

Abstract
Background
Theobroma cacao L. cultivar Matina 1-6 belongs to the most cultivated cacao type. The availability of its genome sequence and methods for identifying genes responsible for important cacao traits will aid cacao researchers and breeders.
Results
We describe the sequencing and assembly of the genome of Theobroma cacao L. cultivar Matina
1-6. The genome of the Matina 1-6 cultivar is 445 Mbp, which is significantly larger than a sequenced Criollo cultivar, and more typical of other cultivars. The chromosome-scale assembly, version 1.1, contains 711 scaffolds covering 346.0 Mbp, with a contig N50 of 84.4 kbp, a scaffold N50 of 34.4 Mbp, and an evidence-based gene set of 29,408 loci. Version 1.1 has 10x the scaffold N50 and 4x the contig N50 as Criollo, and includes 111 Mb more anchored sequence. The version 1.1 assembly has 4.4% gap sequence, while Criollo has 10.9%. Through a combination of haplotype, association mapping and gene expression analyses, we leverage this robust reference genome to identify a promising candidate gene responsible for pod color variation. We demonstrate that green/red pod color in cacao is likely regulated by the R2R3 MYB transcription factor TcMYB113, homologs of which determine pigmentation in Rosaceae, Solanaceae, and Brassicaceae. One SNP within the target site for a highly conserved trans-acting siRNA in dicots, found within TcMYB113, seems to affect transcript levels of this gene and therefore pod color variation.
Conclusions
We report a high-quality sequence and annotation of Theobroma cacao L. and demonstrate its utility in identifying candidate genes regulating traits.
Keywords: Theobroma cacao L., genome, Matina 1-6, haplotype phasing, genetic mapping, pod color, MYB113
Background
The cacao tree (Theobroma cacao L.) is a neotropical species native to Amazonian lowland rainforests [1,2] and is now grown in more than 50 countries throughout the humid tropics. T. cacao is a member of the Malvaceae family, and its beans (seeds), harvested from pods (fruits), are used for the chocolate, confectionery, and cosmetic industries [3]. Cacao production is essential to the livelihoods of 40 to 50 million people worldwide, including the smallholder farmers who cultivate the crop, who number more than 5 million [4]. The crop is often grown in agroforestry-type ecosystems alongside other fruit and commodity crops, thereby providing sustainable economic and environmental benefits to some of the poorest and most ecologically sensitive areas of the world [5]. Cacao-growing regions are also largely centered in important biodiversity hotspots and in proximity to 13 of the world's most biologically diverse regions [6].
Recent molecular analyses [2] have permitted cacao germplasm classification into 10 major clusters or groups: Amelonado, Contamana, Criollo, Curaray, Guiana, Iquitos, Maraňón, Nanay, Nacional, and Purús. Compared with traditional cacao classification schemes, this new system more accurately reflects the genetic diversity available to breeders. The Costa Rican cacao variety, Matina, was named after the Matina river valley, where the variety was first grown and where it has been maintained in its original form [7]. Although some variation in phenotype does exist within the types considered to be Matina, its genotype is known to be largely homozygous [7]. The Matina 1-6 clone is a traditional cultivar that exhibits the Amelonado phenotype and belongs to the Amelonado genetic group. This group shows little genetic diversity compared with the other nine groups [2,8] and, critically, it is the most common cultivated type of cacao worldwide [8-10].
Because of its high yield and disease resistance, the most ubiquitous clone in large cacao plantations in Latin America is CCN 51. It is also the optimal parent in many breeding programs, owing to its favorable combining ability for yield. Unfortunately, it has a rather undesirable flavor profile because of its high acidity and astringency, and also because it lacks desirable floral aromas. The undesirable flavor profile of CCN 51 is particularly pertinent to cacao exporters in Ecuador, who frequently mix CCN 51 with beans from the Nacional variety, which has desirable floral flavors and green pods. However, the adulteration of Nacional beans reduces the overall quality of Ecuadorian exports and consequently the chocolate made from this origin. In an effort to make a distinction between farmers growing CCN 51 (red pod color) from those growing clones with favorable flavor profiles, the aim of the breeding program of the Ecuadorian Cacao Research Institute (INIAP) is to select for high-yielding clones that exhibit desirable floral aromas, which bear green pods and are thus easily differentiable from CCN 51. The ultimate goal of this segregation strategy is to improve the quality of the beans exported from Ecuador.
The color of immature cacao pods, which varies from green (light to dark) to red and purple, is caused by differential accumulation of anthocyanin [7]; mature pods tend to be yellow, orange, red or purple, although some mature pods remain green. Although it is an oligogenic trait [7], pod color in the progeny of crosses cannot be predicted based simply on parental phenotypes, because of the presence of allelic dominance [7]. Identification of genes that regulate pod color therefore constitutes a crucial first step toward the development of a platform for marker-assisted selection (MAS) aimed at the development of high-yielding alternatives to CCN 51. The ability to screen young cacao seedlings with molecular markers and to select only those carrying alleles that result in green pods would greatly reduce the population sizes required for the laborious and expensive phenotypic evaluations of unlinked flavor and yield traits.
The genome sequence of a Criollo genotype (B97-61/B2) was recently reported [11]. Although this cacao type is genetically distinct [1,2], it is a poor representative of the cacao types cultivated worldwide. In an effort to enhance the accuracy and speed of traditional cacao breeding, we sequenced the genome of Matina 1-6, a self-compatible and highly homozygous genotype that is more representative of the cacao cultivated worldwide. The ultimate goal of our sequencing and trait-mapping efforts is the development of tools for MAS in cacao. The genome sequences of over two dozen plant species have been generated in recent years [12]. However, we are aware that there are few concrete examples of strategies that translate these sequence data into knowledge relating specific genes to phenotypic traits. This paucity of solid links between genes and traits has led to criticism of the utility of these genome sequences [13].
A preliminary version of the T. cacao L. Matina 1-6 genome (V0.9) was released to the public in 2010. We describe here our sequencing effort, an improved version of the Matina 1-6 genome (V 1.1), and its use in investigating genotype-phenotype relationships in T. cacao. Our approach to demonstrating the usefulness of this genomes in identifying candidate genes regulating desirable traits involved augmenting the improved Matina 1-6 genome sequence with sequence information from additional genotypes, implementing haplotype phasing for use in linkage mapping, and enhancing these results using association mapping and differential gene-expression analysis. To demonstrate the power of this comprehensive approach, we chose pod-color variation as an example of a trait that could be altered through MAS. Genotypic and phenotypic data from three different segregating populations (MP01, T4 Type 1, and T4 Type 2) were collected for this study. The two T4 mapping populations are part of a population that was recently used to identify multiplequantitative trait locus (QTL) associated with resistance to the frosty pod (Moniliophthora roreri) and black pod (Phytophthora spp.) diseases, with self-incompatibility, and with other horticultural traits, including pod color [14,15]. A single QTL associated with pod color was identified and mapped onto linkage group 4 (chromosome 4) [14]. This pod-color trait was mapped using T4 Type 1 and Type 2 populations, between the microsatellite markers mTcCIR158 (at 8.5 cM) and mTcCIR107 (at 36.4 cM) [14]. In the present study, we further refined this region to a 0.6 Mbp interval on chromosome 4 (20.5 to 21.1Mbp) and identified a candidate gene for pod color that is homologous to a gene that regulates organ pigmentation in Rosaceae, Solanaceae, and Brassicaceae.
Results and discussion
Genome size
The flow cytometry (FCM) estimate of the Matina 1-6 genome size is 445 Mbp; this value approximates the mean determined for 28 different T. cacao genotypes (see Additional file 1, Table S1), including representatives of all 10 structural diversity groups defined by Motamayor et al. [2]. When analyzed by structural group, the genome sizes fall into three statistically distinct sets (see Additional file 1, Table S2). Genotypes within the Nacional group have the largest genomes, whereas most of the other genotypes have genomes similar in size to that of Matina 1-6, a member of the Amelonado group. Cultivars within the Criollo group, including B97-61/B2 (409 Mbp) whose genome was recently published [11], have some of the smallest genomes within the species (see Additional file 1, Table S2).
Assignment of chromosomes to pseudomolecules using fluorescence in situ hybridization
We performed fluorescence in situ hybridization (FISH)-based karyotyping of mitotic T. cacao chromosomes, using two types of probes: genomic repeats (centromeric repeats) for differential chromosome 'painting', and bacterial artificial chromosomes (BACs) for chromosome identification. Centromeric repeat-based FISH has been used to identify chromosomes in soybean [16] and maize [17,18]. We identified sequences homologous to a previously identified candidate centromeric repeat [11] by searching unassembled sequence reads acquired during generation of the Matina 1-6 physical map [19]. A ClustalW alignment (not shown) of the 236 sequences with the highest percentage of identity indicated a length for the Matina 1-6 Cent-Tc repeat of 171 bp (see Additional file 2, Figure S1 for consensus), comparable with the centromere monomer length in other plant species [20-22]. We designed specific oligonucleotide probes that target highly conserved or less well conserved centromeric repeat T. cacao (Cent-Tc) regions (see Additional file 2, Figure S1). Probe OLI-07 was present in the majority (74%) of the aligned Cent-Tc repeats, whereas probe OLI-13 was present in 12.5%. When used together in FISH experiments (see Materials and methods section for details) the two probes differentiated the mitotic chromosomes into several distinct color and intensity subgroups (Figure 1). To identify individual chromosomes, BAC probes (Materials and methods; also see Additional file 1, Table S3) derived from low-repeat content regions of each of the 10 pseudomolecules defined by Matina 1-6 physical mapping [19] were individually mapped to Cent-Tc-labeled chromosomes (Figure 1 and data not shown). We then developed a six-component (two Cent-Tc probes plus four BAC probes) FISH 'cocktail' that simultaneously identified every chromosome in a single mitotic chromosome spread (Figure 1). Because the Matina 1-6 physical map is anchored by high-density molecular markers that relate the map to the sequenced genome, pseudomolecule-derived BAC probes permit the association of molecular-linkage groups and the pseudomolecules with the chromosomes themselves, thereby enabling chromosome numbering based on pseudomolecule numbering, and generating a single, unified cytogenetic map for Matina 1-6.
Figure 1.
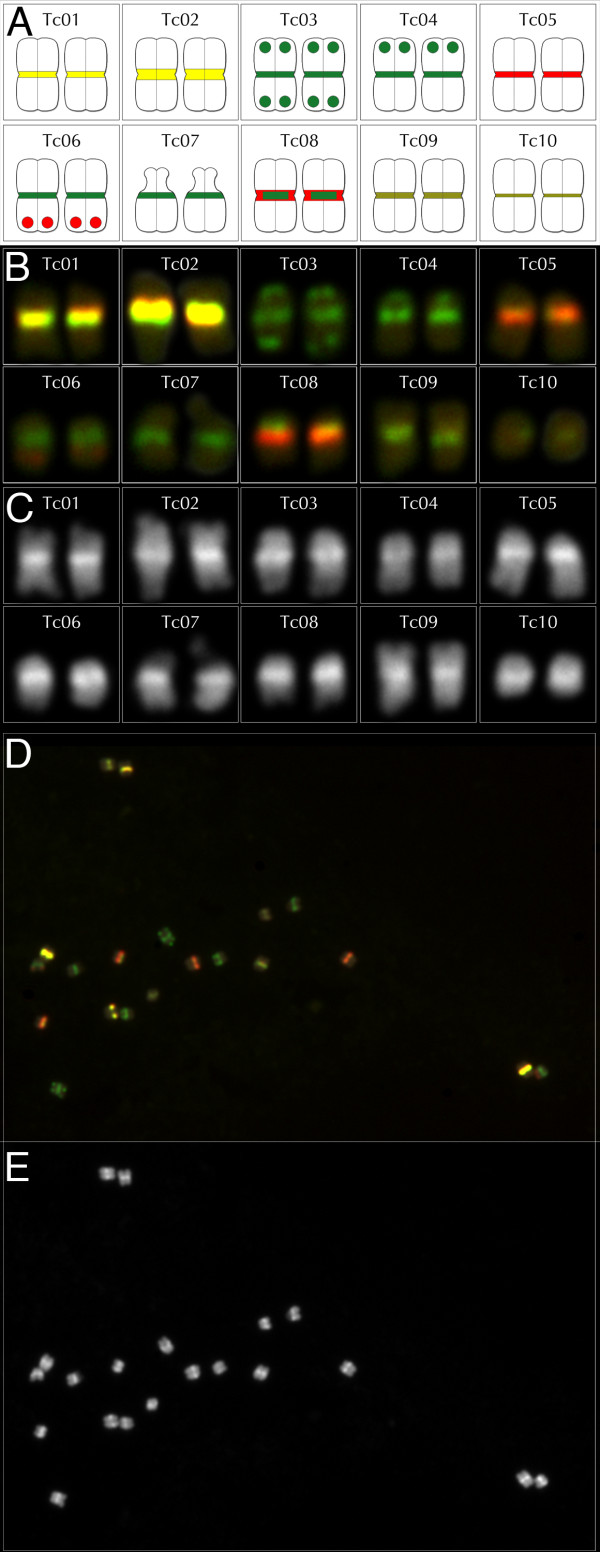
Fluorescence in situ hybridization (FISH)-based karyotype of Theobroma cacao Matina 1-6. A FISH cocktail comprised of two Cent-Tc oligonucleotide probes plus four BAC clones permitted identification of the ten chromosome pairs. (A) Ideogram of the T. cacao Matina 1-6 karyotype. Centromeres are coded in accordance with the color and size of the combined FISH signals for OLI-07 (green pseudo-colored) and OLI-13 (red pseudo-colored). Bacterial artificial chromosome (BAC) probes (red or green) are indicated by paired dots near chromosome termini. The following BACs were used for probes: for Tc03, TcC_Ba057I03 and TcC_Ba027M06; for Tc04, TcC_BB065A03; for Tc06, TcC_Ba018I22. Relative chromosome sizes are not indicated, with the exception of the satellite arm of Tc07, which is shown as a knob. (B) Chromosomes labeled with the FISH cocktail arranged by chromosome number. Chromosomes are discriminated as follows: Tc01 has the second-brightest yellow centromere. Tc02 has the brightest yellow centromere. Tc03, Tc04, Tc06, and Tc07 all have similar centromere labeling (pure green), but are differentiated based on unique BAC probe labeling: Tc03 is labeled at each end by green BAC probes; Tc04 is labeled at one end by a green BAC probe; Tc06 is labeled at one end by a red BAC probe; and Tc09 is not labeled by BAC probes. Tc05 has the second-brightest red centromere; Tc08 has the brightest red centromere with an 'internal' green domain; and Tc09 has the brightest yellow-green centromere and is much longer than Tc10, which has the second-brightest yellow-green centromere. (C) DAPI channel image of chromosomes in (B). The satellite arms of Tc07 are above the centromeres. (D) A FISH image containing a complete chromosome spread. (E) Corresponding DAPI channel image from which chromosomes in (C) were extracted.
