

Abstract
By its very nature, genomics produces large, high-dimensional datasets that are well suited to analysis by machine learning approaches. Here, we explain some key aspects of machine learning that make it useful for genome annotation, with illustrative examples from ENCODE.
Introduction
The complete sequencing of the human genome marked an important milestone in modern biology [1,2], but it also produced a whole new set of challenges in elucidating the functions and interactions of different parts of the genome. A natural first step to tackling these formidable tasks is to construct an annotation of the genome, which is to (1) identify all functional elements in the genome, (2) group them into element classes such as coding genes, non-coding genes and regulatory modules, and (3) characterize the classes by some concrete features such as sequence patterns. Over the years, many experimental and computational methods have been invented to accelerate this annotation process. Among the popular computational methods are those based on the concept of machine learning (Box 1). Originally a branch of artificial intelligence, machine learning has been fruitfully applied to a variety of domains. The basic idea of machine learning is to construct a mathematical model for a particular concept (an element class in the case of genome annotation) based on its features in some observed data. The model can then be applied to identify new instances of the concept in other data [3-5].
In this review, we discuss some key properties of machine learning that make it useful for genome annotation, using some classic problems for illustration. We also describe some examples in the latest work of the ENCODE Project Consortium [6] to highlight some recent trends. We focus on the identification and classification of genomic elements, and do not go into the details of machine learning approaches to functional annotation, such as the predictions of gene expression, gene functions and protein interactions. In addition, due to limited space, we can only include a small portion of the related references in the literature. Readers interested in the application of machine learning in some major classes of genomic elements are referred to the corresponding reviews listed in Table 1. This review is intended to serve as an introduction to machine learning and its use in genome annotation for a general audience, requiring no prior knowledge in these topics. A more general description of the use of machine learning in bioinformatics can be found in Baldi and Brunak [4]. More formal discussions on machine learning can be found in various text books [5,7,8]. An overview of experimental and computational genome annotation approaches can be found in some other reviews [9,10].
Table 1.
Reviews on machine learning methods for identifying some major classes of genomic elements
Key properties of machine learning and their relevance to genome annotation
From expert knowledge to data-driven patterns
One major reason for the popularity of machine learning methods is its ability to automatically identify patterns in data. This is particularly important when the expert knowledge is incomplete or inaccurate, when the amount of available data is too large to be handled manually, or when there are exceptions to the general cases. We use protein binding motifs as an example for this part of the discussion.
Many DNA binding proteins, including transcription factors (TFs), recognize their target DNA regions by some short sequence motifs [37]. The motifs are usually not exact, in that a TF can bind DNA sequences with some differences, albeit with different affinity. When the number of experimentally known binding sites of each TF was limited, it was common for human experts to abstract the binding motifs by some prominent features common to the observed binding sites, such as the most conserved locations within the motifs. The resulting expert knowledge was summarized by simple representations such as consensus sequences.
As high-throughput methods for probing TF binding sites, such as protein binding microarrays [38] and chromatin immunoprecipitation followed by microarrays (ChIP-chip) [39,40] or high-throughput sequencing (ChIP-seq) [27,28] have become popular, it has become easier to collect a large number of sequences that contain binding sites of a TF. Machine learning methods can automatically identify patterns common in these sequences but rare in the genomic background [22]. Due to the large amount of examples available, the resulting models can have richer probabilistic representations with more parameters than what a human expert can easily handle, such as position weight matrices [41] and profile hidden Markov models [42].
In many cases, the exact binding locations of the TF in the input sequences are unknown. One needs to try different combinations of binding locations on these sequences and compare the resulting models. This computationally expensive task can be handled by standard methods such as Gibbs sampling [43] and expectation maximization [44]. There could also be errors in the input data such as false positives, in the form of sequences that do not really contain a binding site of the TF. A human expert could be misled by the false positives and try to form over-fitted models (Box 1) that fit these error cases. A machine learning method with well-controlled complexity, on the other hand, may prefer a model that does not classify the error cases as positives. More generally, each input sequence may contain zero, one, or more occurrences of a motif [45], the input sequences may contain multiple motifs (for example, due to indirect binding [46]), and motif finding can be confounded by repeat sequences. All these complications are more easily handled by automatic methods.
From single data type to integration of heterogeneous data
Machine learning methods are also good at integrating multiple, heterogeneous features. This property allows the methods to detect subtle interactions and redundancies among features, as well as to average out random noise and errors in individual features. We use the identification of cis-regulatory modules (CRMs) as an example to illustrate this property.
A CRM is a DNA regulatory region, usually containing the binding sites of multiple TFs, that regulate a common gene nearby [47], such as cis-acting promoters, enhancers, silencers and insulators. Many types of features have been individually used by previous methods to identify CRMs, including the density and statistical over-representation of TF binding motifs, evolutionary conservation, direct binding signals from ChIP-seq or ChIP-chip data, and biochemical marks such as histone modifications [26]. In general, information related to binding patterns is useful for distinguishing between CRMs and genomic regions with fewer binding sites such as exons; information related to evolutionary constraints is more useful in distinguishing between CRMs and other less conserved regions, such as introns and some intergenic regions; information about histone modifications is useful in distinguishing between different types of regulatory regions and between the active and inactive ones. It was found that no single type of features could perfectly separate CRMs from negative examples [26]. As a result, some recent approaches have started to integrate different types of features by using a machine learning framework [48]. Depending on the mathematical form of the model (Box 1), the different features can be integrated in ways from linear combinations to highly nonlinear ones.
