

Abstract
The systematic description of complex traits, from the organism to the cellular level, is important for hypothesis generation about underlying disease mechanisms. We discuss how intelligent algorithms might provide support, leading to faster throughput.
Introduction
The systematic description of variation has gained increasing importance since the discovery of the causal relationship between a genotype placed in a certain environment and a phenotype [1]. The triumvirate connection of a phenotype, the underlying genotype and the environment in which the genotype is placed plays an important role to enhance our knowledge. Phenotypes can be applied to clinical questions, for example, the genetic origins of diseases [2-4], as well as biological problems, such as the evolution of species over time [5]. For example, PhenomeNET [6] compares phenotypes recorded in mutagenesis experiments in eight different species with the signs and symptoms of human diseases and uses orthology to determine viable gene candidates. Another example for the application of phenotypes is the PhenoScape knowledge base [7], which records phenotypes to answer questions such as ‘How were limbs formed from fins?’ Effective use of phenotype information and an eventual facilitation of translational research [8] requires researchers to achieve a common mindset and build a shared conceptual view on the definition, representation and interoperability of phenotypes. While this need has been previously recognized, it has, however, proven to be a challenging process, even for biological data corresponding to one species [9]. The intrinsic complexity of phenotypes is the most important obstacle in the process of reaching consensus and a common understanding. In general, phenotypes are considered to be observable characteristics, spanning from a molecular to an environmental level [8].
From a conceptual perspective, the comprehensive description of phenotypes covers three dimensions: (1) representation - defining phenotypes in a format that enables machine processing; (2) acquisition - capturing and storing phenotypes to enable large-scale analysis; and (3) processing - devising techniques with specific analytical goals. To these, we can add a fourth orthogonal dimension - interoperability (that is, aligning intraspecies and cross-species representations) - which emerges as a result of the intrinsic interdisciplinarity of the domain. Figure 1 depicts these dimensions together with existing solutions, discussed later in this review, and can be used as a map of the current phenotype technology ecosystem. Representation aims to structure and formalize the knowledge encoded in phenotypes by defining them in a particular context (for example, as part of a taxonomy) and by relating them to other domain-specific concepts (for example, anatomical models). Over the course of the years, ontologies have proven to be the most appropriate framework to unlock the potential contained in phenotype data, with the Human Phenotype Ontology (HPO) [10] and the Mammalian Phenotype Ontology (MP) [11] pioneering the community efforts. Once specific representations have been defined, numerous projects embarked on the challenging acquisition goal. Examples include the International Mouse Phenotyping Consortium (IMPC), aiming to catalogue the entire mouse phenome through systematic gene knockouts [12], or the Deciphering Developmental Disorders (DDD) study [13], unraveling the origins of rarely occurring human genetic diseases by gathering copy number variations (CNVs) as well as point mutations and associated phenotypes from individual patients and their ancestors. Most of these projects follow a traditional manual curation approach. However, with advances in biomedical natural language processing, researchers have also started to look into automating the acquisition process [14,15].
Figure 1.
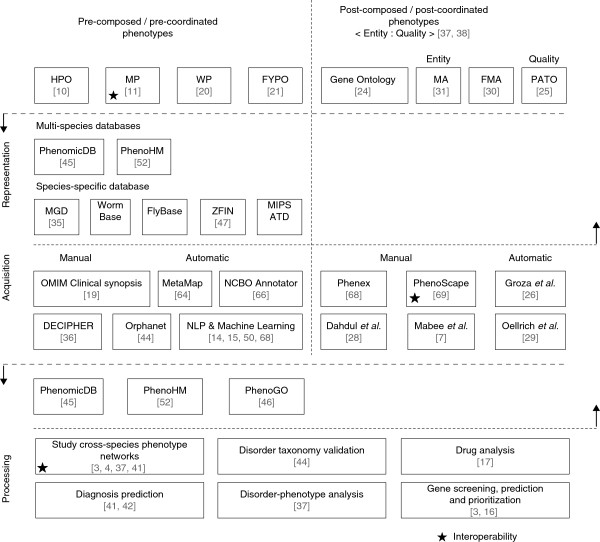
Overview of the four perspectives of phenotype information together with existing corresponding solutions. From a generic perspective, the phenome ecosystem can be described according to three main dimensions, in addition to a fourth orthogonal one: (i) representation - providing the mechanisms required to capture phenotype knowledge; (ii) acquisition - providing the tools for acquiring and storing phenotype data and knowledge; (iii) processing - dealing with the use and application of the phenotype knowledge in diverse contexts; and (iv) interoperability - aiming to bridge the different representations and formalisms describing phenotype knowledge. The figure depicts a map of the current solutions corresponding to each of the four dimensions. Two major strands emerge when targeting the representation and acquisition of phenotypes - that is, focusing on pre-composed or post-composed phenotypes. Also, some approaches deal with multiple dimensions at the same time. DECIPHER, Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources; FMA, Foundation Model of Anatomy; FYPO, Worm Phenotype Ontology; hp, human phenotype; MA, Mouse Adult Gross Anatomy Ontology; MGD, Mouse Genome Database; MIPS, Munich Information Center for Protein Sequence; MP, Mouse Phenotype Ontology; NCBO, National Center for Biomedical Ontology; NLP, natural language processing; OMIM, Online Mendelian Inheritance of Man database; OWL, Web Ontology Language; PATO, Phenotype and Trait Ontology; WP, Worm Phenotype Ontology; ZFIN, Zebrafish Information Network.
