species, as compared to their mainland relatives [13,14], which was again interpreted as reflecting a weakened efficiency of purifying selection due to a smaller long-term Ne.
The signature of a reduced Ne in the islands, although significant, was only observed in approximately 60% of the island/mainland couples [14], perhaps because of a lack of power - just one or two genes were analyzed for each species pair (and see reference [15]). It should also be noted that the dN/dS ratio, used in most of the above-mentioned studies, is not only determined by the strength of purifying selection, but also by the rate of adaptive amino-acid substitutions, which in some species can be far from negligible [16]. If adaptation on islands was a prominent process at the molecular level too, then the dN/dS ratio might be inflated independently of demographic effects. In this case, the ratio of non-synonymous over synonymous polymorphism (πN/πS), not divergence, would be a more appropriate marker of a reduced Ne, since it is much less affected by adaptive evolution than the dN/dS ratio [17]. The availability of within-species variation data is therefore of primary importance to disentangle the adaptive vs. non-adaptive forces driving molecular evolution on islands.
Here we present a population genomic study of the giant Galápagos tortoise, Chelonoidis nigra, an endangered species endemic from the Galápagos archipelago. Mitochondrial DNA (mtDNA) analyses suggested that this insular species has been isolated from the South American continent during millions of years [18,19]. C. nigra is together with the Aldabra tortoise the largest known living species of terrestrial turtles, much larger than its mainland congenerics, and can live well above 100 years. Its new environment, the Galápagos archipelago, is affected by strong climatic fluctuations in space and time, with some islands being generally quite arid, and all of them experiencing long periods of drought associated to ‘El Niño’ southern oscillations [20]. C. nigra is therefore a perfect model for the study of adaptation following island colonization. On the other hand, its large size combined to its endemic status is suggestive of a small long-term Ne for this species, which might have favored non-adaptive evolution through reinforced genetic drift. To test these hypotheses and quantify the relative influence of positive vs. purifying selection, the transcriptomic diversity of five individuals was investigated and compared to similar data gathered in several continental species of turtle.
Results
Datasets
Five C. nigra individuals from three distinct subspecies were analyzed (Table 1, Additional file 1: Figure S1). Besides C. nigra, samples from the congeneric red-footed tortoise C. carbonaria and from the Spanish pond turtle Mauremys leprosa (one individual each) were also collected. The dataset was completed by transcriptome data from the previously published European pond turtle Emys orbicularis (10 individuals) and pond slider Trachemys scripta (three individuals, Table 2).
Table 1.
Specimens sampled in this study
| Species | id a | id2 b | id3 c | Sampled in | mtDNA d | Reads ( n ) | Mbp |
|---|---|---|---|---|---|---|---|
|
C. nigra |
GA05A |
|
SRS509366 |
Rotterdam Zoo (Netherlands) |
clade c, PBL (becki) |
8,995,838 |
793 |
|
C. nigra |
GA05G |
zuz10 or zuz20 |
SRS509367 |
A Cupulatta Corsica (France) |
clade c, PBL (becki) |
10,770,970 |
987 |
|
C. nigra |
GA05H |
zuz10 or zuz20 |
SRS509368 |
A Cupulatta Corsica (France) |
clade c, PBL (becki) |
10,247,396 |
940 |
|
C. nigra |
GA05E |
zuz30 |
SRS509369 |
Zurich Zoo (Switzerland) |
clade d, VA (vandenburghi) |
12,862,334 |
1,143 |
|
C. nigra |
GA05F |
zuz01 |
SRS509370 |
Zurich Zoo (Switzerland) |
clade d, CRU (porteri) |
4,927,381 |
440 |
|
C. carbonaria |
GA05D |
|
SRS509371 |
Montpellier Zoo (France) |
|
9,218,341 |
831 |
| M. leprosa | GA03B | SRS509372 | Banyuls (France) | 9,903,466 | 885 |
Table 2.
Turtle transcriptomic sequence datasets analyzed in this study
| Species | Family | Ind | Tissue | 454 | Illu. | Contigs a | N50 | ORF b | Ref. c |
|---|---|---|---|---|---|---|---|---|---|
|
C. nigra |
Testudinidae |
5 |
Blood |
yes |
yes |
80,693 |
685 |
10,929 |
(1), (2) |
|
C. carbonaria |
Testudinidae |
1 |
Blood |
no |
yes |
11,324 |
536 |
1,236 |
(1) |
|
M. leprosa |
Geoemydidae |
1 |
Blood |
no |
yes |
10,199 |
576 |
1,439 |
(1) |
|
E. orbicularis |
Emydidae |
10 |
Blood |
yes |
yes |
68,096 |
720 |
10,503 |
(3) |
|
T. scripta |
Emydidae |
3 |
Blood, brain |
yes |
yes |
27,235 |
667 |
3,383 |
(3), (4) |
|
C. caretta |
Cheloniidae |
1 |
Blood |
yes |
no |
31,135 |
509 |
5,142 |
(2) |
| P. hilarii | Chelidae | 1 | Blood | yes | no | 45,060 | 563 | 8,066 | (2) |
Turtle phylogenomics
We first analyzed a dataset of 248 orthologous genes in eight turtle species. The tree topology we obtained (Figure 1) was consistent with published Testudines phylogenies [25,26]. Branch lengths in Figure 1 are proportional to time, whereas branch thickness reflects the estimated per million years rate of synonymous substitution, obtained by dividing synonymous branch lengths by absolute divergence time. The three tropical lineages (Phrynops, Pelodiscus, and Chelonoidis) showed a higher synonymous substitution rate than the marine (Caretta caretta) and temperate ones (Emys, Trachemys, Mauremys), consistent with a recent report in turtles of a negative correlation between synonymous substitution rate and latitude in turtles [27].
Figure 1.