Three aspects of data integration by machine learning deserve more discussions. First, the input features could contain redundant information. For example, ChIP-seq signals from TF binding and histone modification experiments can be highly correlated with open chromatin signals [49]. Different machine learning methods handle redundant features in drastically different ways. At one extreme, methods such as the Naïve Bayes classifier [50] assume input features to be independent of each other for each class. If the features are in fact not conditionally independent, the redundant features could be unfavorably granted stronger influence on the predictions than the non-redundant ones, which affect the accuracy of the resulting models for some problems. On the other hand, methods such as decision trees and logistic regression could have one feature masking out the effects of all other similar features. In general it is good to carefully select a set of non-redundant input features based on biological knowledge, perform dimension reduction to remove dependency between features (by methods such as principal components analysis [51]) before the learning process, or test the stability of predictions using different subsets of input features.
Second, if a large number of features are integrated but the amount of training examples is limited (a phenomenon quite common in genome annotation), multiple issues could come up. The training examples may not be sufficient to capture the combination of feature values characteristic of the classes to be modeled. If some features irrelevant to the target concepts are included, they could mislead the modeling process, especially in unsupervised settings. There is also a high risk of over-fitting. Feature selection, dimension reduction, regularization and semi-supervised learning (Box 1) are all practical ways to alleviate the problem.
Third, it could be difficult to combine features of different data types. For example, conservation of a potential CRM region is represented by a numeric score (such as PhastCons [52] and phyloP [53]), the raw sequence of it is represented by a text string, while peaks of binding signals of a particular TF could be represented by a binary variable. One systematic approach to handling mixed data types is to turn each type of data into a numerical similarity matrix between the input regions before integrating them. Kernel methods [54] are one particular branch of machine learning methods that work on similarity (kernel) matrices with some simple mathematical requirements. They have been widely used in integrating different types of data for genome annotation. For example, the kernel between two sequences can be defined by their alignment, BLAST scores or k-mer composition [54,55].
From simple rules to complex functions
Another strength of machine learning is its ability to construct highly complex models needed by some genomic element classes. We use gene finding as an example here.
Eukaryotic genes have a complex structure with exons, introns and splice sites at the transcriptional level, and coding sequences and untranslated regions at the translational level. An early approach to computational gene finding involves homology search using tools such as BLASTX [56] to look for regions of a genome with similar sequences in a database of annotated genes or expressed sequence tags. This approach is similar to the standard machine learning method of predicting the label of an object as the one of its nearest neighbor among the labeled examples, but with a maximum dissimilarity cutoff between them. It suffers from not being able to identify genes with no annotated homologs, and not reporting the detailed sub-elements (exons, introns, and so on) of the genes.
Both issues suggest the need for ab initio methods for finding genes directly from sequences. Some of these methods derived sequence-based features of known genes called content statistics (such as codon usage), and defined rules for classifying genes based on these features [11]. It was found that when the features were combined using non-linear artificial neural network classifiers, the prediction performance was much better than some simple combinations of the features [57], which highlights the need for complex models.
In order to model the detailed structures of eukaryotic genes instead of simply predicting if a region contains a gene or not, machine learning methods based on hidden Markov models [58-60] and generalized hidden Markov models [61-63] have become some of the most popular choices for computational gene finding. These methods consider the observed genomic sequence as the output of some hidden states (the sub-element types or their sub-classes). A complete model is composed of the set of states, and the probabilities of starting a sequence at each state, transition between states and outputting a base/sequence at each state as model parameters. Standard algorithms exist for learning the parameter values of such complex models.
With the advent of RNA-seq [64,65] and other high-throughput experimental methods for identifying RNA transcripts, ab initio gene finding has become less popular. In the current post-transcriptomic era, machine learning has taken on some new roles in gene finding. First, specialized methods that take into account a large number of features and their complex interactions have been designed to model some biological mechanisms not yet fully understood, such as recognizing transcription start sites and determining the splicing events [66-68]. A related problem is to predict complete isoforms and their relative abundance of a gene in a certain sample, using single-end or paired-end short sequencing reads [69]. Second, methods developed for identifying protein-coding genes are now adopted to identifying long non-coding RNAs (lncRNAs) [67], which share some common features with protein-coding genes (such as the presence of introns) but the annotations of which are much less complete and thus there are limited training examples available.
Case study: multi-class whole-genome annotation
An ultimate goal of genome annotation is to identify all types of functional elements and all their occurrences in a genome. How far are we from this goal? Currently there are still likely undiscovered genomic element classes given the rapid discovery of new classes (such as many non-coding RNAs (ncRNAs)) in recent years. Some element classes also have so far very few discovered instances. In terms of machine learning, these two facts imply that currently it is impossible to perform purely supervised learning for all element classes. As a result, in a recent work by the Encyclopedia of DNA Elements (ENCODE) Project Consortium, which aims at delineating all functional elements encoded in the human genome [70], several different approaches have been adopted to confront this grand challenge.