Finally, with the increasing amount of available phenotype data, initial steps have been taken to process and achieve interoperability of data from a range of resources using semantic layers [3,4,16,17]. Interoperability through semantic layers means that ontologies are aligned to each other, for example, through lexical or ontological features, and the aligned ontologies enable the comparison of data being annotated with different ontologies. The integrated data can then be processed and facilitate biological discoveries. For example, PhenoDigm [18] aligns phenotypes from mutagenesis experiments in several species with the signs and symptoms of human diseases through ontological as well as lexical features. Once the phenotypes are aligned, the mutated genes are ranked according to their phenotype similarity with the disease, and the mutated genes exhibiting the highest similarity with the disease constitute candidate genes for this disease. However, interoperability and processing of data cover only a very small subset of all available data and further projects are required to address these aspects.
In this review, we assess the current status of phenotype information technologies, with a focus on the perception of phenotypes in different domains and the influence of this perception on the above-mentioned four dimensions. We highlight the progress made towards extant goals as well as provide a visionary perspective on the next steps required to bridge the existing solutions to facilitate seamless cross-domain research.
Unraveling phenotype structures
Formally represented phenotypes are a key prerequisite in enabling advanced computational methods for functional genomics, cross-species studies or clinical decision support. Over the past decade, research in this area has mostly focused on using ontologies to formalize phenotype descriptions. Ontologies provide an ideal ecosystem to model the inherent diversity of phenotypes, as in addition to supporting multifaceted classification and definition, they also play a major role in interoperability across communities, domains and species. Several efforts have emerged in this direction, each concentrating on a particular organism or domain. Examples of such projects include HPO (initially mined from the Online Mendelian Inheritance in Man (OMIM) database [19]), the Worm Phenotype Ontology (WP) [20], the Fission Yeast Phenotype Ontology (FYPO) [21] and MP [11] (most ontologies are openly available via the National Center for Biomedical Ontology (NCBO) BioPortal [22] or Open Biological and Biomedical Ontologies (OBO) Foundry [23]). Table 1 lists the phenotype annotation resources discussed in this review.
Table 1.
Phenotype annotation resources
DECIPHER, Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources; FMA, Foundation Model of Anatomy; FYPO, Worm Phenotype Ontology; GO, Gene Ontology; HPO, Human Phenotype Ontology; MIPS, Munich Information Center for Protein Sequence; MP, Mouse Phenotype Ontology; MPD, Mouse Phenome Database; NCBO, National Center for Biomedical Ontology; OMIM, Online Mendelian Inheritance of Man database; PATO, Phenotype and Trait Ontology; WP, Worm Phenotype Ontology; ZFIN, Zebrafish Information Network.
The formalization of phenotypes raises several challenges, also common to other domain ontologies, for example, Gene Ontology (GO) [24]. Most of the existing representations define phenotypes as pre-composed/pre-coordinated entities - that is, concepts that externalize as a whole the intrinsic duality of the underlying localization and the defined trait (for example, MP:0008572 - Abnormal Purkinje cell dendrite morphology; or HP:0008905 - Rhizomelic limb shortening). This implicit duality is sometimes made explicit via the structure of the ontology, using multiple inheritance (that is, one concept with multiple parents); for example, HP:0008905 is a descendent of HP:0001507 - Growth abnormality (denoting the focus on the trait) and of HP:0002813 - Abnormality of the limb bone morphology (denoting the focus on the localization - limb bone).
The same aspect significantly increases the complexity of building a realistic, fine-grained phenotype model. Gkoutos et al. [25] note that in order to fully capture knowledge expressed by phenotypes, we require a more precise and fine-grained definition for them or, more concretely, we need to perform an explicit decomposition in their elementary units. This is known as the post-composed/post-coordinated representation of phenotypes (see glossary in Table 2). In contrast to the pre-composed approach, post-composed representations capture and combine the elementary concepts defining the phenotype - that is, the localization of the phenotype (limb in HP:0008905, or dendrite of Purkinje cell in MP:0008572) and the trait associated with it (shortening in HP:0008905, or abnormal morphology in MP:0008572). The result is a composite concept that has a semantics equivalent to the one of the pre-composed entity, but defined at a richer level and enabling novel reasoning and exploration mechanisms. The formal representation of post-composed entities is known as the entity/quality (EQ) formalism, while the resulting concepts are also known as EQ statements.
Table 2.
Glossary of terms
| Term | Description |
|---|---|
| Annotation |
Descriptions that are added to data such as text |
| Curation |
Representation of data (for example, biological data sets) through annotations (for example, through ontologies) |
| Entity/quality |
A conceptual model of a term according to (a) an entity part that denotes an anatomical or process part and (b) a quality part that characterises how the entity is affected |
| Grounding |
To establish the specific reference of a term according to an ontology |
| Mention |
A sequence of words in a text that denotes a term according to some external reference system |
| Pre-composed (pre-coordinated) term |
A term that has been affirmed and defined as a whole without division into its constituent parts |
| Post-composed (post-coordinated) term |
A term that is defined according to the decomposition of its constituent parts and the grounding of those parts in one or more external ontologies |
| Ontology |
A specification of a conceptualization |
| OWL |
The Web Ontology Language is a family of formal languages intended to aid machine understanding of resources on the World Wide Web |
| RDF |
Resource Description Framework is family of specifications for describing resources on the World Wide Web. It is a World Wide Web Consortium standard |
| Semantic Web | A collaborative movement to promote common data formats for data re-use by machines |
Several ontological approaches have been proposed to implement EQ statements [26,27] and, subsequently, tools have been developed to manually [28] or automatically [26,29] construct them. These tools rely on the existence of ontologies that define localization and trait concepts, such as the Foundational Model of Anatomy (FMA) [30] for human, or the Mouse Adult Gross Anatomy Ontology (MA) [31] for mouse, and the species-agnostic Phenotype and Trait Ontology (PATO) [25] for traits.