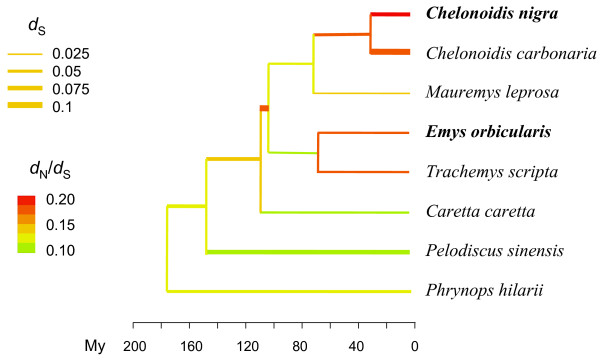
Maximum-likelihood coding sequence analysis of 248 genes in eight turtle species. Branch lengths are proportional to time. Branch thickness: number of synonymous changes per million years per 100 synonymous sites. Colors: branch-specific dN/dS ratio.
Colors in Figure 1 reflect lineage-specific dN/dS ratio. The ratio was highly heterogeneous across lineages, as revealed by the significantly better fit of a model assuming one specific dN/dS ratio per branch, compared to a one-ratio model (2*delta log likelihood = 1,472, 15 degrees of freedom, p-val <10-100). Interestingly, the C. nigra branch was the one showing the highest dN/dS ratio. This appears consistent with the hypothesis of faster non-synonymous evolutionary rate in the islands [14]. However, according to a phylogenetic analysis of five genes in 32 Testudinoidea species [28], the divergence between the C. nigra and C. carbonaria lineages dates back to approximately 30 million years ago, whereas the colonization of the Galápagos archipelago by C. nigra must be younger than 3 to 4 million years, that is, the age of the oldest Galápagos Islands (see Discussion). Whether insularity explains the high dN/dS ratio observed in the C. nigra branch is therefore unclear. Within-species data are clearly more appropriate to specifically address this issue.
C. nigra coding sequence polymorphism
In C. nigra, individual genotypes and single nucleotide polymorphisms (SNPs) were called using a probabilistic approach paying specific attention to the removal of spurious SNPs due to hidden paralogy. Various conditions were tried regarding the stringency of paralog filtering and the minimal number of genotyped individuals required to validate a SNP (Table 3, see Methods). Only contigs for which an ORF longer than 200 base pairs (bp) was predicted and an ortholog was detected in C. carbonaria were included in this (and subsequent) analyses. Between 814 and 1,059 predicted ORFs were analyzed, depending on the settings. They yielded 769 to 1,933 coding SNPs, of which >99.5% were biallelic. Results were reasonably consistent across conditions. Increasing the minimal required number of individuals resulted in a moderate increase in estimated πS, and a moderate decrease in πN/πS ratio. Increasing the paralog filtering stringency resulted in a decreased πS, as expected, but hardly affected the estimated πN/πS. The sampling variance of these estimates was acceptably low: the width of bootstrap confidence intervals was 10% to 15% of the πS average, and 15% to 20% of the πN/πS one. The results corresponding to condition A3 are shown in Figure 2, and compared with a similar analysis conducted in the continental European pond turtle E. orbicularis (Table 3, last line).
Table 3.
Robustness of non-synonymous and synonymous diversity estimates in the giant Galápagos tortoise
| Condition | Ind. ( n ) | Paralogue filter | Genes ( n ) | SNPs ( n ) | π S (%) | π N /π S | F IT | α 0.2 |
|---|---|---|---|---|---|---|---|---|
| A1 |
≥3 |
Stringent |
1,053 |
1,933 |
0.18 ±0.03 |
0.35 ±0.06 |
-0.14 |
-0.30 |
| A2 |
≥4 |
Stringent |
968 |
1,538 |
0.18 ±0.03 |
0.34 ±0.06 |
-0.12 |
-0.34 |
| A3 |
5 |
Stringent |
814 |
1,041 |
0.19 ±0.04 |
0.31 ±0.06 |
-0.10 |
-0.15 |
| B1 |
≥3 |
Very stringent |
1,059 |
1,423 |
0.13 ±0.02 |
0.33 ±0.05 |
-0.11 |
-0.05 |
| B2 |
≥4 |
Very stringent |
974 |
1,131 |
0.14 ±0.02 |
0.31 ±0.05 |
-0.10 |
-0.09 |
| B3 |
5 |
Very stringent |
820 |
769 |
0.14 ±0.02 |
0.28 ±0.05 |
-0.08 |
0.18 |
| Emys | ≥5 | Stringent | 953 | 1,845 | 0.42 ±0.04 | 0.09 ±0.02 | 0.21 | 0.57 |
Figure 2.
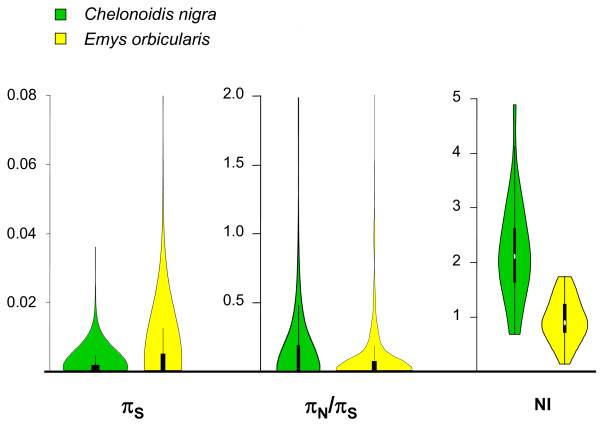
Distribution of population genomic statistics across predicted coding sequences inC. nigraandE. orbicularis. πN/πS distribution: coding sequences for which πS = 0 were omitted. NI distribution: coding sequences for which dS = 0 were omitted.
The average estimated synonymous diversity (πS) in C. nigra was 0.0013 to 0.0019, that is, a very low value, similar to the human one [6]. No positive FIT was detected, meaning that the two alleles of a given individual were not more similar, on average, than two alleles sampled in two distinct individuals. Likewise, no significant FST was detected between the two mitochondrial clades sampled in this study, that is, clade c (three individuals) and clade d (two individuals, Table 1). The estimated average πN/πS ratio in C. nigra was around 0.3. This is higher than the highest πN/πS ratio reported so far (approximately 0.2 in human). The average C. nigra/C. carbonaria dN/dS ratio in this analysis was 0.13 to 0.15. This value is similar to the human-specific and chimpanzee-specific dN/dS ratios [6], but still substantially lower than the C. nigra πN/πS ratio. Consequently, the neutrality index was above unity (Figure 2), even after removal of low-frequency variants, and the proportion of adaptive amino-acid substitution estimated by the McDonald-Kreitman method, α and α0.2, was below zero in C. nigra. When a method accounting for the whole distribution of allele frequencies was used [29], an estimate of 0.13 was obtained, with a confidence interval including zero. The other turtle species for which we have within-species variation data, E. orbicularis, did not show such an extreme population genomic pattern: πS and α were higher, and πN/πS and dN/dS lower, in E. orbicularis than in C. nigra (Figure 2). These results are indicative of an extremely small long-term Ne in the Galápagos tortoise, and suggest that the elevated rate of non-synonymous substitutions observed in C. nigra is in the first place a consequence of increased genetic drift, not accelerated adaptive evolution.