ENCODE has recently produced about 1,600 sets of whole-genome experimental data that cover many types of molecular states and activities, including transcription, long-range DNA interactions, and chromatin features such as histone modifications, protein-DNA binding and open chromatin signals [6]. In one approach to whole-genome annotation, the experimental data were used to perform unsupervised segmentation of the human genome [6,71,72], so that each genomic location was assigned to a segment. The segments were then grouped into clusters in an unsupervised manner. Each resulting cluster was described by the characteristic features of its members. Surprisingly, although the clusters were discovered by an unsupervised procedure, many of them have simple interpretations corresponding to known genomic element classes such as promoters, transcribed regions and enhancers. The segmentation was also able to provide sub-classes of particular element classes, such as enhancers with strong and weak activities in particular cell types, respectively. In general, this method can reveal groups of sequence elements according to the observed data alone without defining the target element classes a priori.
One difficulty in performing this unsupervised clustering was to determine the number of clusters to produce. Having too few clusters would merge elements from different genomic element classes together, while having too many clusters would make the results difficult to interpret. In order to avoid manually defining the number of clusters, in another approach the segments were put onto a two-dimensional toroidal map, where similar segments were put close to each other using the unsupervised Self-Organizing Map method [73]. The resulting map provides a way to study the relationships between different segments and the meanings of each local region on the map without defining the number of clusters and the cluster boundaries [6]. It also provides information about the similarity between different clusters identified by the segmentation method.
The whole-genome segmentation approach has the advantage of requiring no a priori definition of element classes, so that the discovery process is directly based on the observed data. On the other hand, when there is a specific type of genomic elements of interest, customized methods could potentially include more information specific to it. As an example, one important effort of ENCODE was to experimentally validate computationally predicted enhancers using different types of reporter assays [6]. A number of methods had previously been proposed for identifying enhancers in a genome, including both supervised [74,75] and unsupervised [76,77] methods. These methods were constrained by a lack of whole-genome experimental data, and had thus relied on either a relatively small set of experimental features or static information such as genomic sequence and evolutionary conservation. Correspondingly, a specialized pipeline was designed by ENCODE to identify enhancers at the genome scale, utilizing the large amount of experimental data produced [6,78]. Both the predictions from the segmentation approach and the enhancer prediction pipeline were found to achieve reasonable levels of accuracy [6].
Based on the ENCODE experience, one could imagine a potential hybrid approach that combines the benefits of both the unsupervised and supervised approaches described above. First, the segmentation approach is applied to systematically discover genomic element classes from large datasets. Specialized supervised methods can then be designed to provide detailed modeling of each element class using extra domain knowledge and auxiliary data available.
Current challenges and future outlooks
We conclude by discussing some current challenges in applying machine learning to genome annotation and the corresponding outstanding key research problems.
Interpretability of models
As mentioned above, very complex models have been proposed for some difficult genome annotation tasks. For example, a machine learning method involving hundreds of features has been reported to achieve high accuracy in predicting tissue-specific alternative splicing [66]. There are also machine learning methods that make use of the concept of ensemble learning, which combines the predictions of multiple (possibly very complex) models to achieve better performance. Examples include the classical bagging [79] and boosting [80] methods and Random Forests [33], which build multiple models using different subsets of examples or features. For instance, Random Forests were reported to outperform some other machine methods in identifying ncRNAs [81]. In fact, ensemble methods have become a popular choice in public machine learning challenges that involve big datasets, such as the well-known Netflix Prize [82]. They outperformed methods that produced simpler models, which were unable to provide the required 10% accuracy improvement in recommending films when compared with the original method used by Netflix.
These complex models, achieving high prediction accuracy notwithstanding, are in general difficult to interpret. Whether one should use them in genome annotation depends on the exact goal of the project. If the goal is to produce a list of genomic elements as accurately as possible, it would be fine to use complex models as 'black boxes' as long as they can provide the required accuracy. On the other hand, if the goal is to use machine learning as a means to understand the underlying biological mechanisms, one may want to construct models that are more easily interpretable. For example, if one hopes to understand the major features that can help identify 80% of the elements of a certain class, a simple model may suffice, sacrificing the prediction accuracy of the remaining 20% as a tradeoff. It is rarely possible to achieve high accuracy and good interpretability at the same time; thus, it is important to define the goal clearly and select the machine learning method accordingly.
Context specificity and transferability of models
Large-scale genomic projects, such as ENCODE [6], modENCODE [83,84], 1000 Genomes [85] and Roadmap Epigenomics [86], have produced a huge amount of valuable data that cover many aspects of genomes. These datasets offer an unprecedented opportunity to model genomic element classes and the effects of genetic mutations on them. However, a lot of these data are associated with properties specific to the corresponding experiments, such as cell or tissue types, experimental conditions, developmental stages of the animals and the population backgrounds of the sequenced individuals. Care should be taken when using these data to model the active genomic elements in other types of data or to construct general, non-context-dependent models.