Figure 2 depicts MP:0008572 - Abnormal Purkinje cell dendrite morphology - at the three levels of abstraction discussed above, as shown in [27]. The pre-composed form of this concept is represented by the concept itself as defined in the MP. The duality localization-trait is made explicit in its post-composition (the EQ statement) by a set of four concepts defined in various ontologies. In this example, the localization (Purkinje cell dendrite) is represented by an association of two subcellular anatomy ontology [32] concepts (Dendrite and Purkinje cell) via a relation (part of) introduced by the relation ontology [33] altogether forming the entity, while the trait is represented via two PATO concepts (morphology and abnormal) denoting the quality and a modifier of the quality. At a formal level, the post-composed entity is defined with the logical statement: morphology inheres_in some (dendrite part_of some Purkinje_cell) and has_qualifier some abnormal.
Figure 2.
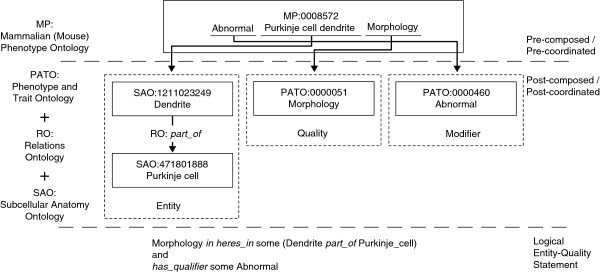
Overview of the three levels of phenotype representation: pre-composed, post-composed and logical entity/quality statements. This figure depicts the high-level process of modeling phenotypes based on diverse levels of logical decomposition. At the most coarse-grained level, we find pre-composed entities, which are usually introduced as monolithic concepts within particular ontologies. In the example, the Mammalian Phenotype Ontology defines MP:0008572 (Abnormal Purkinje cell dendrite morphology). In order to make the trait–localization duality explicit, this entity can be modeled in a post-composed form by an entity/quality association defined by foundational models. Hence, MP:0008572 can be represented by an entity, composed of SAO:1211023249 (Dendrite) and SAO:471801888 (Purkinje cell) - connected via RO:part_of, a quality - defined by PATO:0000051 (Morphology) and, in this particular case, a modifier associated with the quality - PATO:0000460. Subsequently, this model can then be materialized in a logical statement, with the help of additional relationships, such as inheres_in and has_qualifier, as defined in [27].
The formal relations constructing the logical EQ statement are, in this case, inheres_in and has_qualifier (as defined in [27]), while the rest are the concepts introduced by the external ontologies and described above. This example also provides a glimpse of the complex logical formalisms that may emerge from post-composed entities, such as nested definitions of terms (dendrite part_of some Purkinje_cell). From an analysis and exploratory perspective, the EQ formalism provides clear advantages. However, it also features its own series of challenges; two of the most important are the formalization of complex entities and single-term phenotypes. Some representative examples of the former are the definition and representation of phenotypes that involve relationships between several anatomical elements, traits of specific parts of anatomical elements (for example, fingertips or interdigital folds), and traits of spatial, functional and non-functional properties of anatomical elements (for example, mineral density, movement, angles). Single-term phenotype expressions, on the other hand, do not externalize the localization-trait duality in an explicit manner (for example, HP:0010884 - Acromelia). Their semantics can still be encoded using the EQ formalism; however, it requires significant human input and comprehension, because in most cases the localization aspect is vaguely defined (for example, in the case of Acromelia: shortness of the <distal part> of a limb). Even though standardization efforts are ongoing, the representation of phenotypes varies across the different resources from narrative descriptions, over vocabularies and terminologies to ontologies [34]. In order to derive novel genotype-phenotype associations or links between genes and drugs/diseases, this diversity of data needs to be integrated in a coherent manner. Efforts ranging from overarching databases to semantic integration via ontologies are underway, but, currently, none of the existing tools are capable of catering for all phenotype-relevant use cases.
State-of-the-art applications
Given the variety of phenotype descriptions [34] and resources [19,35,36], and the diversity of domains phenotypes are relevant to, existing tools fulfill versatile purposes. In the area of medicine, phenotypes are applied to: (i) screening, predicting or prioritizing genes that are potentially relevant to human genetic disorders [3,4,16] (for example, PhenomeNET showed a potential connection between Tetralogy of Fallot (OMIM:187500) and the mouse gene Adam19 (MGI:3028702) that is supported by other published studies); (ii) analyzing patients with unidentified medical conditions [37] (the authors suggested 431 potential causes, all novel, for 27 CNV disorders); or (iii) finding new ways of treating diseases with existing drugs [17] (for example, PhenomeDrug [38] suggests that tretinoin could be used as therapy for cystic fibrosis (OMIM:219700); this is also reported in the scientific literature). However, all these tools rely on the public availability of phenotype data represented with semantic annotations and diverse semantic similarity metrics [39] to derive associations between phenotypes and genes, diseases or drugs.
PhenomeNET [3] and MouseFinder [16] are two examples of tools that use animal models from Mouse Genome Database (MGD) [35] to identify potential novel gene candidates for heritable diseases contained in OMIM. Both MGD and OMIM use a different ontological phenotype representation [34]; however, interoperability is achieved via logical axioms built on EQ statements (see Unraveling phenotype structures). Figure 3 depicts an example of such cross-species interoperability using EQ statements. Unfortunately, these are available for only a subset of the contained phenotypes and are mostly manually generated [40] in order to ensure correctness. Another difference between the two approaches can be found in the way they process phenotypes. PhenomeNET aims to acquire the entire phenome for a mouse model or a disorder, while MouseFinder focuses on identifying meaningful pairs of phenotypes.
Figure 3.