Sequencing and assembly of the Matina 1-6 genome
In total, 32,460,307 sequence reads (a combination of Sanger and Roche 454 pyrosequencing; see Additional file 1, Table S4), were assembled using a modified version of Arachne (version 20071016 [23]). The resulting assembly was integrated with multiple high-quality genetic maps (see Additional file 1, Table S5) to produce a chromosome-scale assembly. Sanger sequences were obtained from fosmids and from BAC ends selected using the minimum tiling path of a previously reported physical map [19] (see Materials and methods).
The initial assembly generated 1,672 scaffold sequences with a scaffold N50 of 7.4 Mbp, and 91 scaffolds longer than 100 kbp (see Additional file 1, Table S6), giving a total scaffold length of 348.7 Mbp. To generate version 1.1 of the Matina 1-6 genome assembly, we removed contaminating sequences and scaffolds containing exclusively any of the following: unanchored repetitive sequences, unanchored ribosomal (r)DNA sequences, mitochondrial DNA, chloroplast DNA, and sequences less than 1 kbp in length. Next, we integrated the maps and constructed chromosome-scale pseudomolecules (see Materials and methods). Unanchored repetitive sequences were identified from the scaffolds remaining from the construction of pseudomolecules. These are scaffolds composed of more than 95% 24-mers occurring more than four times in scaffolds longer than 50 kb. Unanchored rDNA, mitochondrial, and chloroplast sequences were identified by aligning the remaining scaffolds against the nr/nt nucleotide collection at the National Center for Biotechnology Information (NCBI).
The chromosome-scale assembly contains 711 scaffolds that cover 346.0 Mbp of the genome, with a contig N50 of 84.4 kbp and a scaffold N50 of 34.4 Mb. The resulting final statistics are shown in Table 1. Plots of the marker placements were constructed for all three genetic maps used for the assembly, and for the synteny between exons in the T. cacao Matina 1-6 and Criollo genomes (see Additional file 2, Figure S2). The pseudomolecules comprise 99.2% (346.0 Mbp of 348.7 Mbp) of the assembly. For detailed comparison of the Criollo and Matina scaffold assemblies, see Additional file 1 (Table S7). The genome assembly has been deposited in GenBank (accession number [ALXC01000000]).
Table 1.
Final summary assembly statistics for chromosome-scale assembly of the Matina 1-6 (version 1.1) genome sequence.
| Scaffold total | 711 |
|---|---|
| Contig total | 20,103 |
| Scaffold sequence total | 346.0 Mbp |
| Contig sequence total | 330.8 Mbp (4.4% gap) |
| Scaffold N50, size (number) | 34.4 Mbp (5) |
| Contig N50, size (number) | 84.4 Kbp (1,080) |
Arachne assembly assessment
An initial assessment of the completeness of the Arachne2 euchromatic genome assembly was estimated using 1,015,064 Roche/454 leaf transcript sequence reads [24]. The results of this analysis indicated that 98.9% of these (see Materials and methods) mapped to the 10 chromosomes. Other completeness assessments are described in the annotation results (see below). We also validated the Arachne2 sequence assembly against a 998 kbp reference region of Sanger-sequenced BAC clones from the Matina 1-6 library [25]. A dot-plot comparing the assembled genome and the BAC clone reference region was constructed (see Additional file 2, Figure S3). Overall, the analysis indicated high contiguity across the region; a detailed summary reporting error events and bps affected is presented (see Additional file 1, Table S8).
Evidence-based gene annotation
We prioritized the generation and use of RNA sequencing data from diverse experiments, and from available plant whole-gene sets, for evidence-based annotation. The overall results are shown schematically in Figure 2.
Figure 2.
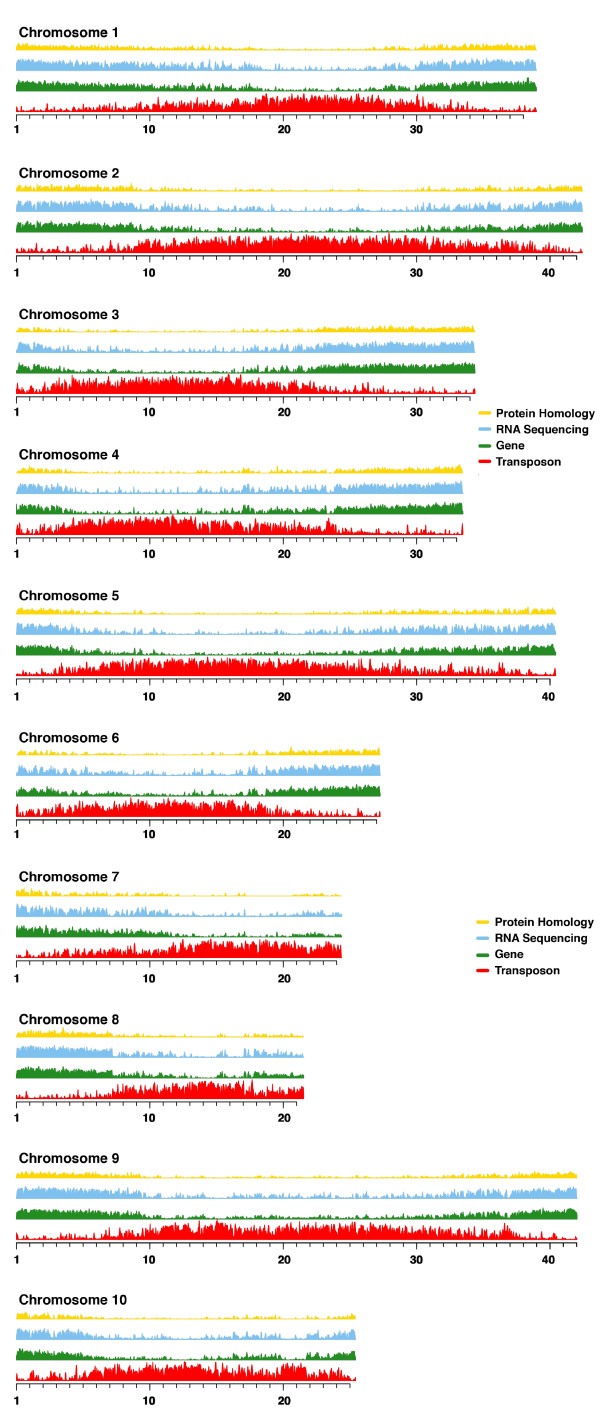
Genomic features of Theobroma cacao Matina 1-6. Shown are overall densities of evidence sets (see Materials and methods) that contributed to T. cacao Matina 1-6 annotation, and the final results as described in the text. Data were plotted for the chromosomes (pseudomolecules) in 50 Kbp sliding windows. Yellow denotes protein homology evidence by alignment to proteins of eight previously annotated plant genomes; blue denotes mapping of transcriptome data from second-generation RNA sequencing; green denotes gene models; red denotes transposons from homology-based and structure-based annotation, as described in the text (see Materials and methods).
Longer transcript read sets (Roche/454, see Materials and methods) were first used to confirm the completeness of genome assembly version 1.1 as above. Seven million reads (see Additional file 1, Table S9) sequenced from leaf, bean, and floral RNA from different genetic backgrounds mapped at rates ranging from 88.25% to 99.25% in Matina 1-6 (version 1.1) and from 85.23% to 97.89% in Criollo (version.1 [11]) genome assemblies. Mapping sequence identities were similar, suggesting that no significant differences were derived from sampling (see Additional file 1, Table S9). The aforementioned reads were also used for gene model annotation together with 1.22 billion shorter paired-end reads from Illumina RNA sequencing (see Additional file 1, Table S10).
We used coding and intron spans from protein alignments of eight annotated plant genomes for homology modeling (see Materials and methods; also see Additional file 1, section 3). The contributions of RNA and protein analyses are displayed independently with final models in Figure 2.
Evidence-supported models represent 29,408 loci with 14,806 alternative transcripts determined from RNA assemblies (see Additional file 1, Table S11). Additionally, about 20,000 elements were annotated with little or no support. These included around 13,000 gene-like regions that overlapped substantially (≥ 33%) with transposon loci (described below) and showed little or no evidence of expression, as well as loci predicted ab initio only, partial gene models, possible non-coding RNAs, and pseudogenes.
If the evidence-supported gene models alone are considered, around 15% of the Matina 1-6 genome (54 Mb) is expressed, and 36 Mbp of this represents protein-coding sequence. Intron size distribution was bimodal, as seen in other plant genes [26] (see Additional file 1, Table S12). The breadth of RNA data and the larger set of plant genome sequences available since our first cacao annotation release (version 0.9) contributed to a more substantiated annotation by every measure. The improved gene models are characterized by longer protein-coding sequence, full expression support through untranslated regions, and fewer fragments relative to both Matina (version 0.9 and Criollo (version 1) (see Additional file 1, Table S13).
Analysis of gene content and orthology
Among the primary cacao protein structures, we found 18,457 unique alignments to UniProt references [27] and 4,903 that were without match in this broadly comprehensive database. Products of 3,426 gene models (5,819 transcripts) were associated with 140 KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway maps [28]. This compares with 125, 124, 123, and 124 currently annotated metabolic pathways for Arabidopsis, poplar, grape, and castor bean, respectively [28]. Cacao genes were categorized using OrthoMCL [29] into 15,523 putative orthologous groups situated among those of 8 other plants used in the annotation, and of these groups, 13,700 contained a single representative. Remarkably, only 43 orthologous groups that were defined based on genes present in the other plant species had no cacao gene representative, the lowest level among the fully sequenced plant genomes analyzed (see Additional file 1, Table S14a). Orthology analyses performed in this study (see Materials and methods; see Additional file 1, section 3) generate group partitions that sometimes divide known phylogenetic clades. Ten (23%) of the groups not represented in cacao were defined by uncharacterized or hypothetical proteins in the other plants. The remainder of the unrepresented groups averaged only one or two copies in the other plants, and generally reflected sequence divergence within plant gene families whose paralogs were indeed present in cacao. For example, although one group in the 2-oxoglutarate and Fe(II)-dependent oxygenase superfamily was not represented in cacao, a number of cacao genes falling into 59 orthologous groups and an additional 8 ungrouped genes were annotated as members of this superfamily. There were 16 calcium-dependent kinase (CDPK) groups and three ungrouped CDPKs found in cacao, although one group that was defined for the other plants was not represented in cacao by this analysis. The over-represented and under-represented gene families for cacao, including these examples are tabulated (see Additional file 1, Table S14b), and the results summarized in a Venn diagram form as shared gene families for five compared plants (see Additional file 1, Figure S4).
The identified proteins were assigned to gene ontology (GO) categories, provided by The Arabidopsis Information Resource (TAIR), and the proportion of genes assigned to each category were found to be relatively close between poplar, cacao, and grape, but significantly different from Arabidopsis (χ2 = 2895.128, degrees of freedom (df) = 78, P < 2.2 ×10-16; see Additional file 1, Table S14c). This initial comparison suggested that cacao has the highest overall similarity to poplar (1.0 correlation, χ2 = 7.2923, df = 25, P= 0.9998), and that Arabidopsis drives most of the differences in gene content per GO category. Although further work will contribute to improve the functional annotation of the proteins identified in Matina 1-6, the initial assessment and functional characterization provided here considerably improves the genomic tools available for cacao, as we describe below for the mapping and identification of genes regulating phenotypic traits.
Lineage-specific genes are undoubtedly among the 703 T. cacao gene clusters [29] that share no apparent orthology with genes in any of the other 8 plants used in this study (see Additional file 1, Table S14a). Ongoing analyses, which incorporate an annotated gene set for cotton, a second member of the Malvaceae family that was recently sequenced [30], will refine this picture, and will also help define genes that are associated with speciation. Hierarchical clustering of cacao gene families, among those of many other plants, is available in Phytozome (version 9 [31]).
To further understand the results from the previously described analyses and to annotate seemingly divergent or uncharacterized unique genes in cacao, we performed additional hidden Markov model (HMM) searches [32]. We annotated 4244 transcripts from 4,085 genes (transposon overlaps excluded) based on expression, that is, association with RNA sequencing evidence alone, as these showed no homology to proteins in the other 8 plant genomes. We then surveyed potentially remote homology that would not have been identified within our annotation standards by searching these cacao proteins against the profile HMM databases Pfam and TIGRFAMS, using HMMER3 [32]. In total, 126 HMM families (E-value 0.005) were found within the no-homology set. We compared these with the set of HMM families obtained from an identical scan of cacao proteins with other plant homology (for groupings of these annotations and the proportion of HMM families between the two sets, see Additional file 1, Table S14d). Although the Pfam database is part of InterPro (used above), probabilistic scoring in the HMMER3 algorithm can exceed the accuracy of finding motifs by single optimal alignments [33,34]. Although only four HMM families were unique to the no-homology set, these were members of clades that were represented in the set of annotated plant homologs (see Additional file 1, Table S14d). Overall, this analysis therefore added modest numbers of additional members to known structural categories, and approximately 3% more definition to the unknown proteins of cacao.
Gene models of the Matina 1-6 (version 1.1) sequence have been deposited in NCBI as (GenBank accession number [ALXC00000000]).
Transposable elements
A significant portion of eukaryotic genomes is composed of transposable elements (TEs) [35-37]. Using a structure-based and homology-based strategy, we identified 8,542 intact TEs in the T. cacao Matina 1-6 genome. Those elements, together with numerous truncated elements and other fragments, cover 41.53% of the assembled genome. The overall results are shown schematically in Figure 2. In a parallel comparative analysis using the same approach, we identified 5,089 intact TE copies in the T. cacao Criollo genome [11], and found only 35.40% of the Criollo genome assembly to be comprised of TEs (see Additional file 1, Table S15). Although this estimate represents a nearly 10% increase in TEs over a previous annotation of Criollo (version 1 [11]), the proportion of TEs in the assembled portion of the Criollo genome is notably less than that in the assembled portion of the Matina 1-6 (version 1.1) genome.