It would be useful for machine learning methods to provide multiple levels of abstractions for the static and context-specific information. For example, when direct binding data of a certain TF × from ChIP-seq experiments are available for one cell type, a model can be constructed to describe the relationships between the ChIP-seq signals and the actual binding sites of TF × in this cell type. However, if in a second cell type ChIP-seq experiments have only been performed for some other TFs but not TF X, the model from the first cell type cannot be directly applied to predict the binding sites of TF × in this second cell type as the feature required by the model is not available. In this situation, the ChIP-seq data for the TFs available in the second cell type could be used to construct a higher-level model that describes some features common to the binding sites of different TFs, such as DNA accessibility. Combining it with non-context-specific static information such as sequence motifs of TF X, it is still possible to construct an accurate model for predicting the binding sites of TF × without ChIP-seq data in the second cell type [87].
A key to providing different levels of abstraction from the same input data is a careful selection of negative examples. In the above example, when constructing the general model for identifying binding sites of any TF, the negative set should contain regions not bound by any TF, including those with no direct ChIP-seq signals and those likely to be depleted of TF binding, such as coding exons. In contrast, when constructing the model for identifying binding sites of a particular target TF based on ChIP-seq data alone, the negative examples should also include binding sites of other TFs in addition to non-TF-binding regions, so that the learned model is specific to the target TF.
Lack of training examples and unbalanced positive and negative sets
For some classes of genomic elements, there are insufficient known examples for supervised machine learning methods to capture the general patterns of the classes. For example, there are few validated enhancers cataloged in databases relative to the expected total number [88]. Many prediction methods have thus relied on a combination of unsupervised learning and manually defined rules [6,76-78]. In the case of ncRNAs, a large portion of the most functionally characterized ones are the short, strongly structured RNAs, which could bias models for identifying ncRNAs towards this subset and render them less able to detect ncRNAs with few known examples and novel ncRNA classes. Moreover, confirmed negative examples are seldom available, but are crucial to most machine learning methods. A related issue is that most genomic element classes occupy only a small portion of the genome, and therefore the ratio of positive to negative regions is very small. Even a highly accurate classifier could have a lot of false positives among its top predictions.
We propose that these issues should be tackled from multiple fronts. First, as explained in Box 1, the concept of semi-supervised learning [36] is potentially capable of combining information about the distributions of known examples and unlabeled data points (Figure 1c). Its application to genomic annotation deserves more investigations.
Figure 1.
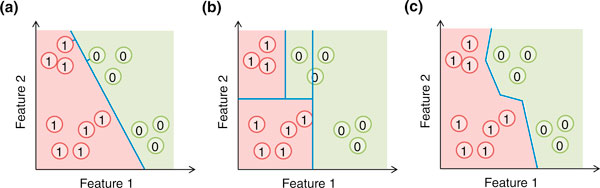
Some commonly used machine learning methods. For illustration, each genomic region is represented by a circle and described by two features. (a) A support vector machine (SVM) forms an affine decision surface (a straight line in the case of two dimensions) in the original feature space or a vector space defined by the similarity matrix (the kernel), to separate the positive and negative examples and maximize the distance of it from the closest training examples (the support vectors, those with a perpendicular line from the decision surface drawn). It predicts the label of a genomic region based on its direction from the decision surface. In the case a kernel is used, the decision surface in the original feature space could be highly non-linear. (b) A basic decision tree uses feature-parallel decision surfaces to repeatedly partition the feature space, and predicts the label of a genomic region based on the partition it falls within. (c) The one-nearest neighbor (1-NN) method predicts the label of a genomic region based on the label of its closest labeled example. In all three cases, the areas predicted to be positive and negative are indicated by the red and green background colors, respectively.
Second, systematic methods for selecting negative examples for genomic annotation should be developed, taking into account the accuracy of the examples and their influence on the models. For instance, extreme cases that are 'very negative' are likely accurate but not too informative. Relevant discussions for the problem of predicting protein physical interactions provide some good references on this topic [89-91]. There is a relatively small set of verified protein physical interactions, a large number of putative interactions from high-throughput experiments such as yeast-two-hybrid and co-immunoprecipitation, and no protein pairs that are confirmed to never interact. The way to choose negative examples could have profound effects on the resulting models.
When confirmed negative examples are scarce or unavailable, certain features indicative of the class label could be intentionally left out from the model training process and used to evaluate the performance of the model learned from the other features. For example, in a recent study for identifying lncRNAs, information useful for predicting protein-coding genes, including sequence conservation, homology to known genes, codon usage and coding potential, was not used in the lncRNA detection pipeline [92]. An a posteriori check of the coding potential of the predicted lncRNAs could serve as an indirect evidence of the prediction accuracy.
Third, when constructing a model for a particular genomic element class, it is generally good to test for the existence of sub-classes, by means of either a model that allows for multiple clusters per class, pre-clustering of training examples and construction of separate models for different clusters, or post-clustering of predictions.
Finally, if experimental validations are performed to confirm the computational predictions, an active learning [93] strategy can be adopted to select predictions that maximize the expected information gain or similar measures [94]. Ideally, the computational prediction and experimental validation phases should be repeated for multiple iterations, in order to facilitate the selection of the most informative examples for validation.
Abbreviations
ChIP-chip: chromatin immunoprecipitation followed by microarrays; ChIP-seq: chromatin immunoprecipitation followed by high-throughput sequencing; CRM: cis-regulatory module; lncRNA: long non-coding RNA; miRNA: microRNA; ncRNA: non-coding RNA; RNA-seq: high-throughput cDNA sequencing; SOM: Self-Organizing Map; TF: transcription factor.