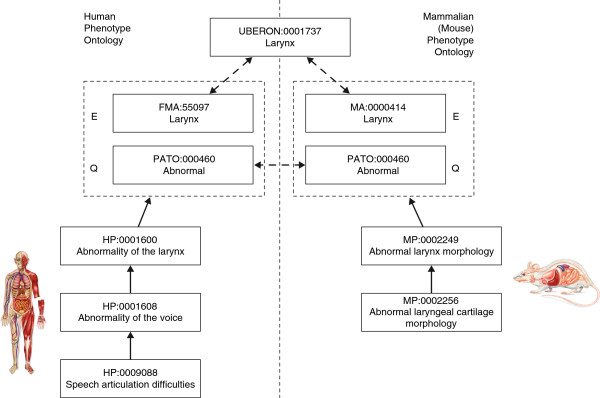
Example of cross-species phenotype interoperability using the entity/quality (EQ) formalism. This example depicts a mapping between two species-oriented concepts (HP:0001600 (Abnormality of the larynx) in human and MP:0002249 (Abnormal larynx morphology) in mouse) denoting the same conceptual phenotype. As opposed to classic mapping mechanisms, where a direct relation would be created between the two concepts, here the mapping is realized by transforming each phenotype in a post-composed format (that is, EQ statements), followed by the alignment of the corresponding elementary items. The quality is captured by a single common entity (PATO:000460 - Abnormal), while the anatomical terms are bridged via a term defined by an overarching anatomical ontology. FMA, Foundation Model of Anatomy; MA, Mouse Adult Gross Anatomy Ontology; MP, Mouse Phenotype Ontology; PATO, Phenotype and Trait Ontology.
Phenotypes have also been used to support clinical diagnosis. Phenomizer [41], for example, uses a semantic scoring mechanism that calculates the similarity of a phenotype with the signs and symptoms of a disease. This procedure is particularly helpful in cases of patients where a diagnosis is difficult due to controversial phenotype information, for example, patients contained in the Database of Chromosomal Imbalance and Phenotype (DECIPHER) [36]. A similar approach has also been followed by Paul et al. [42] with a focus on skeletal dysplasias. Finally, the same mechanism has been applied to study to what extent existing disorder classifications (for example, Orphanet [43]) are grounded in the publicly available phenotype-disorder associations [44]. Koehler et al. [44] have shown that by combining OMIM and Orphanet phenotype data it is possible to re-create to a large extent human-made classifications, thus demonstrating the validity of the classifications as well as the value provided by existing disorder characterizations.
Based on the assumption that species possess orthologous genes and that these genes exhibit identical phenotypes, the systematic assessment of phenotypes and their corresponding genes may reveal new functions when assessed across species. PhenomicDB [45] is a database not only holding textual phenotype descriptions for a number of species but also enabling the comparison of phenotypes across species through text mining, thus enabling the discovery of novel gene functions. PhenoGO [46] also applies text mining but instead of directly identifying gene-phenotype associations, it lists connections between phenotypes and GO annotations. As long as a gene is phenotypically described, a GO profile can be derived based on the assigned phenotypes.
Most of the content in existing biological databases is populated through manual curation of the scientific literature; for example, MGD, OMIM or Zebrafish Information Network [47]. The process of manual curation is, however, time-consuming and labor intense, resulting in huge costs for creating and maintaining the databases. To reduce time, labor and costs, semi-automated solutions gain more and more importance in supporting biocuration. PharmGKB [48], for example, is a database holding information about entities relevant to pharmacogenetics that have been automatically extracted from published literature with text mining. Only parts of the PharmGKB data have been validated through curation efforts.
PharmsPresso [49], another database generated from scientific literature, focuses on the extraction of relations between entities relevant to pharmacogenetics. When assessing phenotypes mentioned in OMIM records, van Driel and colleagues could derive meaningful phenotype clusters, resembling consistent GO annotations and protein-protein interactions [50]. Obtained phenotype information from this study was made available via the MimMiner web interface [51]. PhenoHM [52] allows, similar to PhenomicDB, comparison of phenotype information on a textual level across species and to access orthologous genes via their phenotypes.
In conclusion, even though initial steps have been made in the direction of the integration of phenotype data and text mining phenotype information, no exhaustive solutions have been developed yet to address the arising challenges from the analysis of phenotype data in medical, biological and translational contexts. Challenges include the differences in understanding of what a phenotype constitutes in different domains [53] (for example, the synonymous use of disease, syndrome, trait and phenotype), gaps in the terminologies, vocabularies and ontologies to represent phenotypes (for example, HPO with its 10,000 concepts has a lot of information about skeletal phenotypes but is sparse in other areas), missing annotations in databases (for example, diseases in OMIM are under annotated [54]), and jargon for individual domains (for example, automatically generating clusters from phenotypes from different species leads to mostly a cluster for certain species instead of a mixed cluster that would result from a shared terminology [55]). Most likely, similar to the generic Web environment, there will never be a ‘one size fits all’ solution; however, clearly defining biological and medical solutions will help to identify potential domain-specific breakthroughs, as well as highlight where improvements are required in order to keep pace with all the ongoing phenotype efforts.
Beyond keywords: towards intelligent tool support
Despite the fact that the existing text mining systems still need improvements, systems based on text mining from news articles are now being used to support analysts in detecting infectious disease outbreaks such as pandemic influenza [56]. In experimental biology, groups of researchers have come together to propose shared tasks such as the Natural Language Processing of Biology Text (BioNLP) [57] and BioCreative challenges [58] that support database curators and accelerate the flow of results from the literature back to the scientific community. BioCreative, for example, has led to developments in gene normalization, chemical and drug name recognition, as well as assigning evidence codes to gene function. In the clinical domain, initiatives such as the i2b2 challenge [59] are aimed at helping translate the findings from genomics research into the design of targeted therapies for heritable diseases such as rheumatoid arthritis, hypertension and multiple sclerosis. One common factor linking all of these fields together is the heterogeneous conceptual class of phenotypes.