Retrotransposons, especially long terminal repeat (LTR) retrotransposons, are the predominant class of TEs identified in the cacao genome. Two types of LTRs are found: intact and solo. Solo LTRs are thought to be formed by unequal intra-element homologous recombination (UR) between two closely identical intact LTRs [38]. The 7,545 LTR retrotransposons identified, comprising 5,345 solo LTRs and 2,200 intact LTRs, can be classified into 369 families as follows: 123 copia-like, 233 gypsy-like, and 13 unclassified families (see Additional file 1, Table S16). Of the 333 retrotransposon families identified in Criollo, 277 are common to both Criollo and Matina 1-6 genomes, 56 families are specific to Criollo, and Matina 1-6 contains 92 LTR retrotransposons families not identified in Criollo (see Additional file 1, Table S16). While 96.02% (Criollo) and 98.10% (Matina 1-6) of the intact copies of these retrotransposons belong to these 277 shared families, the copy numbers of elements in each family differ substantially. For example, for Tcr53, which is the largest family in both genomes, 1,124 copies were identified in the Matina 1-6 genome, whereas only 301 copies were detected in the Criollo genome. It should be noted that, because the portions of either the Matina 1-6 or the Criollo sequences that were not assembled into the current genome pseudeomolecules remain unknown, the contribution of TEs to the difference in the contents and sizes of the two genomes could not be assessed more precisely. Nevertheless, four times more copies of Tcr53 elements were detected in Matina 1-6 than in Criollo, which may be explained mainly by the different levels of independent amplification of this family in the two genomes after their divergence. Most of the families that are unique to either genome contained only single copies. DNA transposons represent 8.87% of the Matina 1-6 genome and 7.00% of the Criollo genome. The gypsy-like retrotransposons comprise 79.93% (4.90% out of 6.13%) of the difference in TE coverage between Matina 1-6 and Criollo. These results suggest that retrotransposons may have undergone a recent amplification in the Matina 1-6 genome.
Synteny between Matina 1-6 and Criollo
We further compared the whole-genome sequences of Matina 1-6 (version 1.1) and the Criollo-type B97-61/B2 (version 1 [11]). The results, depicted using Circos [39], indicate very well conserved synteny, as expected, with 271 orthologous regions (ORs) identified (see Additional file 2, Figure S5). The longest OR is a 7.7 Mbp region in chromosome 9 in both Criollo and Matina 1-6, which contains 2,580 matching exons. The mean number of matching exons in each OR is 150, and the mean lengths of the ORs are 681 kbp in Criollo and 717.5 kbp in Matina 1-6. Of the total number annotated in each genome, 87.23 % (28,666 of 32,862) of the Criollo genes and 76.08 % (22,374 of 29,408) of the Matina 1-6 genes are located in the ORs that we identified. The orthology is well conserved at the chromosomal level, but fewer and smaller ORs between non-orthologous chromosomes were also detected (see Additional file 2, Figure S5). Regions on chromosome 2 of Matina 1-6 are orthologous to regions on chromosome 9 of Criollo, for example, and regions on chromosome 3 of Matina 1-6 are orthologous to regions on chromosome 10 of Criollo. In total, 12 ORs between non-orthologous chromosomes were detected and the mean size of the ORs between non-orthologous chromosomes was much smaller than that of the ORs between orthologous chromosomes (see Additional file 1, Table S18).
We analyzed the distribution of the genes associated with transposons in ORs between non-orthologous chromosomes and those between orthologous chromosomes (see Additional file 1, Table S18). We examined the mean span (kb) between genes and between transposons in each region of the two cultivars. In both cultivars, the mean span between transposons was shorter in ORs between non-orthologous chromosomes, even though the mean span between genes was larger in ORs between non-orthologous chromosomes. These results suggest that a much higher proportion of the genes in ORs between non-orthologous regions are associated with transposon activity than those in ORs between orthologous regions. The higher frequency of transposon-related genes in the ORs between non-orthologous chromosomes, along with the smaller size of the ORs between the non-orthologous chromosomes, suggests that those blocks were translocated to non-orthologous regions by transposon activity.
Although more detailed studies are required to discern if the observed differences are inherent to the different assemblies and annotation approaches, we determined whether any disease resistance-related genes (within the leucine-rich repeats: LRR-RLK class) or genes potentially involved with cacao-bean quality [11] reside in ORs (see Additional file 1, Table S19). The genes that reside outside ORs or in ORs between non-orthologous chromosomes could be responsible for the differences in crop quality and disease resistance between the two cultivars. It has been reported that genes encoding proteins involved in plant interaction with biotic and abiotic extrinsic factors are far more likely to have been transposed than those that are involved in relatively stable processes [40].
Many of the LRR-RLK genes, flavonoid biosynthetic pathway genes, lipid biosynthesis genes, and terpenoid synthesis genes identified in T. cacao by Argout et al. [11] are outside ORs (see Additional file 1, Table S19; for the specific gene models involved in this analysis, see Additional file 1, Table S20a). However, this analysis was based on the Criollo annotation [11] and did not use the genes identified through our evidence-based annotation pipeline in Matina 1-6 for these pathways, many of which are also in non-orthologous regions. For example, the LRR-RLK gene motif is found in twice as many genes in non-orthologous regions in the Matina 1-6 assembly as in the Criollo assembly (see Additional file 1, Table S20b).
Mapping pod color
In both T4 mapping populations (see Materials and methods), the progeny segregate in a 1:1 ratio for pod color, suggesting the involvement of a single gene. The common parent in both of these populations, Pound 7, has green pods. The other parents of these crosses, UF 273 Type 1 and Type 2, have red pods (see Additional file 2, Figure S6). In the MP01 mapping population, pod color segregates in a 3:1 red:green ratio, and we infer that both parents of this population, TSH 1188 and CCN 51, are heterozygous for the dominant red allele (see Additional file 2, Figure S6).
Linkage-mapping analyses previously suggested that pod color is regulated by gene(s) located on chromosome 4 [14]. To corroborate this result, we performed Fisher's exact test for each single-nucleotide polymorphism (SNP), using a contingency table of genotype counts and phenotype scores (that is, red and green) and the marker data from the ten linkage groups on the three mapping populations (T4 Type 1, T4 Type 2, and MP01). In all three mapping populations, a large region on chromosome 4 showed significant (based on the Bonferroni correction at α = 0.05) association with pod color (see Additional file 2, Figure S7).
Using haplotypes can be more powerful than using single-marker methods to detect phenotypic effects of low-frequency variants in genome-wide association studies [41,42]. Haplotype-based methods are also useful for inferring the underlying causal genetic basis for various traits in linkage-mapping populations because, as shown here, it is possible to evaluate the parental inheritance of the associated haplotype more efficiently. We therefore determined parental haplotypes for chromosome 4 in individuals from the three mapping populations studied to help us identify candidate genes that are associated with this trait. We resolved the genotypes of the progeny in each of the mapping populations for chromosome 4 into the two corresponding parental haplotypes, using the output of HAPI-UR [43] (see Materials and methods). To investigate the effect from each parental haplotype separately on pod color, we performed Fisher's exact tests separately for each parent, on individuals from each mapping population, using a 2 × 2 contingency table of parental haplotypes for each marker and red and green pod color. This analysis identified a difference between the T4 and MP01 populations; in both T4 populations, haplotypes belonging to one parent, Pound 7, did not affect the color phenotype; whereas in MP01, haplotypes from both parents did affect pod color (Figure 3). The observation that a single haplotype from each parent associates with red pod color in MP01, but not in T4, is consistent with the mendelian model outlined above, and suggests the existence of a potential common allele in the two T4 mapping populations, even though the parents are different. Our results also suggest that red pod color is dominant over green and that both UF 273 parents are heterozygous for the red allele, whereas the common parent, Pound 7, is homozygous for the recessive green allele.
Figure 3.
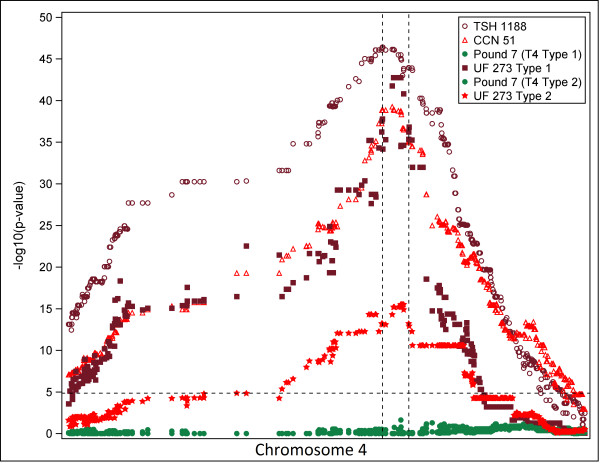
Statistical significance of association of pod color with markers on chromosome 4 of the parental Theobroma cacao haplotypes. The y-value at each marker is -log10(P-value) with the P-value computed using Fisher's exact test for both haplotypes of each parent, taking as input a 2 × 2 contingency table per marker. The segment between the vertical dashed lines is the genomic region most strongly associated with pod color in all three mapping populations. Thresholds denoted by the dashed red line in each plot were calculated using the Bonferroni correction for multiple comparisons at α = 0.05.
We then focused on the small region that we had identified on chromosome 4 (Figure 3) as that most significantly associated with the trait. The locations of the most significant SNP markers vary only slightly between the mapping populations, ranging from 20,987,989 bp to 21,726,996 bp. The haplotype combinations, taking into account SNP 21,126,449 bp, explain the phenotypes in the data with at least 97% accuracy in each population (see Additional file 1, Table S21; see Materials and methods for more details on the computation with Fisher's exact test).
To fine-tune the mapping of the region of interest, we examined trees exhibiting a recombination of the haplotypes within the associated region, and added flanking markers to extend the region to 20.35 to 22.05 Mbp. In each T4 population, there were 8 to 15 trees with recombination events in at least one parental haplotype (Figure 4a,b). The T4 recombinants refined the location of the causal locus to the region between 20,562,635 and 21,726,996 bp on chromosome 4 (Figure 4a,b).
Figure 4.
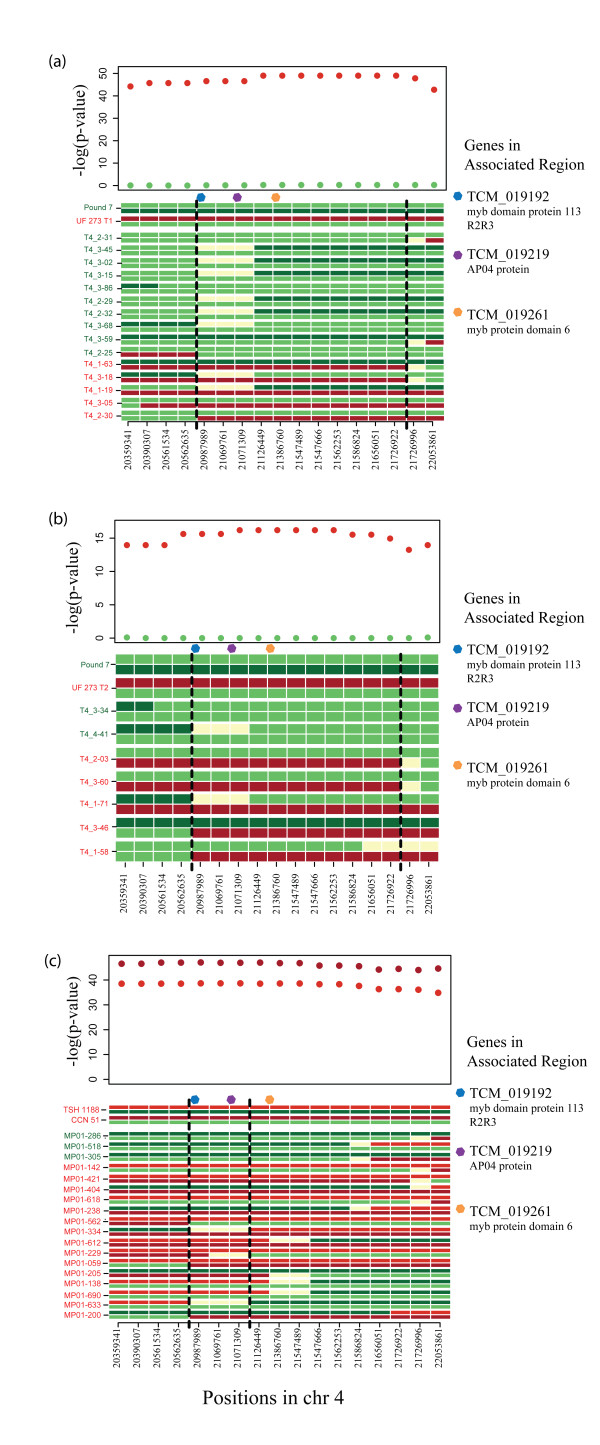
Haplotype analysis of trees exhibiting recombination in the chromosome 4 segment associated with pod color. (a) Recombinant trees from population T4 Type 1; (b) recombinant trees from population T4 Type 2; (c) recombinant trees from population MP01. Maternal and paternal haplotypes are shown at the top of each figure. Tree names are colored according to pod-color phenotype. Red represents haplotypes associated with red pod color, and green represents haplotypes associated with green pod color from the two parents, while the yellow marker values represent uncertainty in the haplotype assignment. The black vertical bars surround the most likely region regulating pod-color variation according to the haplotypes of the recombinants (that is, if a recombinant shows only haplotypes for a given marker associated with green pods, but its phenotype is red, this indicates that the marker is not associated with pod color; this is the case for CATIE 1-63 at marker 22,053,861 in (a). The P-values from the Fisher's exact test are shown above each marker for each parent. The P-values are colored by parental phenotype, with the father always being bright red. The location of three candidate genes is indicated by colored dots above the closest markers.
Using MP01 trees that exhibit recombination in this region, the location of the causal locus was further refined (Figure 4c) to the region between 20,562,635 and 21,126,449 bp on chromosome 4. Indeed, the alleles in TcAPO4 and TcMYB6 associated with red pod color in the CCN 51 and TSH 1188 parents were now also associated with green pod color in the recombinants. This result further supports the hypothesis of a single allele underlying pod color (although there is the possibility that different mutations in the same gene could be responsible).
Candidate genes for pod color
The availability of the high-quality Matina 1-6 reference genome permits straightforward identification of candidate genes within the restricted chromosome region identified. Recombination events identified in the T4 Type 1 and Type 2 mapping populations delimit the region regulating pod-color variation to between 20,562,635 and 21,726,996 bp on chromosome 4 (Figure 4a,b). In this region, three gene models are of particular interest: TCM_019192, homologous to Arabidopsis MYB113; TCM_019219, homologous to the Vitis vinifera APO4 gene; and TCM_019261, homologous to Arabidopsis MYB6. Both MYB genes encode transcription factors of the R2R3 domain class, which are known to be diverse in plants [44]; whereas APO proteins promote photosystem 1 complex stability and associated chlorophyll accumulation [45].
Cacao pod color is determined by the degree of red pigment that is superimposed over the base green color of the pod during development. The green color itself ranges widely, from light to dark green [7]. Because the intensity of pod pigment is influenced by the status of the base green color, we considered the gene homolog to APO4 to be a candidate gene for pod-color determination. It has been hypothesized that pod color could be regulated by one locus with alleles that determine the intensity of the green color acting in concert with a second locus that determines pigmentation [7].