Flanking sequences
To further investigate this hypothesis, we examined the evolution of potential targets of weak selection, namely coding region-flanking sequences. In C. nigra, we extracted 158 5′ UTR and 598 3′ UTR sequences of length above 50 bp, and measured levels of genetic diversity based on sites genotyped in at least four individuals (out of five). In E. orbicularis, we similarly analyzed 89 5′ UTR and 457 3′ UTR sequences, requiring that at least eight individuals (out of 10) were genotyped. Figure 3 summarizes the relative levels of within-species diversity at 5′ UTR, 3′ UTR, synonymous and non-synonymous sites in the two species. In E. orbicularis, the 5′ and especially the 3′ UTR sequences showed a significantly lower amount of diversity than synonymous sites, suggesting that these sequences are under selective pressure. The selective constraint, although detectable, was much less pronounced than for non-synonymous sites, as reflected by the higher πUTR/πS than πN/πS ratio, in line with similar observations made in mammals [12] and birds [30]. The strength of purifying selection on UTR sequences appeared weaker in C. nigra. No significant constraint was detected in 5′ flanking sites, and the estimated level of constraint on 3′ flanking sites was reduced, compared to E. orbicularis. This is consistent with the πN/πS pattern across species, and with the hypothesis of a reduced Ne in the Galápagos tortoise.
Figure 3.
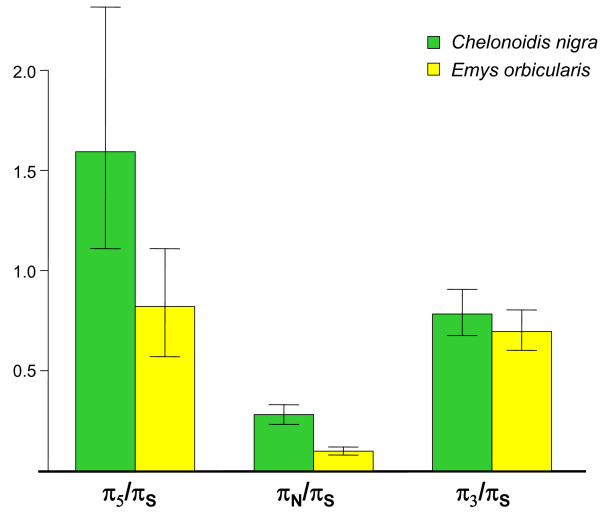
Flanking vs. coding region diversity in C. nigra and E. orbicularis. πS: average nucleotide diversity at synonymous sites; π5: average nucleotide diversity at 5′-UTR sites; π3: average nucleotide diversity at 3′-UTR sites. Very similar results were obtained when we only analyzed predicted cDNAs for which both coding sequence and 3′ UTR sequence were available, and when we restricted the analysis to the 500 first bases of the 3′ UTR regions.
In neither of the two species did we detect a progressive decrease in the amount of selective constraint with increasing distance to the coding sequence, in contrast with the published mammal and bird analyses [12,30]. We suggest that the difference may result from the fact that we are here analyzing cDNA sequences, whereas the two above-cited studies analyzed genomic sequences. Consequently, our flanking regions only include UTR, whereas theirs included both UTR and untranscribed regions, the proportion of which presumably increases as ones moves away from the coding sequence. For this reason, πflanking/πS estimates should not be directly compared between turtles, mammals, and birds across the three studies.
Gene expression analysis
Gene-specific expression profiles were estimated in the five turtle species for which Illumina data were available. Correlation coefficients of log-transformed, normalized expression levels across genes were calculated for each pair of species. The Testudinidae (C. nigra/C. carbonaria) and Emydidae (E.orbicularis/T. scripta) pairs showed the highest level of correlation of gene expression (r2 = 0.78 and r2 = 0.73, respectively). A neighbor-joining analysis of the pairwise correlation matrix produced the ((E. orbicularis, T. scripta), M. leprosa, (C. carbonaria, C. nigra)) topology, in agreement with published turtle phylogenies. Branch lengths revealed no specific pattern in the C. nigra lineage, which did not show an accelerated evolutionary rate of gene expression pattern. Rather, the M. leprosa lineage was the fastest evolving in this analysis (Additional file 2).
Gene ontology analysis
Functional annotation of the C. nigra and E. orbicularis predicted cDNAs was achieved by sequence similarity using the generic GO-slim ‘Biological process’ gene ontology. Roughly two-thirds of the 2,774 annotated coding sequences (ORFs) were associated to metabolic genes, the other ones being associated to transport, regulation, cellular growth and organization, or immunity (Figure 4, Additional file 3: Table S1). For each GO-slim term, the average log-transformed πN/πS ratio was calculated and compared between the two species, in search for terms showing a specific increase or decrease in selective pressure in C. nigra. This was achieved through a z-score analysis accounting for sampling variance and global genomic trends (see Material and methods).
Figure 4.