Two necessary research objectives for intelligent tools are (a) recognizing in text the phrases that form phenotypes and (b) linking them to established pre-composed or post-composed concepts in ontologies. As an example, consider the pre-composed term ‘Abnormal Purkinje cell dendrite morphology’ from Figure 2. This might appear in various forms in free text such as ‘The mice have abnormalities in their Purkinje cell dendritic tree resulting in abnormal morphology’ and ‘Abnormal morphology of dendrites in Purkinje cells’. Success is likely to require a fusion of technologies: prior domain knowledge, natural language processing algorithms and reasoning. We approach this section by briefly surveying the technical issues surrounding these goals and ask if there is a robust technical solution on the near horizon.
Constructing full phenotype vocabularies manually is a daunting task. Despite the success of dedicated phenotype ontologies such as HPO, MP, FYPO and others, the situation regarding pre-composed terminological resources - those in which the term appears without a division into its constituents - is still far from ideal. Such resources have been designed with a focus on classical centralized model databases, such as OMIM or MGD, and specific user communities in mind. However, as Thorisson et al. [60] argue, the centralized database structure sometimes has difficulty in handling complex relationships. In the case of phenotypes, both the concepts and the disciplines that use them are heterogeneous. This makes standards of scope, granularity and compositionality difficult to establish. Moreover, the generation of one pre-composed ontology covering an entire domain (for example, all phenotypes within one particular species) would not be maintainable due to the sheer amount of existing phenotypes.
In time, algorithmic techniques may be developed to fill the gap between pre-composed ontologies and free text variations. One approach to bridging (also called linking [61], normalization [62] and grounding [63]) from text to ontology is to develop automated mapping algorithms. Examples of such applications, currently used on a large scale by the biomedical community, are MetaMap [64], which bridges text and the Unified Medical Language System (UMLS) [65], or the NCBO Annotator [66], which maps textual entries to entities defined by ontologies stored in the NCBO BioPortal. In general, these applications identify term candidates using shallow parsing, generate plausible alternative forms (synonyms) and then match them to the entities forming the knowledge base (for example, the UMLS). Many options and configurations exist, including the ability to include/exclude particular ontologies or semantic groups, or to detect the degree to which variant candidates differ from the original textual form. However, from a user perspective, it is not apparent what weighting to attach to different forms of evidence. Furthermore, one of the major shortfalls of these algorithms is that they match only single constituent phrases, missing coverage in more complex grammatical structures such as striking upslanting of the palpebral fissures, small nose with broad root or short neck with loose skin noted by Schofield et al. [53] in (OMIM:211750). They may also fail in finding associations between closely related but superficially different surface forms such as high blood pressure and hypertension. Finally, a related challenge is in identifying semantic equivalence across ontologies: for example, in cross-species analysis where equivalent phenotypes need to be identified in model organisms; for example, enlarged hind paws in mouse and enlarged feet in human [16]. Specific, phenotype and/or domain-oriented approaches have also been proposed based on data-driven learning, that is, machine learning, from labeled collections of texts and dictionaries (for example, [14,15,67]). In these examples, a software program learns from a small manually annotated data set whether a text span represents a phenotype or not, and can, after learning, be applied to more text to identify phenotype mentions. However, these represent mere pioneering efforts and require additional work in order to become reliable.
Another requirement of intelligent tools is the support of extensions and generation of mappings between ontological resources. As discussed earlier, phenotypes can be considered broadly as being compositional entities. For example, HP:0000365 - High frequency hearing loss consists of an anatomical process, GO:0007605 - Sensory perception of sound, and a quality, PATO:0002018 - Decreased magnitude, indicating an abnormality of the entity. Given the diverse nature of phenotypes, several researchers have suggested providing post-composed terms [27] in which the constituent parts are provided in a federated fashion by reference to external vocabulary systems. So far, production of post-composed terms has been mainly carried out by manual curation [28,68]. Lately, however, several automated approaches have been proposed, each of which relies on natural language processing techniques to convert terms from the pre-composed to the post-composed form [26,29,69].
Current studies for free-text phenotype recognition and normalization appear hampered by a lack of gold standard data used for training and evaluation and there is a danger that inferences about the best methods may be impaired. Developing accurate systems depends crucially on both an open communication across domains, so that a common understanding about phenotypes and the research needs surrounding them can be achieved, as well as on the development of annotation standards. Furthermore, high-quality large-scale data sets are needed for both trainable systems and benchmark evaluation. The process of collecting and publishing such datasets is time-consuming and costly. However, several projects, such as the IMPC, aim to deal with this challenge yet require time until they reach a certain level of maturity. The lack of open data is also apparent in the clinical domain where the desire to develop new patient treatments has to be balanced against ethical concerns about patient privacy. Steady progress is being made alongside the development of de-identification algorithms [70], as well as collaborative initiatives, such as i2b2, which bring together patient data providers and technologists.
Outlook
Even though initial work has been done in phenotype representation, acquisition and application, further steps are required in order to unlock the full potential of phenotype information, which in turn will drive the knowledge discovery process. Phenotype representations have to be harmonized across different species and a balance has to be found between terminologies used in communities and benefits across research domains. The complexity of phenotype information still hinders the development of a consistent formalization and prevents seamless integration of and data mining across diverse resources. The ongoing Linked Open Data efforts (for example, Bio2RDF [71]) provides access to increasing amounts of phenotype data that require a unified representation, which would then facilitate the creation of a broader picture surrounding hypotheses derivation from the data.