Two of the 18 MYB genes on chromosome 4 of the T. cacao Matina 1-6 genome version 1.1_ (see Additional file 1, Table S22), which are localized within the 20,562,635 and 21,726,996 bp interval, are particularly promising candidate genes for pod-color variation in cacao because variants in MYB transcription factors are known to cause variation in berry pigmentation in grapes [46,47]. While MYB6 is less characterized, MYB113 is in a small clade known to act in complexes with basic helix-loop-helix (bHLH) proteins to activate genes encoding enzymes acting in late stages of flavonol biosynthesis. MYB variants cause pigmentation variation in potato, tomato, and pepper [48], as well as in kale [49]. Pigmentation in apple skin and flesh is also regulated by MYB transcription factors [50]. Moreover, when genes encoding the R2R3 MYB transcription factors (cloned from apple) are transformed into tobacco, they induce the anthocyanin pathway, when co-expressed with bHLH proteins [50]. MYB gene evolution is thought to involve duplication events [51]; a single gene cluster of three MYB genes is involved in berry pigmentation in grape [46], and three genes of this type (including MYB113) are tandemly arrayed in Arabidopsis. The gene cluster in grape that is involved in berry-color variation includes the VvMYBA1 gene (homologous to TCM_019192), VvMYBA4 (homologous to TCM_019261), and VvMybA2. Mutations within VvMybA1 or VvMybA2 cause berries to be pigmented white/green instead of the wild-type purple color [52].
As described above, the MP01 recombinants narrow the region of interest to genes located between 20,562,635 and 21,126,449 bp on chromosome 4 (Figure 4c). Within this region, only one of the two MYB transcription factors is present: TCM_019192, which is homologous to VvMybA1. We refer to this gene as TcMYB113, and it encodes a protein with 275 amino acids sharing 61% identity with grape VvMYBA1 (see Additional file 2, Figure S8 for protein-sequence comparison). In the Criollo genome [11], TCM_019192 is annotated as two genes: Tc04_t014240 and Tc04_t014250. This difference may be attributed to the strict evidence-based Matina 1-6 version 1.1 annotation procedures that we used (see above).
Resequencing of additional genotypes and haplotype phasing of resequenced data
The availability of a high-quality reference genome greatly facilitates resequencing data analyses of additional genotypes. Five parents of the mapping populations (MP01: CCN 51 and TSH 1188,; T4 Type 1: Pound 7 and UF 273 Type 1; T4 Type 2: Pound 7 and UF 273 Type 2), and eight additional genomes (KA 2 101, K 82, LCTEEN 141, NA 331, PA 51, mvP 30, mvT 85, and Criollo 13) were resequenced (see Materials and methods). Reads were phased for the region surrounding the pod-color mapping interval (between 20,561,460 and 21,386,830 bp) on chromosome 4 (see Materials and methods). We identified 21,225 SNPs and 1,879 putative insertions/deletions (indels) within this region. Phasing of the reads permitted generation of haplotype sequences and, consequently, identification of the alleles associated with pod color, specifically for the three candidate genes.
The phased resequencing data from 13 genotypes plus the Matina 1-6 (version 1.1) sequence were used to generate clusters based on the haplotypes for the two MYB genes and APO4 (see Additional file 2, Figure S9). The haplotypes for TcMYB113 appear to cluster in agreement with the haplotype they induce or the haplotype of the phenotype of the clones they represent; this is not the case for TcMYB6 or APO4, in which green and red haplotypes are positioned within the same cluster (see Additional file 1, Figure S9). These results corroborate the association between TcMYB113 and pod-color variation across multiple resequenced unrelated samples with diverse genetic backgrounds. The phylogenetic relationships between the haplotypes also indicate that the haplotypes of the Criollo 13 green-pod genotype are closest to the cluster of red-pod-associated haplotypes (see Additional file 2, Figure S9). This suggests that the TcMYB113 alleles that are associated with red pod color in this study originated from the Criollo genetic group. In fact, only a single allele was found for MYB6 and APO4 in the red pod-associated haplotypes from the mapping population parents (see Additional file 2, Figure S9), but their sequence is identical to the one from the resequenced green-pod Criollo 13. This indicates that the red-pod parents of the mapping populations may have inherited a large segment of chromosome 4 from a Criollo genotype, and in fact, the Criollo genetic group was previously identified as the most likely source of the red-pod gene [7].
Sanger sequencing, association mapping, and putative functional effects of polymorphisms found within TcMYB113
The resequencing data shows specific SNPs in TcMYB113 within coding regions in the alleles associated with red pod color, which are at positions 20,878,747, 20,878,891, 20,878,957, 20,879,122, and 20,879,148. We corroborated this result by Sanger sequencing of the three candidate genes studied and by association mapping (see Additional file 1, Table S23). The association-mapping analysis was performed using 73 SNPs generated via Sanger sequencing (see Materials and methods) and 95 other SNPs from chromosomes 1 to 10 in 54 genotypes with green pods and 17 genotypes with red pods from diverse genetic backgrounds. Without accounting for genetic structure, the most significant P-values from the Fisher test were for the following SNPs (from highest to lowest significance): 20,878,891; 20,875,691, and 20,879,148 (see Additional file 1, Table S23). SNPs at these positions permitted differentiation of alleles that are associated with the green-pod Criollo genotypes from those associated with the red-pod trees of Criollo origin (see Additional file 2, Figure S10). The least significant of these (position 20,879,148; see Additional file 1, Table S23) results in an amino-acid change from serine to asparagine at codon 221 of the TcMYB113 protein in the alleles associated with red pod color. This residue occurs outside of the R2R3 DNA binding domain, but within the C-terminal region that varies substantially between MYB family members, making the structural consequence of this substitution difficult to predict. When genetic structure was taken into account in the association-mapping analysis, the significance of the association between this SNP and pod color was considerably lower (see Additional file 1, Table S23). The second most significant SNP (position 20,875,691) was detected via Sanger sequencing in the TcMYB113 5' untranslated region (UTR), located 25 bases upstream of the ATG start site (see Additional file 1, Table S23). The function of this SNP is difficult to infer; however, mutations occurring near translation start sites are known to affect protein-translation rates [53]. The SNP that was most significantly associated with pod color (position 20,878,891) is a synonymous mutation found within the coding region of TcMYB113. Intriguingly, this SNP is positioned within a target site for a dicot trans-acting small interfering RNA (tasiRNA) derived from TAS4 (TRANS-ACTING siRNA 4) TAS4-siR81(-) as identified by Luo et al. [54] (Figure 5; see Additional file 2, Figure S8). TAS-derived siRNAs post-transcriptionally downregulate protein-coding transcripts in a manner similar to microRNA (miRNA)-directed repression [55,56]. MYB113 regulation in Arabidopsis additionally involves miR828, which acts both to cleave TAS4 to generate the interfering small RNA TAS4-siR81(-) [57] and also, independently, to silence MYB113 [54]. We identified not only the TAS4-siR81 site, but also a conserved miR828 target sequence within TcMYB113 (Figure 5), suggesting that these regulatory mechanisms are highly conserved in cacao. However, we detected no polymorphism within the miR828 target sequencewhich overlaps with the highly conserved R3 domain (Figure 5). The activities of both TAS4 and miR828 ultimately regulate anthocyanin biosynthesis through MYBs in Arabidopsis [54]. Conservation of and natural variation in this regulatory loop is yet to be explored in other plants.
Figure 5.
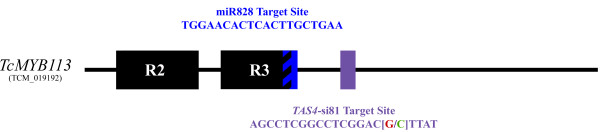
mir828 and TAS4-siR81 (-) sequence targets in TcMYB113. The green base pair indicates the single-nucleotide polymorphism (SNP) (20,878,891) that was most significantly associated with pod-color variation (C is associated with green pods and G is associated with red pods).
Resequencing data of the region 4 kbp upstream of TcMYB113 also revealed SNPs associated with the red pod-inducing haplotypes we had identified (see Additional file 2, Figure S11). However, because these SNPs are also present in the resequenced green-pod genotype Criollo 13, these mutations in the putative region containing the promoter of TcMYB113 are unlikely to be functionally associated with the red-pod phenotype.
Real-time quantitative PCR analysis of candidate genes
Pigmentation intensity has previously been correlated with variation in MYB transcript level [50,58,59]. In parallel with our genetic analyses, we quantified the accumulation of candidate-gene transcripts to assess their involvement with pod color. RNAs from developing young fruits (cherelles) of Pound 7 (a green-pod genotype) and UF 273 Type 1 and Type 2 (red-pod genotypes) were analyzed by quantitative (q)PCR, to compare the relative transcript levels of the T. cacao MYB113, AP04 and MYB6 genes. Relative expression was normalized to leaf cDNA generated from Pound 7. The TcAPO4 and TcMYB6 genes each showed decreased expression in all samples relative to expression in leaf, but there were no differential gene-expression patterns that correlated with pod color. By contrast, TcMYB113 expression in the small cherelle samples for all genotypes showed a modest increase in expression relative to the Pound 7 leaf (Figure 6a). This suggests that TcMYB113 gene expression might be tissue-specific, because anthocyanin accumulates in young leaves, including those of Pound 7 [7]. We found TcMYB113 transcript accumulation to be correlated with color intensity in a variety of samples tested. TcMYB113 transcript levels in large UF 273 cherelles (a clone with red pod color ), were 25-fold higher than that in large Pound 7 cherelles (a clone with green pod color), and 250-fold higher than that in Pound 7 leaves (Figure 6a). Notably higher transcript levels were detected in large cherelles relative to small cherelles (Figure 6a), and this correlated with the increase in pigmentation intensity during the early stages of fruit development.
Figure 6.
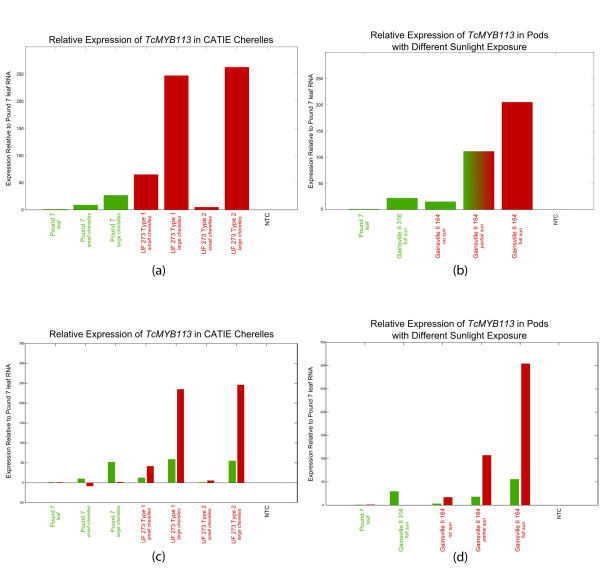
TcMYB113 transcript levels determined by quantitative PCR analysis of RNA from the pericarp of cherelles (young Theobroma cacao fruits) of genotypes with green or red pod color (genotype names are colored accordingly). Bar colors indicate the color of (a,b) the cherelles and (c,d) the alleles analyzed Although standard deviations were calculated, the values obtained were too low to graph at the scale used below. All RNA levels are normalized to control RNA from Pound 7 leaf tissue. (a) TcMYB113 transcript levels in the pericarp of small (10 to 20 mm length) and large (30 to 50 mm length) cherelles of the green-pod genotype Pound 7 and the pericarp of the red-pod genotypes UF 273 Type 1 and Type 2. (b) TcMYB113 transcript levels in the pericarp of green cherelles of the green-pod genotype Gainesville II 316 and the pericarp of green, green plus red. and red cherelles of the red-pod genotype Gainesville II 164. Green cherelles from Gainsville II 164 were obtained from the completely shaded part of the tree sampled. (c) Allele-specific TcMYB113 transcript levels in the pericarp from small (10 to 20 mm length) and large (30 to 50 mm length) cherelles of the green-pod genotype Pound 7 and the pericarp of the red-pod genotypes UF 273 Type 1 and Type 2. (d) TcMYB113 allele-specific transcript levels in the pericarp of green cherelles of the green-pod genotype Gainesville II 316 and inthe pericarp of green (no sun), green plus red (partial sun), and red (full sun) cherelles of the red-pod genotype Gainesville II 164. Green cherelles from Gainsville II 164 were obtained from a completely shaded area of the tree sampled. Red bars in the green-pod Pound 7 is due to background fluorescence.
Interestingly, recent miRNA annotation of apple [60] showed miR828 expression only in flowers and fruit, and not in other tissues examined, suggesting that modulation of MYB expression through non-coding RNAs might be a tissue-specific aspect of conserved anthocyanin biosynthesis regulation.
Because anthocyanin accumulation is influenced by sunlight [58], we also measured transcript levels of TcMYB113 in a genotype (Gainesville II 164) that has red pods at maturity, but exhibits both green and red pigmentation during fruit development according to light exposure. Green cherelles were collected from the shaded branches of a Gainesville II 164 tree, and red cherelles were collected from an area of the same tree that was exposed to sunlight. TcMYB113 transcript levels in Gainesville II 164 were 200-fold and 100-fold higher (relative to the transcript levels in Pound 7 leaf) in the red and the mixed greenred cherelles, respectively, compared with the fully shaded green cherelles (25-fold increase relative to Pound 7 leaf) (Figure 6b). Gainesville II 316 (a green-pod genotype) that had been exposed to full sunlight was used for comparison, and its transcript levels (20-fold increase relative to leaf) were comparable with those of the Gainsville II 164 green cherelles. Together, these results further corroborate the involvement of TcMYB113 in determining pod color in cacao, and are a first indication that anthocyanin production in response to light in pods is mediated by TcMYB113 accumulation.
Given the correlation between differences in TcMYB113 transcript levels and the pod-color variation seen in both qPCR experiments, as well as the association-mapping results presented above, we hypothesize that TAS4 siRNA downregulates TcMYB113 transcripts, which results in pod-color variation. Annotation of TAS and miRNA complements in cacao will require further computational analysis and additional small RNA sequencing, as recently reported for apple [60]. However, we have recently localized a homolog of the conserved TAS4 TCM_019486, also on chromosome 4. An expressed sequence tag from this gene was previously reported [54], and we have directly confirmed expression of this gene and the presence of the siR81 processed segment in small RNA isolated and sequenced from cacao flowers (data not shown).
To further confirm that TAS4 siRNA plays a role in the downregulation of TcMYB113 transcripts, we repeated the experiments described above, but this time using primers specific to the green and red alleles (Figures 6c,d). cDNA from small and large pods of the T4 Type 1 mapping population parents were used as template, as described for the experiments mentioned above. Red-colored bars in the figure represent relative steady-state expression of the red allele, and green bars represent expression of the green allele. These results show a five-fold increase in the expression of the red allele over the green in samples with red pods (UF273 Type 1 and Type 2), whereas in green-colored pods, very little expression of either allele is present. Cumulative expression of both alleles is consistent with the previous expression levels seen for TcMYB113.