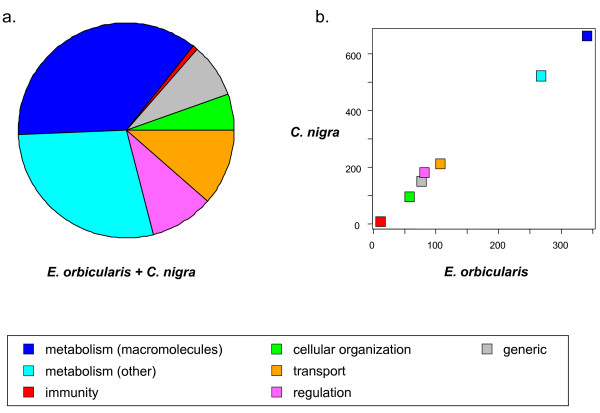
Functional annotation of coding sequences analyzed in C. nigra and E. orbicularis. (a) Pie chart representing the total number of coding sequences from the two species in each category; (b) correlation of category-specific abundance between the two species. Only coding sequences for which we obtained both a GO-slim term annotation and a πN/πS estimate were included. The generic categories used in this figure were defined by grouping terms from the ‘biological process’ GO-slim ontology as indicated in Additional file 3: Table S1, second colon.
Figure 5 plots the term-specific average πN/πS ratio (left panel), and its normalized version (right panel, z-score), in C. nigra versus E. orbicularis. A majority of terms show a πN/πS ratio lower than the genomic average, and therefore a negative z-score. This reflects the fact that coding sequences for which a functional annotation was obtained are more conserved, on average, than the non-annotated ones. Colored circles show the terms for which we detected a significant contrast in πN/πS ratio between the two species, as reflected by the Δz statistics (see Material and methods). Four terms showed a significantly positive Δz, that is, a higher πN/πS ratio in C. nigra than in E. orbicularis relative to genomic averages: ‘RNA metabolic process’, ‘translation’, ‘catabolic process’, ‘nucleotide metabolic process’. Two terms showed the opposite trend, that is, a lower πN/πS ratio in C. nigra: ‘immune system process’ and ‘response to stress’ (Additional file 3: Table S1).
Figure 5.
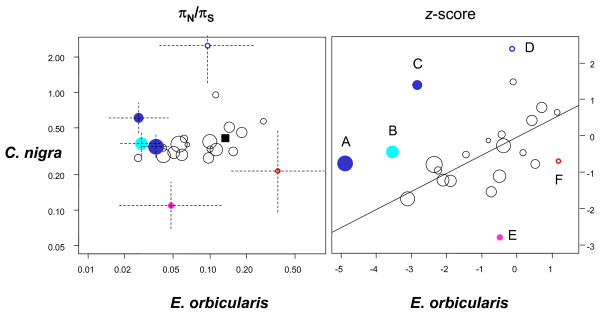
GO term-specific πN/πSanalysis inC. nigraandE. orbicularis. Each circle is for a GO-slim term. Circle surface is proportional to the number of associated coding sequences. Left panel: term-average πN/πS ratio in log scale; black square: genomic averages. Right panel: term-specific z-score. Colored circles: z-score significant at the 10% (open) or 5% (close) level. Blue: macromolecule metabolism; cyan: metabolism (other); purple: response to stress; red: immunity. Dotted lines: standard deviation. Letters refer to Additional file 3: Table S1, first colon.
Discussion
Genome-wide studies have been so far largely restricted to model organisms, that is, domestic or laboratory species. Here, thanks to NGS technologies, we were able to characterize for the first time the transcriptomic variability and population genomics of an endangered, emblematic species, the giant Galápagos tortoise, in absence of prior genomic knowledge.
One specificity of our sample is the lack of geographic information: we do not exactly know from which locality and each island of the Galápagos archipelago the sampled individuals come from. This is a general problem with C. nigra: previous genetic studies have shown that the current location of a turtle is not a reliable indicator of its true origin because of human-made translocations [21,31]. For this reason, these studies concluded that a genetic assignment of lineage of origin is the most trustworthy tool to use. Our sample, which covers a wide range of C. nigra mitochondrial and microsatellite lineages, is appropriate with respect to this criterion. Importantly, our analysis revealed no departure from panmixy in C. nigra. This is the most favorable situation for studying population genomics and molecular evolution, in which any random sample of individuals is expected to provide an unbiased estimate of the species genetic diversity, irrespective of geography. We conclude that this potential concern regarding sampling is unlikely to affect our conclusions, even though they would be worth confirming based on a larger population sample.
Divergence times and substitution rates within the chelonoidis genus
Our phylogenetic analysis revealed a significant heterogeneity in dN/dS ratio across branches, with C. nigra showing the highest dN/dS ratio of all the analyzed turtle lineages. This is consistent with the hypothesis of a reduced population size in the giant Galápagos tortoise. To what extent the high dN/dS ratio in C. nigra is explained by its insularity is unclear, however, and dependent on the timing of divergence within the Chelonoidis genus and colonization of the Galápagos archipelago. The literature is contradictory regarding the divergence date between C. nigra and C. carbonaria. In a phylogenetic analysis of five genes in 32 Testudinoidea species and one fossil calibration, Le and McCord [28] estimated that the C. nigra/C. carbonaria split occurred 29 +/- 7 million years ago. However, from an analysis of mitochondrial DNA within the Chelonoidis genus and a biogeographic calibration, it was suggested that C. nigra diverged from its closest relative C. chilensis as early as 3.2 million years ago [19]. Propagating this estimate to the C. nigra/C. carbonaria pair based on their data yields an estimated age of approximately 5 million years ago for this split, that is, six times younger than the Le and McCord figure.
Our analysis of 248 nuclear genes indicates that the average amount of synonymous divergence between C. nigra and C. carbonaria is 0.052 substitutions per synonymous site (Additional file 2). Assuming a divergence as recent as approximately 5 million years for this split would imply a synonymous substitution rate of approximately 5.10-3 substitution per site per million years in Chelonoidis. This is well outside the range of nuclear synonymous substitution rate recently estimated across 132 turtle species (three nuclear genes) [27], and 20 times as high as the median rate in this study. Such a dramatic increase in substitution rate would be expected to substantially lengthen Chelonoidis branches in molecular phylogenies - an effect that was not conspicuous in references [26] and [28]. In contrast, the 29 million years estimate of Le and McCord, which was used in this study, would imply a plausible synonymous rate estimate of 9.10-4 substitution per site per million years in the Chelonoidis genus. One reason for the unexpectedly recent divergence times inferred by Poulakakis et al. [19] is their assumption that the C. nigra/C. chilensis split occurred at the time of the emergence of the first Galápagos Islands, that is, 2.2 to 4 million years ago. This does not need to be necessarily true: the split might have predated the emergence of the archipelago, assuming that the ancestral species that invaded the islands some Mya has gone extinct in the continent since then [18]. According to this scenario, only about 10% of the C. nigra branch in the tree of Figure 1 would be associated with insularity, which perhaps explains the modest dN/dS increase in this lineage compared to, for example, the C. carbonaria branch.