On a different note, promising first steps have been achieved in the domain of cross-species hypotheses generation. However, the benefits are impacted by both representation and acquisition. With the ever-growing amount of data, manual assessments are at this point infeasible and automated methods to analyze the data are urgently required. Due to a lack of a uniform representation of phenotypes across different domains, integration and consequently knowledge propagation are interrupted. The best benefits can be achieved with a complete and consistent coverage of the up-to-date knowledge about phenotypes and their influencing factors that enable hypotheses generation and derivation of novel findings. From a different perspective, the acquisition of phenotype data could also be tremendously improved through solving mismatched expectations. While a small subset of specific and a large set of generalized solutions exist, cross-community and cross-domain efforts are required to enable a better fit of generalized solutions to existing problems, and specific solutions to be repurposed to other problems. A clear and common understanding about existing problems and possible solutions is required that can only be achieved through open communication. Open communication will allow us to advance research in the field and to derive future solutions that target well-specified, real issues.
Furthermore, automated and supported acquisition of data is only possible with reliable methods. In recent years significant and welcome progress has been made in systematic evaluation of data-driven techniques through shared tasks like BioCreative and BioNLP. On the other hand, text mining progress has sometimes been behind the expectation of user communities due to inaccuracies in system output. This is largely because the language being processed is inherently ambiguous and requires new techniques and resources; for example, cross-domain event extraction, grounding, term decomposition, and harmonized understanding at a document-wide level. Phenotype concept recognition in text is a key non-trivial task that now needs to be addressed. Complex event extraction and normalization involving phenotypes are foundation tasks that need attention from the technical community to deliver working solutions into the hands of users.
Common representation formats for mark-up in text is also important, in particular for phenotype data, and the efforts made over the years by BioCreative and BioNLP should be closely followed. This should be aided by closer dialogue between the text mining, curator and biology communities. Developments in community dialogue on gold standards and system critiques could follow the encouraging model of the User Advisor Group in BioCreative 2011, leading to new approaches for enhancing the user experience.
In conclusion, we believe that improved communication would enable a common understanding across the different research domains and speed-up the development of solutions for most of the existing technical issues. Additional workshops are needed to allow researchers to gather and exchange phenotype resources, including their interpretation, representation, mining and integration. Once a shared mindset has been achieved, all four steps mentioned in this paper will reach a streamlining phase and will hence support translational research attain its real potential.
Abbreviations
BioNLP: Natural language processing of biology text; CNV: Copy number variation; DDD: Deciphering developmental disorders; DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources; EQ: Entity/quality; FMA: Foundation model of anatomy; FYPO: Worm phenotype ontology; GO: Gene ontology; HPO: Human phenotype ontology; IMPC: International mouse phenotyping Consortium; MA: Mouse adult gross anatomy ontology; MGD: Mouse genome database; MP: Mouse phenotype ontology; NCBO: National center for biomedical Ontology; OBO: Open biological and biomedical Ontologies; OMIM: Online Mendelian Inheritance of Man database; PATO: Phenotype and trait ontology; UMLS: Unified Medical language system; WP: Worm phenotype ontology.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors contributed equally to the final version of this manuscript.
Contributor Information
Nigel Collier, Email: collier@ebi.ac.uk.
Anika Oellrich, Email: ao5@sanger.ac.uk.
Tudor Groza, Email: tudor.groza@uq.edu.au.
Acknowledgements
The authors would like to thank Damian Smedley for providing the general idea of Figure 3. Nigel Collier’s research is supported by the European Commission through the Marie Curie International Incoming Fellowship (IIF) programme (Project: Phenominer, Ref: 301806). Tudor Groza’s research is funded by the Australian Research Council (ARC) Discovery Early Career Researcher Award (DECRA) - DE120100508.
References
- Herskowitz IH. Principles of Genetics. New York: Macmillan; 1977. [Google Scholar]
- McGary KL, Park TJ, Woods JO, Cha HJ, Wallingford JB, Marcotte EM. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc Natl Acad Sci U S A. 2010;14:6544–6549. doi: 10.1073/pnas.0910200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R, Schofield PN, Gkoutos GV. PhenomeNET: A whole-phenome approach to disease gene discovery. Nucleic Acids Res. 2011;14:e119. doi: 10.1093/nar/gkr538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington NL, Haendel MA, Mungall CJ, Ashburner M, Westerfield M, Lewis SE. Linking human diseases to animal models using ontology-based phenotype annotation. PLoS Biol. 2009;14:e1000247. doi: 10.1371/journal.pbio.1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabee PM, Ashburner M, Cronk Q, Gkoutos GV, Haendel M, Segerdell E, Mungall C, Westerfield M. Phenotype ontologies: the bridge between genomics and evolution. Trends Ecol Evol. 2007;14:345–350. doi: 10.1016/j.tree.2007.03.013. [DOI] [PubMed] [Google Scholar]
- [ http://phenomebrowser.net/]
- Mabee P, Balhoff JP, Dahdul WM, Lapp H, Midford PE, Vision TJ, Westerfield M. 500,000 fish phenotypes: The new informatics landscape for evolutionary and developmental biology of the vertebrate skeleton. J Appl Ichthyol. 2012;14:300–305. doi: 10.1111/j.1439-0426.2012.01985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freimer N, Sabatti C. The human phenome project. Nat Genet. 2003;14:15–21. doi: 10.1038/ng0503-15. [DOI] [PubMed] [Google Scholar]