Allele-specific transcript levels were also measured in the Gainsville II clone with varying exposure to sunlight. The green-pod Gainsville II 316 showed no steady-state expression of the red allele, and its green-allele expression was similar to that seen in Figure 6d. For the red-pod Gainsville II 164, expression of the red allele was consistently higher than that of the green, and increased with increasing exposure to sunlight. Additionally, green-allele accumulation in this clone, although significantly smaller than that of the red allele, still increased with increasing exposure to sunlight. This suggests that TcMYB113 expression is regulated in response to sunlight exposure.
Together, these experiments further support our hypothesis that the variant in the TAS4-si81 target site on the red allele prevents TAS4 siRNA downregulation of TcMYB113, and allows the accumulation of the TcMYB113 red-allele transcript, whereas the green-allele transcript is degraded.
Conclusions
The high-quality genome sequence of the Matina 1-6 clone (version 1.1 [61]), combined with phenotypic data from the mapping populations, haplotype phasing, phased sequence data from the parents of the mapping populations, and qPCR expression analysis, enabled identification of the T. cacao transcription factor gene TcMYB113 (gene model TCM_019192) as a strong candidate for the gene regulating pod color in cacao. When pod color was evaluated as a qualitative trait, specific SNPs were associated with the absence of anthocyanin in green pods. The correlation between higher transcript levels of TcMYB113 gene and greater red pigmentation in the pericarp suggests that differences in the activity of this gene are likely to explain green/red color differences in cacao pods. This hypothesis is further substantiated by our identification of sequences within TcMYB113 that are indicative of regulation by a highly conserved small RNA network that is involved in anthocyanin biosynthesis across dicot species [54]. One SNP, position 20,878,891 on chromosome 4, which falls within the target site for a highly conserved tasiRNA in dicots: TAS4-siR81(-), is proposed to regulate the degradation of the green-allele transcript and therefore pod-color variation. It would be interesting to explore if siRNA allele-specific degradation also drives the regulation of fruit color in other plants.
The SNPs linked to green pod color can now be added to our set of molecular markers for utilization in MAS strategies [62]. For example, these new markers could be used to positively select for high-yielding, high-aroma, green-pod genotypes in crosses between CCN 51 and cacao clones with desirable aroma profiles. Demonstration of the association of TcMYB113 with pod color illustrates that when the appropriate populations are evaluated and the correct analysis is implemented, it can be straightforward to establish a bridge between a particular genome sequence and a specific phenotypic trait. For highly conserved traits such as anthocyanin pigmentation, for which the MYB transcription factors have been shown to play a major role in Rosaceae [50], Solanaceae [48], Brassicaceae [49] and now Malvaceae, association mapping alone might have provided similar results to those presented here. Nevertheless, we expect other traits to have a more complex pattern of shared variation. For example, we have also been studying self-incompatibility (SI) [15], which is sporogametophytic in T. cacao [63]. This type of SI is not exhibited by any other sequenced plant species.
Within one genetic region that is known to determine cacao SI [15], we detected no homology to gene families that are known to regulate SI in other species [64]. Consequently, for genes that regulate traits more specific to T. cacao (or to related species with insufficient genomic resources), implementing the strategy presented here will be a powerful way to identify candidate genes that regulate crucial oligogenic traits. The key advantage of sequencing an entire genome is that every trait under study will benefit from the genomic and genetic resources already generated (for example, reference genome sequence, resequenced genotypes, and mapping populations), and no significant additional investment will need to be made to identify additional genes or markers for MAS. We are now well poised to implement comparable genetic analyses to identify other genes involved in crucial traits in T. cacao.
Materials and methods
Genome size estimation
T. cacao genome sizes were estimated using a modified version of the FCM protocol described in [65] and the Accuri C6 flow cytometer manual. We used the soybean Glycine max (L.) Merr. var. Williams 82 (G. max W82) as an internal reference standard in all of our FCM runs. G. max W82 has been sequenced [66] and its estimated haploid genome size of 1.1 Gb is sufficiently similar (approximately twice the size) to the expected range of T. cacao genome size values so as to ensure the accuracy of our measurements [67]. T. cacao leaves from most of the studied genotypes were obtained from trees at the US Department of Agriculture Subtropical Horticultural Research Station (Miami, FL, USA). Leaves from genotypes representing the Nacional structural group (Dr Rey Gaston Loor, Instituto Nacional Autónomo de Investigaciones Agropecuarias, Quevedo, Ecuador and the Criollo group cultivar B97-61/B2 )Cocoa Research Unit, University of West Indies, Trinidad and Tobago) were also obtained; Harvested leaves were stored at -80°C until analysis. For each FCM sample run, 100 mg of leaf tissue from a given T. cacao genotype and 100 mg of G. max W82 leaf tissue were finely chopped together, using a double-sided razor (Gillette Super Platinum; Boston, MA, USA ), in 2 ml of woody plant buffer (WPB; [68]) in Petri dishes on ice. Samples were then filtered through a 30 μm filter (CellTrics®; Partec GmBH, Munster, Germany) into a 1.5 ml microcentrifuge tube. To 500 µl of this nuclear suspension, RNase A (Fermentas/Thermo Fisher Scientific Inc., Rockford, IL, USA ) was added to a final concentration of 50 μg/ml and samples were incubated on ice for 30 minutes. Propidium iodide (Sigma-Aldrich, St Louis, MO, USA) was then added to a final concentration of 50 μg/ml [68], and samples were incubated on ice for another 30 minutes.
Samples were then run on a flow cytometer (Accuri C6; BD Biosciences Inc., San Jose, CA, USA). Three technical replicates and one to five biological replicates (representing different leaves from the same tree, based on leaf availability) were analyzed for each genotype. Data were analyzed using FCS Express 4 (Research Edition 2011; De Novo Software, Los Angeles, CA, USA). The T. cacao and G. max W82 peaks were assessed to obtain the 2C mean fluorescence value for each; the peak coefficient of variation was below 5% for all samples. Once the mean fluorescence was obtained, the 2C value for each genotype was determined as described previously [69]. Briefly, the ratio between the 2C mean sample (T. cacao) and 2C standard mean reference (G. max W82) values was calculated and then multiplied by the soybean estimated diploid genome size (2C = 2,230 Mb) [70]. Statistical analyses were conducted using SAS software (version 9.2; SAS Institute, Cary, NC, USA [71]). Analysis of variance (ANOVA) and least squares means analyses were performed using the general linear model (Proc GLM of the SAS software) to estimate genome sizes by cultivar and structural groups, and the post hoc Tukey-Kramer test at 5% significance was used for separation of means.
Mapping populations for the genome assembly
To aid in assembly of the Matina 1-6 genome sequence (see below) three genetic-mapping populations were used to map 5,214 SNPs using anInfinium chip (Illumina Inc., San Diego, CA, USA) and JoinMap® software (version 4.1; Kyazma BV, Wageningen, the Netherlands) [72]. The first mapping population, MP01, is located at the Mars Center for Cocoa Sciences (MCCS; Itajuípe, Bahia, Brazil). It was created by crossing TSH 1188 with CCN 51, and originally contained 598 progeny trees planted in 2004. In total, 3,251 SNP markers, using 461 trees in this population, were mapped. The second mapping population, T4 Type 1, is located at the Tropical Agricultural Research and Higher Education Center (CATIE; Turrialba, Costa Rica), and has been described previously [14,15]. It comprises the progeny of a cross between Pound 7 and UF 273 Type 1. In total, 3,014 SNPs, using 180 trees (Type I) from this population, were mapped. The third population, derived from a KA 2-101 by K 82 cross, is located at the Coconut and Cocoa Institute (Papua New Guinea)A total of 2,534 segregating SNPs, using 340 trees in this population, were used to create the genetic map.
SNP genetic maps
SNP identification was carried out as described previously [24]. Briefly, leaf RNA samples isolated from each of 15 members of a diversity panel of cacao genotypes were sequenced on the GAII platform (Illumina) and aligned to the T. cacao Matina 1-6 leaf transcriptome [24]. A variant report was generated that displayed all SNPs identified from the 15 genotypes, and standard filtering protocols were applied [24]. The SNPs were further filtered before being selected for inclusion on a custom SNP chip (Infinium; Illumina). Passing the additional filter required an Illumina Infinium Assay Design Tool score of greater than 0.9, a minor allele frequency of greater than 0.2, and heterozygosity for that SNP in at least one genotype in the diversity panel. In addition, only single bead-type SNPs (A/C, A/G, T/C, T/G) were chosen; where possible, two SNPs per transcript locus were selected. The final list of 6,000 SNP sequences was submitted to Illumina for Infinium SNP Chip production and subsequent genotyping. After validation by Illumina, a total of 5,214 bead types remained, which were then used to genotype the mapping populations. For details on DNA sample preparation for SNP fingerprinting, see Additional file 1.
All members of the three mapping populations described above were genotyped for the 5,214 SNPs, but not all of the SNPs segregated in the mapping populations. The genetic linkage maps were created using JoinMap® version 4.1 [72]) We excluded from further analyses any SNPs showing segregation distortion (χ2 values greater than 10), SNP loci that were absent from more than 10% of the trees, and any individual trees missing more than 10% of the SNP markers. The resulting SNP genotype data were transformed manually to produce the final maps using JoinMap version 4.1 [72] with the maximum likelihood (ML) mapping algorithm and default settings. These genetic maps and the physical map of T. cacao Matina 1-6 [19] were used to order the sequence assembly and to map pod color after haplotype phasing of chromosome 4 (see below).
Mapping populations for pod-color mapping
Phenotypic data for pod color, described as a binary green versus red trait, were collected in the MP01 and T4 Type 1 mapping populations mentioned above (see Additional file 2, Figure S6). An additional population issued from a cross between Pound 7 and UF 273 Type 2 (called T4 Type 2) was also studied; this comprised 68 trees (see Additional file 2, Figure S6), which were successfully fingerprinted using the same type of Illumina Infinium chip described above. For the total number of markers and trees used for genotype and haplotype (chromosome 4) mapping in these populations, see Additional file 1 (Tables S24-S26).
Genome sequencing
DNA was isolated from Matina 1-6 leaf nuclei as reported previously [24], and prepared into a variety of libraries for whole-genome shotgun sequence (WGS), and for cloning for BAC and fosmid end-sequencing [19,25].
For the bulk of the shotgun data, we used pyrosequencing (454 Life Sciences/Roche, Bradford, CT, USA) of both linear and paired libraries. Linear libraries included 7.3 times the assembled coverage from the GS FLX Titanium XL platform (454 Life Sciences) and 8.23 times the assembled sequence from GS-XLR (454 Life Sciences) reads. We sequenced eight paired recombination libraries (see Additional file 1, Table S4) on a GS-XLR instrument (454 Life Sciences/Roche), which comprised four libraries with an insert size of 3 kb, one with an insert size of 6 kb, and three with an insert size of 8 kb; fter removal of duplicate paired reads, these three sets yielded respectively 2.5, 0.9 and 2.3 times the assembled sequence coverage.
We also sequenced the insert ends of three fosmid libraries and three independent BAC libraries to add an additional 0.4 times sequence coverage from the Sanger platform in order to increase the contiguity of the assembled WGS sequence. Methods detailing BAC selection for end-sequencing have been described previously [19]. Briefly, a dense array of BACs for end-sequencing was selected using an in-house Perl script to iterate through the T. cacao physical map contigs and to select BACs with flanking ends that were approximately 7 kbp apart. BAC DNA was purified after standard alkaline lysis miniprep methods [73], and used as template for BAC end-sequencing. The BAC end-sequencing reactions were performed in 96-well format plates using a commercial product (BigDye, version 3.1; Applied Biosystems, Foster City, CA, USA), and the flanking ends were sequenced using the T7 and M13 reverse priming sites located on the BAC vector. DNA sequence was collected on a genetic analyzer (ABI3730xl; Applied Biosystems). Preparation and sequencing of the 998 kbp Sanger reference genomic segment has been described in detail elsewhere [25] (also see Additional file 1, Supplemental Materials and methods). [For details of the preparation and end-sequencing of fosmids, see Additional file 1 Additional materials and methods.
Genome assembly
In total, 32,460,307 sequence reads (for clone size breakdowns, see Additional file 1, Table S4) were assembled using a modified version of Arachne (version 20071016 [23]) with parameters maxcliq1 250, lap_ratio = 0.8, max_bad_look = 2000 and BINGE_AND_PURGE = True ( for scaffold and contig total, see Additional file 1, Table S4). A subsequent filtering step was applied to remove contigs with less than 3 reads and those shorter than 150 bp. This produced 1,672 scaffold sequences with a scaffold L50 of 7.4 Mb and 91 scaffolds greater than 100 kb, for a total scaffold length of 348.7 Mb. We identified contaminants using Megablast [74] to screen against Genbank NT, and blastx [74] to screen against a set of known microbial genes. Scaffolds were also removed if they consisted of repetitive sequences (greater than 95% 24-mers that occurred four or more additional times in scaffolds larger than 50 kb; total of 267 scaffolds), contained only unanchored rDNA sequences (1 scaffold), contained mitochondrial or chloroplast DNA (3 scaffolds), or were less than 1 kbp in length (634 scaffolds).
Map integration and chromosome-scale pseudomolecule construction was carried out using a combination of marker maps, BAC/fosmid joins, and additional information related to specific genes in functional regions. Markers from the genetic maps were aligned to the assembly using BLAT [75], and the best placement, based on base-pair identity and marker coverage, was selected to position the marker. Scaffolds were broken if they contained linkage-group discontiguity coincident with an area of low BAC/fosmid coverage. Initially, three separate sets of joins (one for each marker map) were generated using an automated chromosome construction algorithm. Briefly, the goal of the automated process is to attempt to find the best order using the marker position midpoints for each scaffold; orientation is then generated using the map locations on the scaffold. Consensus between the three maps was assessed, and in the event of a conflict between two or more maps, priority was given to the MP01 map. One additional scaffold was added with the information provided by the functional region genes. Synteny between the T. cacao Matina 1-6 genome and the T. cacao Criollo genome (see below) sequences was used to fine-tune the breakpoints. In total, 47 breaks were identified, and a subset of the broken scaffolds was combined using 103 joins to form 10 pseudomolecule chromosomes. Map joins are denoted with 10,000 Ns. All scaffold orientation was reliably determined using either BAC/fosmid joins or map positions; significant telomeric sequences at the end of the chromosomes (TTTAGGG repeats) were correctly oriented in the final assembly by adjusting scaffold orientation, if necessary. Plots of the marker placements for all three maps and the synteny between exons in the T. cacao Matina 1-6 and Criollo genomes were created (see Additional file 2, Figure S2). Exons (46,140) were extracted from previous work [76] for this comparison. The pseudomolecules contained 346.0 Mbp (99.2%) of the total 348.7 Mbp of the assembly. The final assembly contained 711 scaffolds covering 346.0 Mbp of the genome with a contig N50 length of 84.4 kbp and a scaffold N50 length of 34.4 Mbp.