Competing interests
The authors declare that they have no competing interests.
Box 1: A primer on machine learning
We first consider a basic setting of machine learning for binary classification, and later describe variations of it commonly encountered in genome annotation. Suppose we want to identify enhancers in a genome. We divide up the genome into a list of genomic regions X = (x1, x2, ..., xN). Each region xi has a corresponding binary label yi, where yi = 1 if xi is an enhancer, and yi = 0 if not. Each region is described by a set of features xi = (xi1, xi2, ..., xim). For example, xi1 could be the evolutionary conservation of xi among several close species, xi2 could be the average ChIP-seq [27,28] signal of the active enhancer mark H3K27ac (histone 3 lysine 27 acetylation) among the bases within the region from a certain experiment, and so on. The goal of machine learning is to find a function f (called a model) such that f(xi) is close to yi, that is, to tell if a region is an enhancer from some observed features alone.
To find a suitable f, we need to (1) decide on a mathematical form of f; (2) find known positive and negative examples that can help estimate the parameters of f; and (3) actually estimate the parameters of f, in a way that it likely predicts the labels of regions accurately, even for regions for which the corresponding labels are unknown.
For task 1, many families of f and their corresponding algorithms for learning the parameters have been studied. The popular ones include artificial neural networks [29], Bayesian networks [30], decision trees [31], k-nearest neighbors [32], random forests [33] and support vector machines [34]. They differ in the form and complexity of their models. Some examples are shown in Figure 1. Predictions are made based on the mathematical form of f and the parameters learned from the examples, such as the location of orientation of the decision surface of a SVM (Figure 1a).
Task 2 could be quite tricky for some element classes (see the corresponding discussions in the main text). Task 3 can be further sub-divided into two sub-tasks, that of finding a model to fit the training examples, and of ensuring the model to be able to predict the labels of unseen regions correctly. The first sub-task can be achieved by finding parameter values of f that minimize a loss function, such as the sum of squared errors of the n examples, . Since the parameter values are determined according to the observed data, the process is described as 'learning' a model from the data. The second sub-task is achievable only if one makes certain assumptions about the models and examples. It is usually assumed that the observed examples and unobserved instances of each type of functional elements share the same distribution of feature values, and that when two models can fit the observed examples equally well, the simpler one (for example, one with a smaller number of parameters or a smaller total magnitude of the parameter values) is likely to generalize better to unobserved instances. A model too specific to the observed data, usually characterized by a high complexity of the model, may over-fit the data; that is, may capture patterns that are only true for the observed examples. To avoid over-fitting, some machine learning methods control the complexity of the models by model pruning [35] or regularization [3], with the observed examples fitting less well to the model as a tradeoff. Some other methods produce multiple models on different subsets of data to identify reliable patterns that appear frequently in these models (see main text for more discussions). Procedure-wise, over-fitting is detected by building a model based on a subset of the examples (the training set), and evaluating its accuracy based on another subset not involved in training (the testing set). An over-fitted model would have good training accuracy but poor testing accuracy. The process is usually repeated with different portions of data treated as the training set in turn to compute the average accuracy in a cross-validation procedure.
Setting variation 1: Binary classification, multi-class classification and regression
When we have a pre-defined set of discrete values for the labels, we have a classification problem with each value corresponding to a class and f is called a classifier. The simplest case of which, when there are only two classes, is called a binary classification problem. A more complex example of classification is to distinguish enhancers (yi = 1) from promoters (yi = 2) and other regions (yi = 0). There are also situations in which the labels can take on continuous values. The corresponding machine learning problem is called a regression problem and f is called an estimator or a regressor. In this review we focus on classification problems as the goal of genome annotation is to identify DNA sequences belonging to each element class. However, it should be noted that, in practice, many classifiers output a continuous value fj(xi) that indicates how much a region xi appears to belong to the class j. For instance, probabilistic methods formally define fj(xi) as the data likelihood Pr(xi|yi = j) or posterior probability Pr(yi = j|xi). Classification can be performed by assigning each region xi to the class j with the largest value of fj(xi) among all classes.
Setting variation 2: Supervised, unsupervised and semi-supervised learning
In the basic setting, the model is constructed from observed examples with known labels, which is called the supervised learning setting (Figure 2a). Sometimes we do not predefine a set of classes, but want to identify natural clusters of genomic regions according to their distribution of feature values alone. This is called the unsupervised learning problem (Figure 2b). For example, in addition to enhancers and promoters, there are also other types of regulatory elements such as silencers and insulators. One may not want to predefine the set of regulatory element classes but rather to discover them from the observed data, assuming that the instances of each class share similar feature values. There are also situations in which we want to determine the model from both data with and without labels. This semi-supervised learning setting [36] could be very useful when training examples are limited or are available only for some classes. For example, if there are few experimentally validated enhancers and high-confidence negative examples, one may want to first use the available examples to roughly define the area in the feature space that belongs to each class, and then use the distribution of feature values of unlabeled genomic regions to estimate the boundaries of the areas (Figure 2c).
Figure 2.