A typical low-Ne species
The average synonymous diversity in C. nigra was very low (πS = 0.0013 to 0.0019), much lower than in the similarly analyzed European pond turtle E. orbicularis. Among vertebrates, levels of synonymous diversity below our C. nigra estimate have only been reported in the common marmoset Callithrix jacchus (πS = 0.0012), and Homo sapiens (πS = 0.0012) [6]. Noticeably, this top-three list of low-diversity champions includes very diverse species in terms of current abundance - the geographically restricted Galápagos tortoise, the relatively abundant, locally invasive common marmoset, and the highly successful humans.
Besides its low genome-average level of genetic polymorphism, we found an extremely high ratio of non-synonymous to synonymous diversity in C. nigra. Our πN/πS estimate (0.28 to 0.32) is the highest value reported so far from genome-wide data, suggesting that purifying selection against mildly deleterious non-synonymous variants is less effective in C. nigra than in most living species. As compared to published values, our estimate might be slightly inflated by the absence of a reference genome in C. nigra, and the de novo prediction of ORF we performed. However, we note that a similar analysis performed in E. orbicularis did not yield such an elevated πN/πS ratio. Divergence analysis consistently revealed a high dN/dS ratio in C. nigra., similar to the one reported in human (Figure 1, Table 3). The analysis of flanking regions also revealed a reduced efficiency of purifying selection as compared to E. orbicularis.
Therefore, for all the population genomic indicators we considered, C. nigra showed the expected characteristics of a low-population sized species, that is, a reduced amount of genetic diversity and a weakened efficiency of selective effects. This is consistent with the idea that C. nigra has experienced a reduced long-term Ne as a consequence of its isolation in a restricted geographic area [14]. This result, if confirmed from other case studies, would make island endemic species a promising model for the analysis of the Ne effect on genome evolution. Besides the variable we examined in this study, one may think to investigate, for example, the complexity of the transcriptome, proteome and interactome in island vs. mainland species, to test the suggestion that these features primarily evolve through the long-term accumulation of neutral or slightly deleterious elements and interactions [9].
From a conservation point of view, the report of a small long-term Ne and of elevated πN/πS and dN/dS ratio suggests that the giant Galápagos tortoise suffers from a particularly heavy load of deleterious mutations, both fixed and polymorphic, as compared to the other turtles of this study. This might be a matter of worry. However, our own species, H. sapiens, is similarly affected by a substantial load of deleterious mutations [32], which have apparently not hampered its ecological success. Obviously, the giant Galápagos tortoise deserves to be protected irrespective of its level of genetic diversity and mutation load.
Our data did not reveal any departure from panmixy in C. nigra. No nuclear population structure between the two sampled mitochondrial lineages was found, and no significant excess of individual homozygosity was detected. This result is in apparent contradiction with the report of significant amounts of genetic differentiation between numerous pairs of C. nigra populations, based on a much larger sample of tortoises and 10 microsatellite loci [33]. The two studies are very different in terms of locus sampling, population sampling, and data type, and more data and analyses would be required to identify the reasons for this discrepancy. We note that the E. orbicularis analysis revealed a substantial excess of homozygosity and significant FST between populations, suggesting that our approach has some power to detect population differentiation when effective. At any rate, the lack of genetic differentiation genome-wide that we report between entities that recently reached species level [34] calls for a re-examination of the taxonomy in this group.
Functional population transcriptomics
The McDonald-Kreitman approach did not reveal any evidence for adaptive amino-acid substitution in the C. nigra lineage. The πN/πS ratio was higher than the dN/dS ratio, and this was still true after low-frequency variants were accounted for. This negative result, however, might be explained by a recent drop of Ne in C. nigra, which would have inflated the πN/πS ratio to a value well above its average level during the divergence between C. carbonaria and C. nigra. A global analysis of gene expression level did not reveal any specific pattern in the C. nigra lineage, in which the global rate of gene expression evolution did not appear to be accelerated, as compared to other turtle species. We note that these results about gene expression levels should be taken with caution because the physiological state of the sampled individuals was not properly controlled, and probably very different between species - for instance, E. orbicularis individuals were caught from the wild, whereas C. nigra individuals were sampled in zoos.
The GO-term specific πN/πS analysis we conducted identified four terms for which the πN/πS ratio is significantly higher in C. nigra than in E. orbicularis relative to the genomic average, that is, evidence for relaxed selective pressure in the Galápagos tortoise (Figure 5, Additional file 3: Table S1). These terms correspond to basic metabolic functions of the cell, including ‘translation’ and ‘RNA metabolic process’. We note that RNA translation is amongst the most energy-consuming cellular functions. The increased amount of functional constraint on these genes in E. orbicularis might be related to the necessity of substantial energetic savings in this temperate species, which enters into a deep hypometabolic state during wintering [35,36].
Two GO-slim terms, on the other hand, yielded evidence for reduced relative πN/πS ratio in C. nigra, namely ‘immune system process’ and ‘response to stress’. ‘Immune system process’ is the only GO-slim term for which the C. nigra average (0.22) is below the E. orbicularis one (0.36). A high non-synonymous to synonymous ratio in immunity genes is typical and interpreted as reflecting the effect of balancing selection, that is, a selective force favoring the diversity of pathogen-responding alleles. Observing a relatively low πN/π value for immunity genes in C. nigra is therefore suggestive of a reduced pathogenic/parasitic diversity in this insular species. This is in agreement with the hypothesis of a shift from antibody-mediated, acquired immunity to cell-mediated, innate immunity in insular species [37].