Gene annotation and orthology analysis of the Matina 1-6 genome
RNA was isolated from T. cacao Matina 1-6 leaves, beans, and pistils, and used to prepare long normalized or short-paired read libraries, which were then sequenced to build independent datasets of expression evidence that were used for genome annotation (for details of the read sets, see Additional file 1, Tables S9 and S10; each has been deposited in the NCBI SRA within BioProject 51633).
Approximately 7 million reads from the 454 Titanium sequencing and approximately 1 billion paired-end reads from the Illumina sequencing were pre-assembled as separate evidence sets from leaf, bean, and floral collections, and mapped to the Matina 1-6 genome reference. Transcriptome assemblies from each collection were deposited in the NCBI TSA within BioProject 51633. Assemblies of shorter reads (54 to 112 nucleotides (nt)) were combined with those of longer reads (average 300 to 450 nt), and final transcript assemblies were refined using PASA [77].
Plant protein sequences from Arabidopsis thaliana [78], V. vinifera [79], Populus trichocarpa [80], G. max [81], Ricinus communis [82], Fragaria vesca [83], Solanum tuberosum [84] and Sorghum bicolor [85] (a total of 297,061 protein sequences) were aligned using tblastn (P≤ 1 × 10-5) [74] to a repeat-soft-masked version of the T. cacao Matina 1-6 genome assembly, and then refined using Exonerate [86] to yield putative protein-encoding gene models. Local alignments of the same gene were joined and extended to the best-matching complete gene alignment using a previously published method [86]. Mapped transcript assemblies, protein-coding gene models, and ab initio predictions were combined to form several preliminary annotation sets; the model with the best evidence, based on a consensus among three or more plant protein-coding gene models, was selected for each locus. The ab initio predictions were primarily made using AUGUSTUS software [87], which was trained for T.cacao gene parameters using full-length Matina 1-6 leaf cDNAs assembled de novo (cacao genome version 0.9) [61]. Models were corrected for any coding sequence (CDS) errors found, and some UTRs were extended using PASA software [77]. If a minimum model length overlap with RNA sequencing or a protein homolog evidence alignment was 66% or greater, the gene model was classified as 'strong'. Models with lesser support were retained with the assumption that they might improve with additional evidence. Classes categorized as 'medium' and 'weak' represented minimum evidence overlaps of 33% and 5%, respectively.
UniProt [27] reference plant genes, a non-redundant set of 345,650 proteins representing 2,365,369 plant genes from 14,970 species, was used for annotation, in addition to the full proteomes from the 8 plant species in addition to Matina 1-6 mentioned above. Analysis of orthology among the nine plant gene sets was performed using OrthoMCL [29] For further details, see Additional file 1 (sections 3.5 to 3.7).
HMMER3 [32] was used to search cacao proteins against the profile HMM databases Pfam and TIGRFAMS, using a maximum sequence E-value of 0.005 and retaining only matches above the HMMER3 noise cut-off point. The set of HMM families was reduced to one instance per gene by retaining only the longest CDS of each gene model and confirming no loss of data when alternative transcript CDS were removed. HMM family counts that were found among 4,085 proteins with RNA evidence but with no ortholog called among 8 whole-plant genomes (see Materials and methods) were compared with those of the complete Matina 1-6 T. cacao gene annotation and grouped as described in Additional file 1, Table S14d.
Identification and annotation of transposable elements
TEs were categorized based on previously described criteria [88]. Structure characteristics and sequence homology were used for identification and annotation as described previously [66]. De novo identification of LTR retrotransposons was first determined by integrating results from the programs LTR_STRUC [89] and LTR_FINDER [90]. After manual inspection, confirmed intact elements were then used to search against the entire T. cacao Matina 1-6 genome, in the manner described by Ma et al. [91], in order to identify additional intact elements and solo LTRs. Classification of superfamilies (copia-like, gypsy-like, and unclassified retro-elements) and families relied on homology with reverse transcriptases and 5' and 3' direct LTRs. Long interspersed nuclear elements and several DNA transposons (including Tc1-Mariner, hAT, Mutator, PIF-Harbinger, and CACTA) were identified by a previously described two-step strategy [92]. Autonomous elements were first identified using BLASTX [74] to search the assembled genome sequence for entries in the Repbase [93] protein database. The upstream and downstream sequences of the candidate hits were then extracted to clear boundaries and to define the terminal inverted repeats (TIRs) and target site duplications. The second step was to use the TIRs from the autonomous members to detect non-autonomous elements. Hilitron elements were identified by the HelSearch 1.0 program [94] and by manual inspection. Miniature inverted-repeat TEs (MITEs) were identified by using two previously published methods, MUST [95] and MITE-Hunter [96]. Elements typical of each category were selected and mixed together as a database for RepeatMasker [97], which was then used to find additional truncated fragments.
Synteny between the Matina 1-6 and Criollo genomes
Comparison of the whole-genome sequences of Matina 1-6 (version 1.1) and Criollo (version 1.0 [11]) was carried out using the Mercator program [98]. This program identifies orthologous regions (ORs) using BLAT-similar anchor pairs in a modified k-way reciprocal best-hit algorithm, and produces matches of a single region in the Matina 1-6 genome to a single orthologous region in the Criollo genome. The individual ORs identified using Mercator and aligned by MAVID [99] can be searched and explored using the graphic synteny viewer, GBrowse_syn [100] in the Cacao Genome Database [101].
Preparation of T. cacao Matina 1-6 chromosomes and FISH karyotyping
Mitotic chromosomes from T. cacao seedling root tips were prepared using a combination of methods for efficient release of metaphase chromosomes from their cytoplasmic milieu [102], efficient mitotic arrest [103], and FISH [18]. After beans were extracted from two or three pods, the seed coats were manually removed using a scalpel. The beans were then surface-sterilized by two sequential treatments in a 20% solution of commercial bleach (5.25% sodium hypochlorite) for 10 minutes each, followed by three rinses in sterile distilled, deionized (SDD) water. Beans were then treated in 10 mmol/l hydrochloric acid, again followed by three rinses in SDD water, and were then planted in previously sterilized, 1-liter plastic beakers containing perlite soaked in a 1× solution of all-purpose plant food (Miracle Gro®; Scotts, Marysville, OH, USA. The beakers were covered with aluminum foil and incubated for 1 to 3 weeks at around 28°C in a greenhouse or incubator. Chromosome spreads were prepared from root tips using a protocol that we developed (see Additional file 1, Materials and methods) by combining and modifying two previously described methods [102,104]. For details on chromosome preparations for FISH, see Additional file 1. FISH was carried out precisely as described by Gil et al. [104], except that 100 ng/µl of sheared salmon sperm DNA (Stratagene Corp., La Jolla, CA, USA) was used for the slide-blocking step.
Haplotype mapping
To infer haplotypes from genotype data, we took the consensus phasing results from 101 runs of HAPI-UR (window size of 75 and Ne of 1000) [43]. HAPI-UR does not explicitly relate progeny haplotypes to their respective parental haplotypes. Thus, to assign the haplotype of each progeny to its respective parental haplotype (or haplotypes, if the progeny was recombinant), we first identified which parental haplotype each progeny was most similar to, based on the number of pairwise nucleotide differences (Hamming distance) between them. Each one of the two haplotypes from each progeny was assigned to whichever one of the four parental haplotypes showed the shortest distance of the four pairwise comparisons. There were no instances of the two haplotypes of a progeny being assigned to the same parent. Next, parental assignments for recombinant haplotypes (that is, where the distance to any given parent haplotype was >0) were resolved by identifying which of the two parental haplotypes was represented in the progeny haplotype at each heterozygous marker. Parental assignments at homozygous markers were then inferred by examining the assignments made at the closest heterozygous positions to either side. If the two closest heterozygous assignments agreed, then that assignment was used at the homozygous position; if not, then no assignment was made. By using this method, we implicitly sought to minimize the number of recombination events between the two parental strands as represented in each recombinant progeny haplotype. Parental assignments identified in this way were used in the haplotype-phenotype association test (Figure 3) and to identify haploblocks from the four parental haplotypes on recombinant progenies (Figure 4). We also resolved the haplotypes and haploblock (parental) assignments using additional software (iXora [105]) developed by our group and obtained identical results. iXora is a phasing and trait association method that allows precise inference of haplotypes of F1 progeny from mapping and breeding populations derived from non-inbred parents.
Statistical tests for genotype-phenotype and haplotype-phenotype association
To test the statistical significance of genotype-phenotype and haplotype-phenotype associations, we applied Fisher's exact test independently for each marker using SAS software (SAS Institute, Cary, NC, USA) [71]. The input to the test is a contingency table in which columns denote the phenotypes (red or green pods), and rows denote the genotypes or the haplotypes. We applied the test for the genotypes at each SNP marker from the 6K SNP chip, resulting in a 3 × 2 or 2 × 2 contingency table (see Additional file 2, Figure S7), and also for each parent (using both haplotypes), resulting in a 2 × 2 contingency table per marker for each parent (Figure 3). The test on the parents separately demonstrates each parental effect on the phenotype.
Thresholds for significance were calculated using Bonferroni correction for multiple comparisons at α = 0.05.
Resequencing the parents of the mapping populations and additional genotypes
The parents of the mapping population plus eight other genotypes were sequenced as paired libraries to 112 × 2 nt in length using Illumina GAIIx to a median coverage ranging from 15 to 29 fold per individual genome. Prior to mapping, reads were assessed for overall quality; median base composition and median quality plots along the reads were used to determine the usable parts of the reads. We hard-trimmed the reads before mapping in order to eliminate positions at the beginning of the reads with biased base composition, and at the end of the reads if the median quality fell below 25 (Phred-like quality scores). We mapped the reads to the Matina 1-6 (version 1.1) genome using bwa [106], with dynamic trimming to drop sections of the reads with qualities lower than 25 (Phred-like quality scores). The mapped reads were analyzed to remove PCR duplicates, and indexed for further analyses using Samtools (version 0.1.17) [107]. Local realignment and base-quality recalibration were performed on indexed BAM files, using GATK [108], following best-practice recommendations [109]. The aligned reads in the interval corresponding to the 0.5 Mbp regionto which pod color was mapped using SNP array data were merged and extracted, and then calls were made on that specific region using the Unified Genotyper routine of GATK [108].
Phasing of the resequenced data
We identified haplotypes for the region associated with pod color using the algorithm implemented in the program fastPHASE [110]. In this model, the different haplotypes identified in a given population are assumed to emanate from a small number (K) of possible haplotype clusters. Thus, every haplotype can be represented as a mosaic of small haplotype blocks sampled from each of the clusters. This discretized structure is modeled as an HMM with K states.
The algorithm proceeds in two steps. First, the parameters driving the emission probabilities (probability that a given allele 1 will be present in a given haplotype cluster) and the transition probabilities (probability of transitioning from one haplotype cluster to the next) are estimated using an expectation-maximization algorithm. Second, the most likely configurations of adjacent heterozygous sites are obtained through a Monte Carlo simulation in which possible diplotypes are sampled based on their relative likelihoods, as estimated by the forward-backward algorithm of the HMM. Using the phased SNPs, we recreated the alleles for the candidate genes from the resequenced data, and generated FASTA files.
Phylogeny of candidate genes
The phylogenetic relationships between alleles were inferred under ML. The translated amino-acid sequences were used, and the reconstruction made under a Jones-Taylor-Thornton model of amino-acid substitution with 500 bootstrap replications, using the program MEGA (version 5; [111]).
Association mapping for pod color variation
Association mapping was performed on 54 green-pod and 17 red-pod genotypes fingerprinted through Sanger sequencing and the Fluidigm EP-1 platform (192.24 platform; Fluidigm Corporation, San Francisco, CA, USA). The Sanger sequence of the three candidate genes studied generated a total of 73 SNPs. A subset (n = 95) of the SNP markers used to generate genetic maps, distributed through the 10 T. cacao chromosomes, were converted to SNP-type assays and fingerprinted in accordance with the manufacturer's protocol, using the Fluidigm EP-1 platform. The Fisher's exact test was performed to test the association between pod color and genotypes at each of the 168 (73 + 95) positions. Population structure and hidden relatedness that are unaccounted for can lead to the identification of spurious signals of association between predictors (that is, SNPs) and response variables (phenotype of interest) [112,113]. Therefore, we performed association analyses while accounting for hidden relationships by estimating a kinship matrix between individuals using markers other than those on chromosome 4 that we had putatively identified as being associated with pod color in the mapping populations. We then used the kinship matrix to identify SNPs that were significantly associated with pod color, using a linear mixed model that incorporates the genetic relatedness estimated in our kinship matrix, as implemented in the EMMA package [114].
Real-time qPCR expression analysis
Leaf tissue from a Pound 7 cacao tree was collected directly into liquid nitrogen and stored at -80ºC before RNA extraction. Developing cherelles were collected from Pound 7, UF 273 Type 1, and UF 273 Type 2, and placed in liquid nitrogen until RNA extraction. Cherelles were considered small if they were 10 to 20 mm long, and large if 30 to 50 mm long. Cherelles from the genotypes Gainesville II 316 (green pods) and Gainesville II 164 (red pods) were collected, and RNA was isolated as above. Gainesville II 316 cherelles were exposed to full sunlight and were all green in color. Cherelles from Gainesville II 164 were collected from areas of the tree that were unexposed (green cherelles), partially exposed (green-red cherelles), or fully exposed (red cherelles) to sunlight. Prior to RNA extraction, the pericarp of cherelles was removed with a vegetable peeler; RNA was then extracted from 1 g of pericarp tissue using previously described methods [24,115]. RNA was quantified spectrophotometrically, and all samples were normalized to 800 ng/µl before they were reverse transcribed into cDNA (SuperScript® VILO™ cDNA Synthesis Kit Invitrogen, Carlsbad, CA, USA). The resulting cDNA was diluted eight-fold and used in qPCR reactions consisting of 12.5 µl SybrGreen MasterMix (ABI, Foster City, CA, USA), 2.25 µl 10 mmol/l forward primer, 2.25 µl 10 mmol/l reverse primer, 6 µl water, and 2 µl cDNA. Primers for qPCR were designed with Primer3 [116], ensuring that at least one primer of each primer pair spanned two exons (for primer sequences used for qPCR analysis, see Additional file 1, Table S27). Thermocycling was performed (7300 Realtime PCR system; ABI) using standard conditions (1 cycle of 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute). Ct values were determined and compared using the -ΔΔCt method [117]. qPCR amplifications were performed in triplicate and averaged. TcActin was used as the internal standard gene, and all values are presented relative to Pound 7 leaf expression. Real-time qPCR analysis was performed for the three candidate genes (TcMYB113, TcAPO4, and TcMYB6).