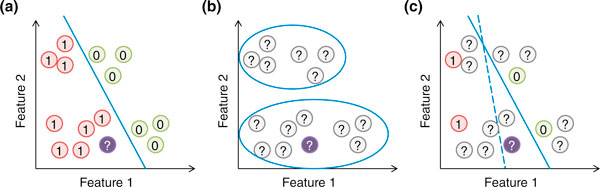
Supervised, unsupervised and semi-supervised learning. (a) In supervised learning, the model (blue line) is learned based on the positive and negative training examples, and the genomic region without a known class label (purple circle) is classified as positive according to the model. (b) In unsupervised learning, all examples are unlabeled, and they are grouped according to the data distribution. (c) In semi-supervised learning, information of both labeled and unlabeled examples is used to learn the parameters of the model. In this illustration, a purely supervised model (dashed blue line) classifies the purple object as negative, while a semi-supervised model that avoids cutting at regions with a high density of genomic regions (solid blue line) classifies it as positive.
Setting variation 3: Instances with independent versus dependent labels
We have been implicitly assuming that the label of each genomic region can be determined by its own set of features alone. In genome annotation, this is often unrealistic for two reasons. First, it is usually hard to define the exact span of a region. Biologically it could be fuzzy to define exactly where a functional element starts and ends (as in the case of an enhancer), and even if the span could be formally defined (as in the case of an RNA transcript), it is usually not known prior to machine learning. One may therefore consider each base separately and predict whether it overlaps a functional element or not. Second, the class labels for neighboring genomic regions/bases are not independent. For example, if a base is within an intron, the next base should be either within an intron or a splice site. In this situation, the label of a base should be predicted according its own features as well as other bases. There are standard methods for this kind of learning tasks, such as hidden Markov models.
Contributor Information
Kevin Y Yip, Email: kylyip@gmail.com.
Chao Cheng, Email: Chao.Cheng@Dartmouth.edu.
Mark Gerstein, Email: mark.gerstein@yale.edu.
Acknowledgements
KYY is partially supported by the Hong Kong Research Grants Council General Research Fund CUHK418511.
References
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C. et al. Initial sequencing and analysis of the human genome. Nature. 2001;14:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N. et al. The sequence of the human genome. Science. 2001;14:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Alpaydin E. Introduction to Machine Learning. Cambridge, Massachusettes: The MIT Press; 2004. [Google Scholar]
- Baldi P, Brunak S. Bioinformatics: The Machine Learning Approach. 2. Cambridge, Massachusettes: MIT Press; 2001. [Google Scholar]
- Mitchell T. Machine Learning. New York: McGraw Hill; 1997. [Google Scholar]
- ENCODE Project Consortium. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, Khatun J, Lajoie BR, Landt SG, Lee BK, Pauli F, Rosenbloom KR, Sabo P, Safi A, Sanyal A, Shoresh N, Simon JM, Song L, Trinklein ND, Altshuler RC, Birney E, Brown JB, Cheng C, Djebali S, Dong X. et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;14:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Mostafa YS, Magdon-Ismail M, Lin H-T. Learning from Data. AMLBook; 2012. [Google Scholar]
- Murphy KP. Machine Learning: A Probabilistic Perspective. The MIT Press; 2012. [Google Scholar]
- Brent MR. Steady progress and recent breakthroughs in the accuracy of automated genome annotation. Nat Rev Genet. 2008;14:62–73. doi: 10.1038/nrg2220. [DOI] [PubMed] [Google Scholar]
- Yandell M, Ence D. A beginner's guide to eukaryotic genome annotation. Nat Rev Genet. 2012;14:329–342. doi: 10.1038/nrg3174. [DOI] [PubMed] [Google Scholar]
- Stormo GD. Gene-finding approaches for eukaryotes. Genome Res. 2000;14:394–397. doi: 10.1101/gr.10.4.394. [DOI] [PubMed] [Google Scholar]
- Zhang MQ. Computational prediction of eukaryotic protein-coding genes. Nat Rev Genet. 2002;14:698–709. doi: 10.1038/nrg890. [DOI] [PubMed] [Google Scholar]
- Brent MR. Genome annotation past, present, and future: how to define an ORF at each locus. Genome Res. 2005;14:1777–1786. doi: 10.1101/gr.3866105. [DOI] [PubMed] [Google Scholar]
- Livny J, Waldor MK. Identification of small RNAs in diverse bacterial species. Curr Opin Microbiol. 2007;14:96–101. doi: 10.1016/j.mib.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Dinger ME, PK C, MT R, MJ S. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;14:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonatto M, Barozzi I, Natoli G. Non-coding transcription at cis-regulatory elements: computational and experimental approaches. Methods. 2013. doi:10.1016/j.ymeth.2013.03.021. [DOI] [PubMed]
- Bentwich I. Prediction and validation of microRNAs and their targets. FEBS Lett. 2005;14:5904–5910. doi: 10.1016/j.febslet.2005.09.040. [DOI] [PubMed] [Google Scholar]
- Berezikov E, Cuppen E, Plasterk RHA. Approaches to microRNA discovery. Nat Genet. 2006;14:S2–S7. doi: 10.1038/ng1794. [DOI] [PubMed] [Google Scholar]
- Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;14:469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- Martin JA, Zhong W. Next-generation transcriptome assembly. Nat Rev Genet. 2011;14:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- Hu J, Li B, Kihara D. Limitations and potentials of current motif discovery algorithms. Nucleic Acids Res. 2005;14:4899–4913. doi: 10.1093/nar/gki791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'haeseleer P. How does DNA sequence motif discovery work. Nat Biotechnol. 2006;14:959–961. doi: 10.1038/nbt0806-959. [DOI] [PubMed] [Google Scholar]