‘Response to stress’ is the other GO-slim term for which a significant reduction in πN/πS ratio was detected in C. nigra, most likely reflecting an increased selective constraint on these genes in the giant Galápagos tortoise. Seven of the 31C. nigra coding sequences associated to this category have homologues that encode for heat-shock proteins (for example, dnaJ), that is, chaperone proteins involved in the response to various kinds of environmental stress, such as temperature, infection, and starvation, among others (Additional file 4: Table S2). This might be related to the highly fluctuating climate of the Galápagos Islands, and the long, recurrent periods of aridity that animals must face [38]. Besides heat-shock proteins, a majority of the C. nigra coding sequences associated to the ‘response to stress’ term have homologues related to the control of oxidative stress (for example, peroxydase), DNA replication/repair (for example, photolyase), and apoptosis (for example, bcl2-associated transcription factor, Additional file 4: Table S2), that is, functions that have been linked to the regulation of ageing and longevity [39]. Our results suggest that the giant Galápagos tortoises, which can live well above 100 years in a warm, mutagenic environment, are undergoing a particularly strong selective pressure with respect to the management of oxidative molecular species and DNA damage.
Conclusions
Our analyses of transcriptomic diversity in C. nigra revealed that molecular evolution in the giant Galápagos tortoise is strongly influenced by an increased rate of genetic drift, which weakens the efficiency of natural selection and creates a substantial mutation load. The relaxation of purifying selection affects most genes, regulatory regions, and functions, with the exception of genes involved in response to stress, on which selective pressure has been reinforced, presumably in response to the new environmental conditions and elongated life span in this species. This study points to insular species as a promising model to explore further the effect of genetic drift on genomic patterns, and its relationship with gene function and adaptation.
Material and methods
Sampling and sequencing
Blood from five adult C. nigra, one C. carbonaria and one M. leprosa individuals were sampled from public European zoological collections (Table 1). The sampling and animal handling have been done by veterinarians and staff of the Montpellier Zoo, Zurich Zoo, Rotterdam Zoo, and A Cupulatta Zoo according to the Code of Practice and Code of Ethics established by the European Association of Zoos and Aquarias. RNA was extracted and cDNA libraries were sequenced using Genome Analyzer II (Illumina®, see Additional file 2). Sequence reads were deposited in the NCBI Sequence Read Archive database as bioproject PRJNA230239, biosample accession SRS509366-SRS509372 (see Table 1).
Transcriptome assembly and genotype calling
Contigs (predicted cDNAs) were assembled following reference [40]. Illumina reads were mapped to the contigs using BWA [41]. In C. nigra, single-nucleotide polymorphisms (SNPs) and genotypes were called using the method described in reference [42]. Dubious SNPs potentially resulting from hidden paralogy were cleaned using the method introduced in reference [22]. This method calculates the likelihood under a two-locus model (that is, assuming that two distinct genes have contributed reads which were erroneously assembled in a single contig), and discard SNPs that reveal a significant increase in likelihood, as compared to the one-locus model. Two stringency levels were tried in this analysis: stringent (likelihood ratio test P value <0.01) and very stringent (likelihood ratio test P value <0.05).
Polymorphism analysis
For each ORF, the following statistics were calculated: per-site synonymous (πS) and non-synonymous (πN) diversity in C. nigra, per-individual heterozygosity, overall heterozygote deficiency (FIT), number of synonymous (pS) and non-synonymous (pN) segregating sites in C. nigra, number of synonymous (dS) and non-synonymous (dS) fixed differences between C. nigra and C. carbonaria, neutrality index NI = (pN/pS)/(dN/dS), neutrality index calculated after removing SNPs for which the minor allele frequency was below 0.2 (NI0.2), estimated fraction of adaptive amino-acid substitutions α = 1-NI, and its corrected version α0.2 = 1-NI0.2. An additional estimate of α, called αEWK, was calculated according to a method based on the inferred site frequency spectrum [29], that is, the distribution of minor allele frequency across SNPs. A similar analysis was conducted in the European pond turtle E. orbicularis based on a sample of 10 individuals, using the pond slider T. scripta as outgroup, reproducing a previously published analysis [22]. In both C. nigra and E. orbicularis, we extracted 5′ and 3′ UTR sequences from contigs in which a start and/or a stop codon had been recovered. UTR sequences of length 50 bp or more available in at least four (C. nigra) or eight (E. orbicularis) individuals were selected. UTR sequence polymorphism was estimated using the nucleotide diversity index, separately averaged across 5′ UTR and 3′ UTR sequences.
Gene ontology analysis
The generic GO-slim ontology [23] was used to explore the distribution of the πN/πS ratio across functional categories of genes in C. nigra vs. E. orbicularis, in search for gene sets showing a specific increase/decrease of selective pressure in one of the two species. GO-slim is a contracted version of the Gene Ontology database including a relatively small number of high-level, little-overlapping terms. A term-averaged log-transformed πN/πS ratio was calculated for each species and each term, and compared between species. A resampling procedure was designed to quantify, for each term, the significance of the normalized difference in πN/πS ratio between the two species (see Additional file 2).
Competing interests
The author declare that they have no competing interests.
Authors’ contributions
EL contributed to conceive the project and performed the population genomic analyses. YC contributed to conceive the project and generated the data. AB and VC contributed analytical tools and helped with data analysis. JR performed the phylogenomic analyses. BN contributed to conceive the project and performed the flanking sequence analyses. JML conceived and performed the gene ontology analysis. NG conceived the project and wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Mitochondrial DNA genealogy in C. nigra. The tree was built based on a 705-long fragment of the control region from 89C. nigra individuals. Sequence accession numbers and island of origin of each turtle are provided. The three major clades identified by Caccone et al. [18] are indicated. The five individuals analyzed in this study appear in red.
Details about sampling, sequencing, assembly, SNP calling, gene expression, gene ontology, and phylogenomic analyses.
Contrasting GO-slim term-specific selective pressure between C. nigra and E. orbicularis. list of GO-slim ‘Biological process’ terms, with the number of associated contigs, their average πN/πS ratio and its normalized version (z-score) in C. nigra vs. E. orbicularis.
C. nigra and E. orbicularis coding sequences associated to GO-slim terms ‘immune process’ and ‘response to stress’. list of predicted contigs associated to the ‘immune process’ and ‘response to stress’ terms in either C. nigra or E. orbicularis, their functional annotation, length, coverage, πN, and πS.
Contributor Information
Etienne Loire, Email: etienne.loire@univ-montp2.fr.