Allele-specific qPCR was performed for TcMYB113 using a TaqMan-based method. TaqMan probes specific to each allele were designed for the most significantly associated SNP marker (position 20,878,891), and primers (see Additional file 1, Table S27) flanking the probe region were designed using Primer Express (version 3.0[116]). TcActin was used as the internal standard gene, and all values are presented relative to Pound 7 leaf expression. To ensure continuity of the assay, a TaqMan probe was also designed to the actin control gene (see Additional file 1, Table S27). Reactions were identical to those described for qPCR above, except: 12.5 µl TaqMan 2× master mix (Genoptyping Master Mix; ABI), 2.25 µl primer1 (10 mmol/l), 2.25 µl primer 2 (10 mmol/l), 0.5 µl probe 1 (10 mmol/l), 0.5 µl probe 2 (10 mmol/l), 5 µl water, and 2 µl cDNA (100 ng/µl). Thermocycling was performed using the conditions described above. Allele-specific qPCR amplifications were performed in triplicate and averaged, and Ct values were determined and compared using the -ΔΔCt method [114].
Abbreviations
bHLH: basic helix-loop-helix; BAC: Bacterial artificial chromosome; CATIE: Centro Agronómico Tropical de Investigación y Enseñanza; CDPK: calcium-dependent kinase; CDS: Coding sequence; Cent-Tc: centromeric repeat T. cacao; df: degrees of freedom; FCM: Flow cytometry; FISH: Fluorescence in situ hybridization; GO: gene ontology; HMM: Hidden Markov model; indel: insertion/deletion; LTR: long terminal repeat; MAS: Marker-assisted selection; MCCS: MARS Center for Cocoa Sciences: MITE: Miniature inverted-repeat transposable element; NCBI: National Center for Biotechnology Information; NT: Nick translation; miRNA: MicroRNA; nt: Nucleotides; OR: Orthologous region; qPCR: quantitative polymerase chain reaction; rDNA: ribosomal DNA; SDD: Sterile distilled deionized; SI: self-incompatibility; siRNA: Small interfering RNA; SNP: Single-nucleotide polymorphism; tasiRNA: Trans-acting small interfering RNA; TE: Transposable element; TIR: terminal inverted repeat; UR: unequal intra-element homologous recombination; UTR: Untranslated region; WGS: Whole-genome sequencing; WPB: Woody plant buffer.
Competing interests
NH, FU, and LP are employees of IBM Research; JCM, JPM, HS, and RJS are employees of MARS Incorporated; OC and FU are partially funded by MARS Incorporated; and other authors may have received indirect funding from MARS Incorporated,.
Authors' contributions
JCM, KM, JS, HS, CB, RS, LP, and DNK designed the research. JCM, KM, JS, NH, DL, OC, SDF, PZ, SR, FU, CS, JJ, RP, BES, JCS, FAF, GMM, FA, WP, JPM, DM, JM, RS, DM, DG, LP, and DNK performed the research and analyzed data. JCM, KM, JS, NH, DL, OC, SF, PZ, SR, FU, CS, JJ, MZ, WP, JM, RS, DM, DG, LP, and DNK wrote the article. All authors read and approved the final manuscript.
Accession numbers
All data contributing to this genome initiative has been deposited in NCBI under BioProject PRJNA51633, and the genome accession number is [ALXC00000000]. The genome version described in this paper is the first version, accession number [ALXC01000000].
Supplementary Material
This PDF document contains Supplemental Tables S1 to S28, Supplemental Results and Supplemental Materials and methods.
This PDF document contains Supplemental Figures S1 to S11.
Contributor Information
Juan C Motamayor, Email: juan.motamayor@effem.com.
Keithanne Mockaitis, Email: kmockait@indiana.edu.
Jeremy Schmutz, Email: jschmutz@hudsonalpha.org.
Niina Haiminen, Email: nshaimin@us.ibm.com.
Donald Livingstone III, Email: dliving3@gmail.com.
Omar Cornejo, Email: ocornejo@gmail.com.
Seth D Findley, Email: sethdfindleyphd@gmail.com.
Ping Zheng, Email: ping23621827@gmail.com.
Filippo Utro, Email: futro@us.ibm.com.
Stefan Royaert, Email: Stefan.Royaert@gmail.com.
Christopher Saski, Email: SASKI@clemson.edu.
Jerry Jenkins, Email: jjenkins@hudsonalpha.org.
Ram Podicheti, Email: mnrusimh@indiana.edu.
Meixia Zhao, Email: zhao185@purdue.edu.
Brian E Scheffler, Email: Brian.Scheffler@ARS.USDA.GOV.
Joseph C Stack, Email: juan.motamayor@effem.com.
Frank A Feltus, Email: FFELTUS@clemson.edu.
Guiliana M Mustiga, Email: juan.motamayor@effem.com.
Freddy Amores, Email: famores.ec@gmail.com.
Wilbert Phillips, Email: wphillip@catie.ac.cr.
Jean Philippe Marelli, Email: jean-philippe.marelli@effem.com.
Gregory D May, Email: gdm@ncgr.org.
Howard Shapiro, Email: howard.shapiro@effem.com.
Jianxin Ma, Email: maj@purdue.edu.
Carlos D Bustamante, Email: cdbustam@stanford.edu.
Raymond J Schnell, Email: ray.schnell@effem.com.
Dorrie Main, Email: dorrie@wsu.edu.
Don Gilbert, Email: gilbertd@cricket.bio.indiana.edu.
Laxmi Parida, Email: parida@us.ibm.com.
David N Kuhn, Email: david.kuhn@ars.usda.gov.
Acknowledgements
We greatly appreciate the expertise and support of Dr Peter Bretting (USDA-ARS) and Alan Benett (UC Davis) over the course of this project. We are very grateful to Dr Belinda Martineau and Dr Sean Myles for their editorial contributions to this manuscript. We also thank Andrew Kaminsky for his valuable logistical contributions to the project; Cecile Tondo, Barbara Freeman. and Dayana Rodezno (USDA-ARS) for their technical contribution; Zach Smith and James Ford (Indiana University CGB) for WGS; Dr David Galbraith (University of Arizona) for technical advice on FCM; Dr Rey Gaston Loor (Instituto Nacional Autónomo de Investigaciones Agricolas y Pecuarias, Ecuador), Mrs Iraima Chacon (Corpozulia, Venezuela) and Dr Pathmanathan Umaharan (Cocoa Research Unit, Trinidad) for T. cacao leaf samples; Valdevino Santana do Carmo, Daniela V Silva. and Samuel MJ Branco for their contribution in the collection of field data; Dr. Laurent Brechenmacher (University of Missouri-Columbia) for G. max W82 seedlings, and Patrice Albert (University of Missouri-Columbia) and Richard Wolfe (Leica Microsystems) for technical advice.
This work was supported in part by the National Science Foundation (grant No. 0640462 to DGG), which provided genomics computational resources via TeraGrid, XSEDE, and also by the National Center for Genome Analysis.
References
- Motamayor JC, Risterucci AM, Lopez PA, Ortiz CF, Moreno A, Lanaud C. Cacao domestication I: the origin of the cacao cultivated by the Mayas. Heredity. 2002;14:380–386. doi: 10.1038/sj.hdy.6800156. [DOI] [PubMed] [Google Scholar]
- Motamayor JC, Lachenaud P, da Silva e Mota JW, Loor R, Kuhn DN, Brown JS, Schnell RJ. Geographic and genetic population differentiation of the Amazonian chocolate tree (Theobroma cacao L). PLoS ONE. 2008;14:e3311. doi: 10.1371/journal.pone.0003311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueira A AL. In: Biotechnology of fruit and nut crops. Litz RE, editor. CAB International Biosciences: Wallingford, UK; 2005. Theobroma cacao (Cacao). pp. 639–670. [Google Scholar]
- Foundation TWC. The World Cocoa Foundation. http://www.worldcocoafoundation.org/learn-about-cocoa/
- Guiltinan MJ VJ, Zhang D, Figueira A. In: Genomics of Tropical Crop Plants. Moore PH & Ming R, editor. Springer New York; 2008. Genomics of Theobroma cacao, "The Food of the Gods". pp. 146–170. [Google Scholar]
- Piasentin F K-RL. Biodiversity conservation and cocoa agroforests. Gro Cocoa. 2004;14:7–8. [Google Scholar]
- Bartley BGD. The genetic diversity of cacao and its utilization. Wallingford, UK: CABI Publishing; 2004. [Google Scholar]
- Motamayor JC, Risterucci AM, Heath M, Lanaud C. Cacao domestication II: progenitor germplasm of the Trinitario cacao cultivar. Heredity. 2003;14:322–330. doi: 10.1038/sj.hdy.6800298. [DOI] [PubMed] [Google Scholar]
- Efombagn I, Motamayor J, Sounigo O, Eskes A, Nyassé S, Cilas C, Schnell R, Manzanares-Dauleux M, Kolesnikova-Allen M. Genetic diversity and structure of farm and GenBank accessions of cacao ( Theobroma cacao L.) in Cameroon revealed by microsatellite markers. Tree Genetics & Genomes. 2008;14:821–831. doi: 10.1007/s11295-008-0155-z. [DOI] [Google Scholar]
- Aikpokpodion P, Motamayor J, Adetimirin V, Adu-Ampomah Y, Ingelbrecht I, Eskes A, Schnell R, Kolesnikova-Allen M. Genetic diversity assessment of sub-samples of cacao, Theobroma cacao L. collections in West Africa using simple sequence repeats marker. Tree Genetics & Genomes. 2009;14:699–711. doi: 10.1007/s11295-009-0221-1. [DOI] [Google Scholar]
- Argout X, Salse J, Aury J-M, Guiltinan MJ, Droc G, Gouzy J, Allegre M, Chaparro C, Legavre T, Maximova SN, Abrouk M, Murat F, Fouet O, Poulain J, Ruiz M, Roguet Y, Rodier-Goud M, Barbosa-Neto JF, Sabot F, Kudrna D, Ammiraju JSS, Schuster SC, Carlson JE, Sallet E, Schiex T, Dievart A, Kramer M, Gelley L, Shi Z, Berard A. et al. The genome of Theobroma cacao. Nature Genetics. 2011;14:101–108. doi: 10.1038/ng.736. [DOI] [PubMed] [Google Scholar]
- Ranjan A, Ichihashi Y, Sinha N. The tomato genome: implications for plant breeding, genomics and evolution. Genome Biology. 2012;14:167. doi: 10.1186/gb-2012-13-8-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer C. Yet-another-genome syndrome. http://blogs.discovermagazine.com/loom/2010/04/02/yet-another-genome-syndrome/
- Brown JS, Phillips-Mora W, Power EJ, Krol C, Cervantes-Martinez C, Motamayor JC, Schnell RJ. Mapping QTLs for resistance to frosty pod and black pod diseases and horticultural traits in Theobroma cacao L. Crop Sci. 2007;14:1851–1858. doi: 10.2135/cropsci2006.11.0753. [DOI] [Google Scholar]
- Royaert S, Phillips-Mora W, Arciniegas Leal A, Cariaga K, Brown J, Kuhn D, Schnell R, Motamayor J. Identification of marker-trait associations for self-compatibility in a segregating mapping population of Theobroma cacao L. Tree Genetics & Genomes. 2011;14:1159–1168. doi: 10.1007/s11295-011-0403-5. [DOI] [Google Scholar]
- Findley SD, Cannon S, Varala K, Du J, Ma J, Hudson ME, Birchler JA, Stacey G. A fluorescence in situ hybridization system for karyotyping soybean. Genetics. 2010;14:727–744. doi: 10.1534/genetics.109.113753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert PS, Gao Z, Danilova TV, Birchler JA. Diversity of chromosomal karyotypes in maize and its relatives. Cytogenet Genome Res. 2010;14:6–16. doi: 10.1159/000314342. [DOI] [PubMed] [Google Scholar]
- Kato A, Lamb JC, Birchler JA. Chromosome painting using repetitive DNA sequences as probes for somatic chromosome identification in maize. Proc Natl Acad Sci USA. 2004;14:13554–13559. doi: 10.1073/pnas.0403659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saski CA, Feltus FA, Staton ME, Blackmon BP, Ficklin SP, Kuhn DN, Schnell RJ, Shapiro H, Motamayor JC. A genetically anchored physical framework for Theobroma cacao cv. Matina 1-6. BMC Genomics. 2011;14:413. doi: 10.1186/1471-2164-12-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananiev EV, Phillips RL, Rines HW. Chromosome-specific molecular organization of maize (Zea mays L.) centromeric regions. Proc Natl Acad Sci USA. 1998;14:13073–13078. doi: 10.1073/pnas.95.22.13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Zapater JM, Estelle MA, Somerville CR. A highly repeated DNA-sequence in Arabidopsis thaliana. Molecular & General Genetics. 1986;14:417–423. doi: 10.1007/BF00331018. [DOI] [Google Scholar]
- Ma JX, Wing RA, Bennetzen JL, Jackson SA. Plant centromere organization: a dynamic structure with conserved functions. Trends in Genetics. 2007;14:134–139. doi: 10.1016/j.tig.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, B J, Gnerre S, Mauceli E, Lindblad-Toh K, Mesirov JP, Zody MC, Lander ES. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Research. 2003;14:91–96. doi: 10.1101/gr.828403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn DN, Livingstone D, Main D, Zheng P, Saski C, Feltus FA, Mockaitis K, Farmer AD, May GD, Schnell RJ. Identification and mapping of conserved ortholog set (COS) II sequences of cacao and their conversion to SNP markers for marker-assisted selection in Theobroma cacao and comparative genomics studies. Tree Genetics & Genomes. 2012;14:97–111. doi: 10.1007/s11295-011-0424-0. [DOI] [Google Scholar]
- Feltus FA, Saski CA, Mockaitis K, Haiminen N, Parida L, Smith Z, Ford J, Staton ME, Ficklin SP, Blackmon BP, Cheng CH, Schnell RJ, Kuhn DN, Motamayor JC. Sequencing of a QTL-rich region of the Theobroma cacao genome using pooled BACs and the identification of trait specific candidate genes. BMC Genomics. 2011;14:379. doi: 10.1186/1471-2164-12-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BB, Brendel V. Genomewide comparative analysis of alternative splicing in plants. Proc Natl Acad Sci USA. 2006;14:7175–7180. doi: 10.1073/pnas.0602039103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The universal protein resource (UniProt) http://www.uniprot.org
- KEGG for linking genomes to life and the environment. http://www.kegg.jp [DOI] [PMC free article] [PubMed]
- Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;14:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S, Zou C, Li Q, Yuan Y, Lu C, Wei H, Gou C, Zheng Z, Yin Y, Zhang X, Liu K, Wang B, Song C, Shi N, Kohel RJ, Percy RG, Yu JZ, Zhu YX, Wang J, Yu S. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012. [DOI] [PubMed]
- Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, Rokhsar DS. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Research. 2012;14:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Accelerated profile HMM searches. PLoS Computational Biology. 2011;14:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol. 1994;14:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Kaul S, Koo HL, Jenkins J, Rizzo M, Rooney T, Tallon LJ, Feldblyum T, Nierman W, Benito MI, Lin XY, Town CD, Venter JC, Fraser CM, Tabata S, Nakamura Y, Kaneko T, Sato S, Asamizu E, Kato T, Kotani H, Sasamoto S, Ecker JR, Theologis A, Federspiel NA, Palm CJ, Osborne BI, Shinn P, Conway AB, Vysotskaia VS, Dewar K. et al. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;14:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Wu J, Kanamori H, Katayose Y, Fujisawa M, Namiki N, Mizuno H, Yamamoto K, Antonio BA, Baba T. The map-based sequence of the rice genome. Nature. 2005;14:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- Vogel JP, Garvin DF, Mockler TC, Schmutz J, Rokhsar D, Bevan MW, Barry K, Lucas S, Harmon-Smith M, Lail K, Tice H, Grimwood J, McKenzie N, Huo NX, Gu YQ, Lazo GR, Anderson OD, You FM, Luo MC, Dvorak J, Wright J, Febrer M, Idziak D, Hasterok R, Lindquist E, Wang M, Fox SE, Priest HD, Filichkin SA, Givan SA. et al. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;14:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- Devos KM, Brown JK, Bennetzen JL. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002;14:1075–1079. doi: 10.1101/gr.132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, S J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Research. 2009;14:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M, Lyons E, Pedersen B, Alam M, Ming R, Lisch D. Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res. 2008;14:1924–1937. doi: 10.1101/gr.081026.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR. Efficient multilocus association testing for whole genome association studies using localized haplotype clustering. Genet Epidemiol. 2007;14:365–375. doi: 10.1002/gepi.20216. [DOI] [PubMed] [Google Scholar]