- Smedley D, Swertz MA, Wolstencroft K, Proctor G, Zouberakis M, Bard J, Hancock JM, Schofield P. Solutions for data integration in functional genomics: a critical assessment and case study. Brief Bioinform. 2008;14:532–544. doi: 10.1093/bib/bbn040. [DOI] [PubMed] [Google Scholar]
- Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;14:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Goldsmith C, Eppig J. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005;14:R7. doi: 10.1186/gb-2005-6-5-p7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SD, Moore MW. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm Genome. 2012;14:632–640. doi: 10.1007/s00335-012-9427-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [ http://www.ddduk.org/]
- Khordad M, Mercer RE, Rogan P. Advances in Artificial Intelligence. 24th Canadian Conference on Artificial Intelligence: May 25–27 2011. St John’s, Canada. New York: Springer; 2011. Improving phenotype name recognition; pp. 246–257. [Google Scholar]
- Collier N, Tran MV, Le HQ, Oellrich A, Kawazoe A, MartinHall-May, Rebholz-Schuhmann D. Proceedings of the 24th International Conference on Computational Linguistics: December 8–15, 2012. Mumbai. Mumbai: The COLING 2012 Organizing Committee, Indian Institute of Technology; 2012. A hybrid approach to finding phenotype candidates in genetic texts; pp. 647–662. [Google Scholar]
- Chen CK, Mungall CJ, Gkoutos GV, Doelken SC, Koehler S, Ruef BJ, Smith C, Westerfield M, Robinson PN, Lewis SE, Schofield PN, Smedley D. MouseFinder: candidate disease genes from mouse phenotype data. Hum Mutat. 2012;14:858–866. doi: 10.1002/humu.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R, Oellrich A, Rebholz-Schuhmann D, Schofield PN, Gkoutos GV. Proceedings of the Pacific Symposium on Biocomputing: January 3–7, 2012. Hawaii. Singapore: World Scientific; 2012. Linking pharmgkb to phenotype studies and animal models of disease for drug repurposing; pp. 388–399. [PubMed] [Google Scholar]
- [ http://www.sanger.ac.uk/resources/databases/phenodigm/]
- Amberger J, Bocchini C, Hamosh A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM) Hum Mutat. 2011;14:564–567. doi: 10.1002/humu.21466. [DOI] [PubMed] [Google Scholar]
- Schindelman G, Fernandes JS, Bastiani CA, Yook K, Sternberg PW. Worm Phenotype Ontology: Integrating phenotype data within and beyond the C. elegans community. BMC Bioinformatics. 2011;14:32. doi: 10.1186/1471-2105-12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood V, Harris MA, McDowall MD, Rutherford K, Vaughan BW, Staines DM, Aslett M, Lock A, Baehler J, Kersey PJ, Oliver SG. PomBase: a comprehensive online resource for fission yeast. Nucleic Acids Res. 2012;14:D695–D699. doi: 10.1093/nar/gkr853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy NF, Shah NH, Whetzel PL, Dai B, Dorf M, Griffith N, Jonquet C, Rubin DL, Storey MA, Chute CG, Musen MA. BioPortal: Ontologies and integrated data resources at the click of a mouse. Nucleic Acids Res. 2009;14:W170–W173. doi: 10.1093/nar/gkp440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B, Ashburner M, Rosse C, Bard J, Bug W, Ceusters W, Goldberg LJ, Eilbeck K, Ireland A, Mungall CJ, Consortium TO, Leontis N, Rocca-Serra P, Ruttenberg A, Sansone SA, Scheuermann RH, Shah N, Whetzel PL, Lewis S. The OBO Foundry: coordinated evolution of ontologies to support biomedical data integration. Nat Biotechnol. 2007;14:1251–1255. doi: 10.1038/nbt1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, AP APD, Dolinski K, Dwight S, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;14:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkoutos GV, Mungall C, Doelken S, Ashburner M, Lewis S, Hancock J, Schofield P, Koehler S, Robinson PN. Proceedings of the 31st Annual International Conference of the IEEE EMBS: September 2–6, 2009; Minneapolis. Piscataway, NJ: IEEE; 2009. Entity/quality-based logical definitions for the human skeletal phenome using PATO; pp. 7069–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groza T, Hunter J, Zankl A. Decomposing phenotype descriptions for the human skeletal phenome. Biomed Inform Insights. 2013;14:1–14. doi: 10.4137/BII.S10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall CJ, Gkoutos GV, Smith CL, Haendel MA, Lewis SE, Ashburner M. Integrating phenotype ontologies across multiple species. Genome Biol. 2010;14:R2. doi: 10.1186/gb-2010-11-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahdul WM, Balhoff JP, Engeman J, Grande T, Hilton EJ, Kothari C, Lapp H, Lundberg JG, Midford PE, Vision TJ, Westerfield M, Mabee PM. Evolutionary characters, phenotypes and ontologies: curating data from the systematic biology literature. PLoS One. 2010;14:e10708. doi: 10.1371/journal.pone.0010708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oellrich A, Grabmueller C, Rebholz-Schuhmann D. Proceedings of the 2012 OMBL Workshop: 2012 Dresden. Germany: University of Leipzig, Institute for Medical Informatics, Statistics and Epidemiology (IMISE); 2012. Automatically transforming pre-to post-composed phenotypes: EQ-lising HPO and MP; pp. E1–E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosse C, Mejino JLV. A reference ontology for biomedical informatics: the Foundational Model of Anatomy. J Biomed Inform. 2003;14:478–500. doi: 10.1016/j.jbi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Hayamizu TF, Mangan M, Corradi JP, Kadin JA, Ringwald M. The Adult Mouse Anatomical Dictionary: a tool for annotating and integrating data. Genome Biol. 2005;14:R29. doi: 10.1186/gb-2005-6-3-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson SD, Fong LL, Gupta A, Condit C, Bug WJ, Martone ME. A formal ontology of subcellular neuroanatomy. Front Neuroinform. 2007;14:3. doi: 10.3389/neuro.11.003.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B, Ceusters W, Klagges B, Kohler J, Kumar A, Lomax J, Mungall CJ, Neuhaus F, Rector A, Rosse C. Relations in biomedical ontologies. Genome Biol. 2005;14:R46. doi: 10.1186/gb-2005-6-5-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oellrich A, Rebholz-Schuhmann D. A classification of existing phenotypical representations and methods for improvement. Proceedings of the 2010 OMBL Workshop: Mannheim, Germany. 2010. pp. J1–J4.