- Zambelli F, Pesole G, Pavesi G. Motif discovery and transcription factor binding sites before and after the next-generation sequencing era. Brief Bioinform. 2013;14:225–237. doi: 10.1093/bib/bbs016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Cote A, Norel R, Annala M, Zhao Y, Riley TR, Saez-Rodriguez J, Cokelaer T, Vedenko A, Talukder S. DREAM5 Consortium. Bussemaker HJ, Morris QD, Bulyk ML, Stolovitzky G, Hughes TR. Evaluation of methods for modeling transcription factor sequence specificity. Nat Biotechnol. 2013;14:126–134. doi: 10.1038/nbt.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DC, Taylor J, Elnitski L, Chiaromonte F, Miller W, Hardison RC. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 2005;14:1051–1060. doi: 10.1101/gr.3642605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Teichmann SA, Down TA. Assessing computational methods of cis-regulatory module prediction. PLoS Comput Biol. 2010;14:e1001020. doi: 10.1371/journal.pcbi.1001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;14:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;14:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Widrow B, Lehr MA. 30 years of adaptive neural networks: perceptron, Madaline, and backpropagation. Proc IEEE. 1990;14:1415–1442. doi: 10.1109/5.58323. [DOI] [Google Scholar]
- Pearl J. Bayesian networks: a model of self-activated memory for evidential reasoning. The 7th Conference of the Cognitive Science Society; 15-17 August 1985; University of California, Irvine. 1985. pp. 329–334.
- Quinlan JR. Induction of decision trees. Mach Learn. 1986;14:81–106. [Google Scholar]
- Cover TM, Hart P. Nearest neighbor pattern classification. IEEE Trans Inf Theory. 1967;14:21–27. [Google Scholar]
- Breiman L. Random forests. Mach Learn. 2001;14:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Cortes C, Vapnik VN. Support-vector networks. Mach Learn. 1995;14:273–297. [Google Scholar]
- Quinlan JR. C4.5 Programs for Machine Learning. Morgan Kaufmann; 1993. [Google Scholar]
- Chapelle O, Schölkopf B, Zien A. Semi-supervised learning. Cambridge, Massachusettes: The MIT Press; 2006. [Google Scholar]
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Peter W. Molecular Biology of the Cell. 4. New York: Garland Science; 2002. [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, He FS, Estep III PW, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol. 2006;14:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA. Genome-wide location and function of DNA binding proteins. Science. 2000;14:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;14:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Stormo GD, Fields DS. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem Sci. 1998;14:109–113. doi: 10.1016/S0968-0004(98)01187-6. [DOI] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;14:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Series B Stat Methodol. 1977;14:1–38. [Google Scholar]
- Bailey TL, Williams N, Misleh C, Li WW. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;14:W369–W373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, Rando OJ, Birney E, Myers RM, Noble WS, Snyder M, Weng Z. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;14:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Davidson EH. Gene regulatory networks for development. Proc Natl Acad Sci USA. 2005;14:4936–4942. doi: 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won K-J, Ren B, Wang W. Genome-wide prediction of transcription factor binding sites using an integrated model. Genome Biol. 2010;14:R7. doi: 10.1186/gb-2010-11-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;14:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingos P, Pazzani M. Thirteenth International Conference (ICML '96); July 3-6, 1996; Bari, Italy. Morgan Kaufmann; 1996. Beyond independence: conditions for the optimality of the simple Bayesian classifier. pp. 105–112. [Google Scholar]
- Jolliffe IT. Principal Component Analysis. 2. New York: Springer; 2002. [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;14:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;14:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schölkopf B, Tsuda K, Vert JP. Kernel Methods in Computational Biology. Cambridge, Massachusettes: The MIT Press; 2004. [Google Scholar]
- Leslie CS, Eskin E, Cohen A, Weston J, Noble WS. Mismatch string kernels for discriminative protein classification. Bioinformatics. 2004;14:467–476. doi: 10.1093/bioinformatics/btg431. [DOI] [PubMed] [Google Scholar]
- Guigó R, Agarwal P, Abril JF, Burset M, Fickett JW. An assessment of gene prediction accuracy in large DNA sequences. Genome Res. 2000;14:1631–1642. doi: 10.1101/gr.122800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uberbacher EC, Mural RJ. Locating protein-coding regions in human DNA sequences by a multiple sensor-neural network approach. Proc Natl Acad Sci USA. 1991;14:11261–11265. doi: 10.1073/pnas.88.24.11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Mian IS, Haussler D. A hidden Markov model that finds genes in E. coli DNA. Nucleic Acids Res. 1994;14:4768–4778. doi: 10.1093/nar/22.22.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R, Eddy SR, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, United Kingdom: Cambridge University Press; 1988. [Google Scholar]