Ylenia Chiari, Email: yle@yleniachiari.it.
Aurélien Bernard, Email: aurelien.bernard@univ-montp2.fr.
Vincent Cahais, Email: vincent.cahais@univ-montp2.fr.
Jonathan Romiguier, Email: jonathan.romiguier@gmail.com.
Benoît Nabholz, Email: benoit.nabholz@gmail.com.
Joao Miguel Lourenço, Email: jlourenco@univ-montp2.fr.
Nicolas Galtier, Email: nicolas.galtier@univ-montp2.fr.
Acknowledgments
We thank Henk Zwartepoorte, Willem Schaftenaar, Samuel Furrer, Pierre Moisson, Cedric Libert, Carmen Palacios, Olivier Vernau, the Diergaarde Blijdorp - Rotterdam Zoo, Zurich Zoo, A Cupulatta, and Montpellier Lunaret Zoo for their support and help with the sampling. This work was supported by European Research Council grant 232971 ‘PopPhyl’ and Fundação para a Ciência e Tecnologia (Portugal), postdoctoral fellowships SFRH/BDP/73515/2010.
References
- Grant PR. Evolution on Islands. Oxford: Oxford University Press; 1998. [Google Scholar]
- Van Valen L. A new evolutionary law. Evol Theor. 1973;14:1–33. [Google Scholar]
- Bazin E, Glémin S, Galtier N. Population size does not influence mitochondrial genetic diversity in animals. Science. 2006;14:570–571. doi: 10.1126/science.1122033. [DOI] [PubMed] [Google Scholar]
- Frankham R. How closely does genetic diversity in finite populations conform to predictions of neutral theory? Large deficits in regions of low recombination. Heredity. 2012;14:167–178. doi: 10.1038/hdy.2011.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker MR, Bentzen P. Positive relationships between genetic diversity and abundance in fishes. Mol Ecol. 2010;14:4852–4862. doi: 10.1111/j.1365-294X.2010.04822.x. [DOI] [PubMed] [Google Scholar]
- Perry GH, Melsted P, Marioni JC, Wang Y, Bainer R, Pickrell JK, Michelini K, Zehr S, Yoder AD, Stephens M, Pritchard JK, Gilad Y. Comparative RNA sequencing reveals substantial genetic variation in endangered primates. Genome Res. 2012;14:602–610. doi: 10.1101/gr.130468.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabholz B, Glémin S, Galtier N. The erratic mitochondrial clock: variations of mutation rate, not population size, affect mtDNA diversity across mammals and birds. BMC Evol Biol. 2009;14:54. doi: 10.1186/1471-2148-9-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press; 1983. [Google Scholar]
- Lynch M. The Origins of Genome Architecture. Sunderland: Sinauer Associates, Inc.; 2007. [Google Scholar]
- Spielman D, Brook BW, Frankham R. Most species are not driven to extinction before genetic factors impact them. Proc Natl Acad Sci USA. 2004;14:15261–15264. doi: 10.1073/pnas.0403809101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev SI, Montoya-Burgos JI, Popadin K, Parand L, Margulies EH, Antonarakis SE. National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program. Life-history traits drive the evolutionary rates of mammalian coding and noncoding genomic elements. Proc Natl Acad Sci USA. 2007;14:20443–20448. doi: 10.1073/pnas.0705658104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Lercher MJ, Eyre-Walker A. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol. 2005;14:e42. doi: 10.1371/journal.pbio.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K, Seger J. Elevated rates of nonsynonymous substitution in island birds. Mol Biol Evol. 2001;14:874–881. doi: 10.1093/oxfordjournals.molbev.a003869. [DOI] [PubMed] [Google Scholar]
- Woolfit M, Bromham L. Population size and molecular evolution on islands. Proc Biol Sci. 2005;14:2277–2282. doi: 10.1098/rspb.2005.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SD, Gillman LN, Ross HA, Keeling DJ. Slower tempo of microevolution in island birds: implications for conservation biology. Evolution. 2009;14:2275–2287. doi: 10.1111/j.1558-5646.2009.00717.x. [DOI] [PubMed] [Google Scholar]
- Bierne N, Eyre-Walker A. The genomic rate of adaptive amino acid substitution in Drosophila. Mol Biol Evol. 2004;14:1350–1360. doi: 10.1093/molbev/msh134. [DOI] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;14:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccone A, Gibbs JP, Ketmaier V, Suatoni E, Powell JR. Origin and evolutionary relationships of giant Galápagos tortoises. Proc Natl Acad Sci USA. 1999;14:13223–13228. doi: 10.1073/pnas.96.23.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulakakis N, Russello M, Geist D, Caccone A. Unravelling the peculiarities of island life: vicariance, dispersal and the diversification of the extinct and extant giant Galápagos tortoises. Mol Ecol. 2012;14:160–173. doi: 10.1111/j.1365-294X.2011.05370.x. [DOI] [PubMed] [Google Scholar]
- Restrepo A, Colinvaux P, Bush M, Correa-Metrio A, Conroy J, Gardener MR, Jaramillo P, Steinitz-Kannan M, Overpeck J. Impacts of climate variability and human colonization on the vegetation of the Galápagos Islands. Ecology. 2012;14:1853–1866. doi: 10.1890/11-1545.1. [DOI] [PubMed] [Google Scholar]
- Russello MA, Hyseni C, Gibbs JP, Cruz S, Marquez C, Tapia W, Velensky P, Powell JR, Caccone A. Lineage identification of Galápagos tortoises in captivity worldwide. Anim Cons. 2007;14:304–311. doi: 10.1111/j.1469-1795.2007.00113.x. [DOI] [Google Scholar]