- Schaid DJ. Evaluating associations of haplotypes with traits. Genet Epidemiol. 2004;14:348–364. doi: 10.1002/gepi.20037. [DOI] [PubMed] [Google Scholar]
- Williams AL, Patterson N, Glessner J, Hakonarson H, Reich D. Phasing of many thousands of genotyped samples. Am J Hum Genet. 2012;14:238–251. doi: 10.1016/j.ajhg.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Martin C. Multifunctionality and diversity within the plant MYB-gene family. Plant Mol Biol. 1999;14:577–585. doi: 10.1023/A:1006319732410. [DOI] [PubMed] [Google Scholar]
- Amann K, Lezhneva L, Wanner G, Herrmann RG, Meurer J. ACCUMULATION OF PHOTOSYSTEM ONE1, a member of a novel gene family, is required for accumulation of [4Fe-4S] cluster-containing chloroplast complexes and antenna proteins. Plant Cell. 2004;14:3084–3097. doi: 10.1105/tpc.104.024935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier-Level A, Le Cunff L, Gomez C, Doligez A, Ageorges A, Roux C, Bertrand Y, Souquet JM, Cheynier V, This P. Quantitative genetic bases of anthocyanin variation in grape (Vitis vinifera L. ssp. sativa) berry: a quantitative trait locus to quantitative trait nucleotide integrated study. Genetics. 2009;14:1127–1139. doi: 10.1534/genetics.109.103929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This P, Lacombe T, Cadle-Davidson M, Owens CL. Wine grape (Vitis vinifera L.) color associates with allelic variation in the domestication gene VvmybA1. Theor Appl Genet. 2007;14:723–730. doi: 10.1007/s00122-006-0472-2. [DOI] [PubMed] [Google Scholar]
- De Jong WS, Eannetta NT, De Jong DM, Bodis M. Candidate gene analysis of anthocyanin pigmentation loci in the Solanaceae. Theor Appl Genet. 2004;14:423–432. doi: 10.1007/s00122-003-1455-1. [DOI] [PubMed] [Google Scholar]
- Zhang B, Hu Z, Zhang Y, Li Y, Zhou S, Chen G. A putative functional MYB transcription factor induced by low temperature regulates anthocyanin biosynthesis in purple kale ( Brassica Oleracea var. acephala f. tricolor). Plant Cell Reports. 2012;14:281–289. doi: 10.1007/s00299-011-1162-3. [DOI] [PubMed] [Google Scholar]
- Lin-Wang K, Bolitho K, Grafton K, Kortstee A, Karunairetnam S, McGhie T, Espley R, Hellens R, Allan A. An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biology. 2010;14:50. doi: 10.1186/1471-2229-10-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanhui C, Xiaoyuan Y, Ku H, Meihua L, Jigang L, Zhaofeng G, Zhiqiang L, Yunfei Z, Xiaoxiao W, Xiaoming Q, Yunping S, Li Z, Xiaohui D, Jingchu L, Xing-Wang D, Zhangliang C, Hongya G, Li-Ji Q. The MYB transcription factor superfamily of Arabidopsis: expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol Biol. 2006;14:107–124. doi: 10.1007/s11103-005-2910-y. [DOI] [PubMed] [Google Scholar]
- Fournier-Level A, Lacombe T, Le Cunff L, Boursiquot JM, This P. Evolution of the VvMybA gene family, the major determinant of berry colour in cultivated grapevine (Vitis vinifera L.). Heredity (Edinb) 2010;14:351–362. doi: 10.1038/hdy.2009.148. [DOI] [PubMed] [Google Scholar]
- Bate N, Spurr C, Foster GD, Twell D. Maturation-specific translational enhancement mediated by the 5′-UTR of a late pollen transcript. The Plant Journal. 1996;14:613–623. doi: 10.1046/j.1365-313X.1996.10040613.x. [DOI] [PubMed] [Google Scholar]
- Luo QJ, Mittal A, Jia F, Rock CD. An autoregulatory feedback loop involving PAP1 and TAS4 in response to sugars in Arabidopsis. Plant Mol Biol. 2012;14:117–129. doi: 10.1007/s11103-011-9778-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa M, Peragine A, Park MY, Poethig RS. A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev. 2005;14:2164–2175. doi: 10.1101/gad.1352605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen E, Xie Z, Gustafson AM, Carrington JC. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;14:207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006;14:3407–3425. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takos A, Jaffe F, Jacob S, Bogs J, Robinson S, Walker A. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006;14:1216–1232. doi: 10.1104/pp.106.088104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, Zhao M, Leavitt JM, Lloyd AM. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. The Plant Journal. 2008;14:814–827. doi: 10.1111/j.1365-313X.2007.03373.x. [DOI] [PubMed] [Google Scholar]
- Xia R, Zhu H, An YQ, Beers EP, Liu Z. Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol. 2012;14:R47. doi: 10.1186/gb-2012-13-6-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacao Genome Database. http://www.cacaogenomedb.org
- Schnell RJ, Brown JS, Kuhn DN, Cervantes-Martinez C, Olano CT, Motamayor JC. Why would we breed cacao in Florida? Proc Fla State Hort Soc. 2005;14:189–191. [Google Scholar]
- Cope FW. The mechanism of pollen incompatibility in Theobroma cacao. L Heredity. 1962;14:157–182. doi: 10.1038/hdy.1962.14. [DOI] [Google Scholar]
- Iwano M, Takayama S. Self/non-self discrimination in angiosperm self-incompatibility. Curr Opin Plant Biol. 2012;14:78–83. doi: 10.1016/j.pbi.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Galbraith DW. Simultaneous flow cytometric quantification of plant nuclear DNA contents over the full range of described angiosperm 2C values. Cytometry A. 2009;14:692–698. doi: 10.1002/cyto.a.20760. [DOI] [PubMed] [Google Scholar]
- Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L. et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;14:178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- Johnston JS, Bennett MD, Rayburn AL, Galbraith DW, Price HJ. Reference standards for determination of DNA content of plant nuclei. Am J Bot. 1999;14:609–613. doi: 10.2307/2656569. [DOI] [PubMed] [Google Scholar]
- Loureiro J, Rodriguez E, Dolezel J, Santos C. Two new nuclear isolation buffers for plant DNA flow cytometry: a test with 37 species. Ann Bot. 2007;14:875–888. doi: 10.1093/aob/mcm152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doležel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry. Nature Protocols. 2007;14:2233–2243. doi: 10.1038/nprot.2007.310. [DOI] [PubMed] [Google Scholar]
- Arumuganathan K, Earle E. Nuclear DNA content of some important plant species. Plant Molecular Biology Reporter. 1991;14:208–218. doi: 10.1007/BF02672069. [DOI] [Google Scholar]
- SAS Institute. http://www.sas.com/
- Van Ooijen JW. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genetics Research. 2011;14:343–349. doi: 10.1017/S0016672311000279. [DOI] [PubMed] [Google Scholar]
- Sambrook J, F EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1989. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;14:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;14:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CocoaGen DB. http://cocoagendb.cirad.fr/gbrowse/download.html
- Haas BJ. Analysis of alternative splicing in plants with bioinformatics tools. Curr Top Microbiol Immunol. 2008;14:17–37. doi: 10.1007/978-3-540-76776-3_2. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. ftp://ftp.arabidopsis.org/home/tair/Sequences/blast_datasets/TAIR10_blastsets/
- Jaillon O, Aury J, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. ftp://ftp.jgi-psf.org/pub/JGI_data/phytozome/v7.0/Vvinifera/ [DOI] [PubMed]
- Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen G-L, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). ftp://ftp.jgi-psf.org/pub/JGI_data/phytozome/v7.0/Ptrichocarpa/ [DOI] [PubMed]
- Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Genome sequence of the palaeopolyploid soybean. ftp://ftp.jgi-psf.org/pub/JGI_data/phytozome/v7.0/Gmax/ [DOI] [PubMed]
- Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, Redman J, Chen G, Cahoon EB, Gedil M, Stanke M, Haas BJ, Wortman JR, Fraser-Liggett CM, Ravel J, Rabinowicsz PD. Draft genome sequence of the oilseed species Ricinus communis. http://castorbean.jcvi.org/downloads.php [DOI] [PMC free article] [PubMed]
- Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, Jaiswal P, Mockaitis K, Liston A, Mane SP, Burns P, Davis TM, Slovin JP, Bassil N, Hellens RP, Evans C, Harkins T, Kodira C, Desany B, Crasta OR, Jensen RV, Allan AC, Michael TP, Setubal JC, Celton J-M, Rees DJG, Williams KP, Holt SH, Rojas JJR, Chatterjee M, The genome of woodland strawberry (Fragaria vesca). https://strawberry.plantandfood.co.nz/gbrowse/navbar/strawberry/download.html [DOI] [PMC free article] [PubMed]
- Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Genome sequence and analysis of the tuber crop potato. http://potatogenomics.plantbiology.msu.edu/index.html
- The Sorghum bicolor genome and the diversification of grasses. ftp://ftp.jgi-psf.org/pub/JGI_data/phytozome/v7.0/Sbicolor/ [DOI] [PubMed]
- Slater GSC, Birney E. Automated generation of heuristics for biological sequence comparison. Bmc Bioinformatics. 2005;14:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. BioInformatics. 2003;14:ii215–ii225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH. A unified classification system for eukaryotic transposable elements. Nature Reviews Genetics. 2007;14:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- McCarthy EM, McDonald JF. LTR_STRUC: a novel search and identification program for LTR retrotransposons. BioInformatics. 2003;14:362–367. doi: 10.1093/bioinformatics/btf878. [DOI] [PubMed] [Google Scholar]
- Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research. 2007;14:W265–W268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Devos KM, Bennetzen JL. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004;14:860–869. doi: 10.1101/gr.1466204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holligan D, Zhang XY, Jiang N, Pritham EJ, Wessler SR. The transposable element landscape of the model legume Lotus japonicus. Genetics. 2006;14:2215–2228. doi: 10.1534/genetics.106.062752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;14:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Yang LX, Bennetzen JL. Structure-based discovery and description of plant and animal Helitrons. Proc Natl Acad Sci USA. 2009;14:12832–12837. doi: 10.1073/pnas.0905563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhou FF, Li GJ, Xu Y. MUST: A system for identification of miniature inverted-repeat transposable elements and applications to Anabaena variabilis and Haloquadratum walsbyi. Gene. 2009;14:1–7. doi: 10.1016/j.gene.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Han YJ, Wessler SR. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Research. 2010;14 doi: 10.1093/nar/gkq862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RepeatMasker Open-3.0. http://www.repeatmasker.org
- Mercator: Multiple Whole-Genome Orthology Map Construction. http://www.biostat.wisc.edu/~cdewey/mercator/
- Bray N, Pachter L. MAVID: constrained ancestral alignment of multiple sequences. Genome Res. 2004;14:693–699. doi: 10.1101/gr.1960404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay SJ, Vergara IA, Stajich JE. Current Protocols in Bioinformatics. John Wiley & Sons, Inc; 2002. Using the Generic Synteny Browser (GBrowse_syn). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criollo vs Matina Synteny. http://www.cacaogenomedb.org/gb-private/gbrowse_syn/tc_criollo_vs_tc_matina/
- Andras SC, Hartman TP, Marshall JA, Marchant R, Power JB, Cocking EC, Davey MR. A drop-spreading technique to produce cytoplasm-free mitotic preparations from plants with small chromosomes. Chromosome Res. 1999;14:641–647. doi: 10.1023/A:1009288119851. [DOI] [PubMed] [Google Scholar]
- Kato A. Air drying method using nitrous oxide for chromosome counting in maize. Biotechnic & Histochemistry. 1999;14:160–166. doi: 10.3109/10520299909047968. [DOI] [PubMed] [Google Scholar]
- Gill N, Findley S, Walling JG, Hans C, Ma J, Doyle J, Stacey G, Jackson SA. Molecular and chromosomal evidence for allopolyploidy in soybean. Plant physiology. 2009;14:1167–1174. doi: 10.1104/pp.109.137935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utro F, Haiminen N, Livingstone D, III, Cornejo OE, Royaert S, Schnell RJ, Motamayor JC, Kuhn DN, Parida L. iXora: Exact haplotype inferencing and trait association. BMC Genetics. in press . [DOI] [PMC free article] [PubMed]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. BioInformatics. 2010;14:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. BioInformatics. 2009;14:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;14:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;14:491–501. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006;14:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution. 2011;14:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;14:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Rosenberg NA, Donnelly P. Association mapping in structured populations. Am J Hum Genet. 2000;14:170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. Efficient control of population structure in model organism association mapping. Genetics. 2008;14:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Puryear J, Cairney J. A simple and efficient method for isolating RNA from pine trees. Plant Molecular Biology Reporter. 1993;14:113–116. doi: 10.1007/BF02670468. [DOI] [Google Scholar]
- Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;14:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;14:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This PDF document contains Supplemental Tables S1 to S28, Supplemental Results and Supplemental Materials and methods.
This PDF document contains Supplemental Figures S1 to S11.