- Lukashin AV, Borodovsky M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1997;14:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp D, Haussler D, Reese MG, Eeckman FH. The Fourth International Conference on Intelligent Systems for Molecular Biology. Menlo Park, California: AAAI Press; 1996. A generalized hidden Markov model for the recognition of human genes in DNA. [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;14:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Schweikert G, Zien A, Zeller G, Behr J, Dieterich C, Ong CS, Philips P, De Bona F, Hartmann L, Bohlen A, Krüger N, Sonnenburg S, Rätsch G. mGene: Accurate SVM-based gene finding with an application to nematode genomes. Genome Res. 2009;14:2133–2143. doi: 10.1101/gr.090597.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;14:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The Transcriptional landscape of the yeast genome defined by RNA Sequencing. Science. 2008;14:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ. Deciphering the splicing code. Nature. 2010;14:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- Rose D, Hiller M, Schutt K, Hackermüller J, Backofen R, Stadler PF. Computational discovery of human coding and non-coding transcripts with conserved splice sites. Bioinformatics. 2011;14:1894–1900. doi: 10.1093/bioinformatics/btr314. [DOI] [PubMed] [Google Scholar]
- Down TA, Hubbard TJ. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 2002;14:458–461. doi: 10.1101/gr.216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;14:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;14:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;14:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods. 2012;14:473–476. doi: 10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohonen T. Self-organized formation of topologically correct feature maps. Biol Cybern. 1982;14:59–69. doi: 10.1007/BF00337288. [DOI] [Google Scholar]
- Fernández M, Miranda-Saavedra D. Genome-wide enhancer prediction from epigenetic signatures using genetic algorithm-optimized support vector machines. Nucleic Acids Res. 2012;14:e77. doi: 10.1093/nar/gks149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Karchin R, Beer MA. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011;14:2167–2180. doi: 10.1101/gr.121905.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006;14:47–59. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Loots GG, Nobrega MA, Ovcharenko I. Predicting tissue-specific enhancers in the human genome. Genome Res. 2007;14:201–211. doi: 10.1101/gr.5972507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip KY, Cheng C, Bhardwaj N, Brown JB, Leng J, Kundaje A, Rozowsky J, Birney E, Bickel P, Snyder M, Gerstein M. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;14:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Bagging predictors. Mach Learn. 1996;14:123–140. [Google Scholar]
- Freund Y, Schapire RE. The Second European Conference on Computational Learning Theory. London: Springer Verlag; 1995. A decision-theoretic generalization of on-line learning and an application to boosting. pp. 23–37. [Google Scholar]
- Lu ZJ, Yip KY, Wang G, Shou C, Hillier LW, Khurana E, Agarwal A, Auerbach R, Rozowsky J, Cheng C, Kato M, Miller DM, Slack F, Snyder M, Waterston RH, Reinke V, Gerstein MB. Prediction and characterization of noncoding RNAs in C. elegans by integrating conservation, secondary structure, and high-throughput sequencing and array data. Genome Res. 2011;14:276–285. doi: 10.1101/gr.110189.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Netflix Prize. http://www.netflixprize.com/
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, Alves P, Chateigner A, Perry M, Morris M, Auerbach RK, Feng X, Leng J, Vielle A, Niu W, Rhrissorrakrai K, Agarwal A, Alexander RP, Barber G, Brdlik CM, Brennan J, Brouillet JJ, Carr A, Cheung MS, Clawson H, Contrino S. et al. Integrative Analysis of the Caenorhabditis elegans Genome by the modENCODE Project. Science. 2010;14:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- modENCODE Consortium. Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF, Washietl S, Arshinoff BI, Ay F, Meyer PE, Robine N, Washington NL, Di Stefano L, Berezikov E, Brown CD, Candeias R, Carlson JW, Carr A, Jungreis I, Marbach D, Sealfon R, Tolstorukov MY, Will S, Alekseyenko AA, Artieri C, Booth BW. et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;14:1787–1797. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;14:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;14:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Shou C, Yip KY, Gerstein MB. Genome-wide analysis of chromatin features identifies histone modification sensitive and insensitive yeast transcription factors. Genome Biol. 2011;14:R111. doi: 10.1186/gb-2011-12-11-r111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;14:D88–92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Hur A, Noble WS. Choosing negative examples for the prediction of protein-protein interactions. BMC Bioinformatics. 2006;14:S2. doi: 10.1186/1471-2105-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Marcotte EM. Revisting the negative example sampling problem for predicting protein-protein interactions. Bioinformatics. 2011;14:3024–3028. doi: 10.1093/bioinformatics/btr514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R, Gerstein M. Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Curr Opin Microbiol. 2004;14:535–545. doi: 10.1016/j.mib.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Nam JW, Bartel DP. Long noncoding RNAs in C. elegans. Genome Res. 2012;14:2529–2540. doi: 10.1101/gr.140475.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settles B. Active Learning Literature Survey. Madison: University of Wisconsin-Madison; 2010. [Google Scholar]
- Du J, Rozowsky JS, Korbel JO, Zhang ZD, Royce TE, Schultz MH, Snyder M, Gerstein M. A supervised hidden markov model framework for efficiently segmenting tiling array data in transcriptional and ChIP-chip experiments: systematically incorporating validated biological knowledge. Bioinformatics. 2006;14:3016–3024. doi: 10.1093/bioinformatics/btl515. [DOI] [PubMed] [Google Scholar]