- Chiari Y, Cahais V, Galtier N, Delsuc F. Phylogenomic analyses support the position of turtles as the sister group of birds and crocodiles (Archosauria) BMC Biol. 2012;14:65. doi: 10.1186/1741-7007-10-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayral P, Melo-Ferreira J, Glémin S, Bierne N, Carneiro M, Nabholz B, Lourenço JM, Alves PC, Ballenghien M, Faivre N, Belkhir K, Cahais V, Loire E, Bernard A, Galtier N. Reference-free population genomics from Next-Generation transcriptome data and the vertebrate-invertebrate gap. PLoS Genet. 2013;14:e10003457. doi: 10.1371/journal.pgen.1003457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat Genet. 2000;14:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barley AJ, Spinks PQ, Thomson RC, Schaeffer HB. Fourteen nuclear genes provide phylogenetic resolution for difficult nodes in the turtle tree of life. Mol Phylogenet Evol. 2010;14:1189–1194. doi: 10.1016/j.ympev.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Lourenço JM, Claude J, Galtier N, Chiari Y. Dating cryptodiran nodes: origin and diversification of the turtle superfamily Testudinoidea. Mol Phylogenet Evol. 2012;14:496–507. doi: 10.1016/j.ympev.2011.10.022. [DOI] [PubMed] [Google Scholar]
- Lourenço JM, Glémin S, Chiari Y, Galtier N. The determinants of the molecular substitution process in turtles. J Evol Biol. 2013;14:38–50. doi: 10.1111/jeb.12031. [DOI] [PubMed] [Google Scholar]
- Le M, McCord WP. Phylogenetic relationships and biogeographical history of the genus Rhinoclemmys Fitzinger, 1835 and the monophyly of the turtle family Geoemydidae (Testudines: Testudinoidea) Zool J Linn Soc. 2008;14:751–767. doi: 10.1111/j.1096-3642.2008.00413.x. [DOI] [Google Scholar]
- Eyre-Walker A, Keightley PD. Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol Biol Evol. 2009;14:2097–2108. doi: 10.1093/molbev/msp119. [DOI] [PubMed] [Google Scholar]
- Künstner A, Nabholz B, Ellegren H. Evolutionary constraint in flanking regions of avian genes. Mol Biol Evol. 2011;14:2481–2489. doi: 10.1093/molbev/msr066. [DOI] [PubMed] [Google Scholar]
- Russello MA, Poulakakis N, Gibbs JP, Tapia W, Benavides E, Powell JR, Caccone A. DNA from the past informs ex situ conservation for the future: an “extinct” species of Galápagos tortoise identified in captivity. PLoS One. 2010;14:e8683. doi: 10.1371/journal.pone.0008683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci USA. 2010;14:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofi C, Milinkovitch MC, Gibbs JP, Caccone A, Powell JR. Microsatellite analysis of genetic divergence among populations of giant Galápagos tortoises. Mol Ecol. 2002;14:2265–2283. doi: 10.1046/j.1365-294x.2002.01617.x. [DOI] [PubMed] [Google Scholar]
- Turtle Taxonomy Working Group. Turtles of the world, 2012 update: annotated checklist of taxonomy, synonymy, distribution, and conservation status. In: Rhodin AGJ, Pritchard PCH, van Dijk PP, Saumure RA, Buhlmann KA, Iverson JB, Mittermeier RA, editor. Conservation Biology of Freshwater Turtles and Tortoises: A Compilation Project of the IUCN/SSC Tortoise and Freshwater Turtle Specialist Group. Chelonian Research Monographs No. 5. Lunenberg, MA: Chelonian Research Foundation; 2012. pp. 000.243–000.328. doi:10.3854/crm.5.000.checklist.v5.2012. [Google Scholar]
- Milton SL, Prentice HM. Beyond anoxia: The physiology of metabolic downregulation and recovery in the anoxia-tolerant turtle. Comp Biochem Phys A. 2007;14:277–290. doi: 10.1016/j.cbpa.2006.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramyan J, Badenhorst D, Biggar KK, Borchert GM, Botka CW, Bowden RM, Braun EL, Bronikowski AM, Bruneau BG, Buck LT, Capel B, Castoe TA, Czerwinski M, Delehaunty KD, Edwards SV, Fronick CC, Fujita MK, Fulton L, Graves TA, Green RE, Haerty W, Hariharan R, Hillier LH, Holloway AK, Janes D, Janzen FJ, Kandoth C, Kong L, de Koning J, Li Y. et al. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 2013;14:R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson J. Are there differences in immune function between continental and insular birds? Proc Biol Sci. 2006;14:2267–2274. doi: 10.1098/rspb.2006.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant PR, Grant BR, Keller LF, Petren K. Effects of El Nino events on Darwin’s finch productivity. Ecology. 2000;14:2442–2457. [Google Scholar]
- Hulbert AJ, Pamplona R, Buffenstein R, Buttemer WM. Life and death: metabolic rate, membrane composition and life span of animals. Physiol Rev. 2007;14:1175–1213. doi: 10.1152/physrev.00047.2006. [DOI] [PubMed] [Google Scholar]
- Cahais V, Gayral P, Tsagkogeorga G, Melo-Ferreira J, Ballenghien M, Weinert L, Chiari Y, Belkhir K, Ranwez V, Galtier N. Reference-free transcriptome assembly in non-model animals from next generation sequencing data. Mol Ecol Resources. 2012;14:834–845. doi: 10.1111/j.1755-0998.2012.03148.x. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;14:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagkogeorga G, Cahais V, Galtier N. The population genomics of a fast evolver: high levels of diversity, functional constraint and molecular adaptation in the tunicate Ciona intestinalis. Genome Biol Evol. 2012;14:740–749. doi: 10.1093/gbe/evs054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mitochondrial DNA genealogy in C. nigra. The tree was built based on a 705-long fragment of the control region from 89C. nigra individuals. Sequence accession numbers and island of origin of each turtle are provided. The three major clades identified by Caccone et al. [18] are indicated. The five individuals analyzed in this study appear in red.
Details about sampling, sequencing, assembly, SNP calling, gene expression, gene ontology, and phylogenomic analyses.
Contrasting GO-slim term-specific selective pressure between C. nigra and E. orbicularis. list of GO-slim ‘Biological process’ terms, with the number of associated contigs, their average πN/πS ratio and its normalized version (z-score) in C. nigra vs. E. orbicularis.
C. nigra and E. orbicularis coding sequences associated to GO-slim terms ‘immune process’ and ‘response to stress’. list of predicted contigs associated to the ‘immune process’ and ‘response to stress’ terms in either C. nigra or E. orbicularis, their functional annotation, length, coverage, πN, and πS.