- Bult CJ, Kadin JA, Richardson JE, Blake JA, Eppig JT. the Mouse Genome Database Group. The Mouse Genome Database: enhancements and updates. Nucleic Acids Res. 2009;14:D586–D592. doi: 10.1093/nar/gkp880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan GJ, Bragin E, Chatzimichali EA, Corpas M, Bevan AP, Wright CF, Carter NP, Hurles ME, Firth HV. DECIPHER: web-based, community resource for clinical interpretation of rare variants in developmental disorders. Hum Mol Genet. 2012;14:R37–R44. doi: 10.1093/hmg/dds362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doelken SC, Koehler S, Mungall CJ, Gkoutos GV, Ruef BJ, Smith C, Smedley D, Bauer S, Klopocki E, Schofield PN, Westerfield M, Robinson PN, Lewis SE. Phenotypic overlap in the contribution of individual genes to CNV pathogenicity revealed by cross-species computational analysis of single-gene mutations in humans, mice and zebrafish. Dis Model Mech. 2013;14:358–372. doi: 10.1242/dmm.010322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [ http://phenomebrowser.net/phenomedrug.html]
- Pesquita C, Faria D, Falcao AO, Lord P, Couto FM. Semantic similarity in biomedical ontologies. PLoS Comp Biol. 2009;14:e1000443. doi: 10.1371/journal.pcbi.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S, Bauer S, Mungall CJ, Carletti G, Smith CL, Schofield P, Gkoutos GV, Robinson PN. Improving ontologies by automatic reasoning and evaluation of logical definitions. BMC Bioinformatics. 2011;14:418. doi: 10.1186/1471-2105-12-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S, Schulz MH, Krawitz P, Bauer S, Dolken S, Ott CE, Mundlos C, Horn D, Mundlos S, Robinson PN. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;14:457–464. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul R, Groza T, Zankl A, Hunter J. Proceedings of the 11th International Semantic Web Conference, November 11–15 2012; Boston. New York: Springer; 2012. Semantic similarity-driven decision support in the skeletal dysplasia domain; pp. 164–179. [Google Scholar]
- [ http://www.orpha.net/]
- Köhler S, Doelken SC, Rath A, Aymé S, Robinson PN. Ontological phenotype standards for neurogenetics. Hum Mutat. 2012;14:1333–1339. doi: 10.1002/humu.22112. [DOI] [PubMed] [Google Scholar]
- Groth P, Pavlova N, Kalev I, Tonov S, Georgiev G, Pohlenz HD, Weiss B. PhenomicDB: a new cross-species genotype/phenotype resource. Nucleic Acids Res. 2007;14:D696–D699. doi: 10.1093/nar/gkl662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam LT, Mendonca EA, Li J, Blake J, Friedman C, Lussier YA. PhenoGO: an integrated resource for the multiscale mining of clinical and biological data. BMC Bioinformatics. 2009;14:S8. doi: 10.1186/1471-2105-10-S2-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague J, Bayraktaroglu L, Clements D, Conlin T, Fashena D, Frazer K, Haendel M, Howe DG, Mani P, Ramachandran S, Schaper K, Segerdell E, Song P, Sprunger B, Taylor S, Slyke CEV, Westerfield M. The Zebrafish Information Network: the zebrafish model organism database. Nucleic Acids Res. 2006;14:D581–D585. doi: 10.1093/nar/gkj086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonagh EM, Whirl-Carrillo M, Garten Y, Altman RB, Klein TE. From pharmacogenomic knowledge acquisition to clinical applications: the PharmGKB as a clinical pharmacogenomic biomarker resource. Biomark Med. 2011;14:795–806. doi: 10.2217/bmm.11.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten Y, Altman RB. Pharmspresso: a text mining tool for extraction of pharmacogenomic concepts and relationships from full text. BMC Bioinformatics. 2009;14:S6. doi: 10.1186/1471-2105-10-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Driel MA, Bruggeman J, Vriend G, Brunner HG, Leunissen JA. A text-mining analysis of the human phenome. Eur J Hum Genet. 2006;14:535–542. doi: 10.1038/sj.ejhg.5201585. [DOI] [PubMed] [Google Scholar]
- [ http://www.cmbi.ru.nl/MimMiner/cgi-bin/main.pl]
- Sardana D, Vasa S, Vepachedu N, Chen J, Gudivada RC, Aronow BJ, Jegga AG. PhenoHM: human-mouse comparative phenome-genome server. Nucleic Acids Res. 2010;14:W165–W174. doi: 10.1093/nar/gkq472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield PN, Gkoutos GV, Gruenberger M, Sundberg JP, Hancock JM. Phenotype ontologies for mouse and man: bridging the semantic gap. Dis Mod Mech. 2010;14:281–289. doi: 10.1242/dmm.002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oti M, Huynen MA, Brunner HG. The biological coherence of human phenome databases. Am J Hum Genet. 2009;14:801–808. doi: 10.1016/j.ajhg.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth P, Weiss B, Pohlenz HD, Leser U. Mining phenotypes for gene function prediction. BMC Bioinformatics. 2008;14:136. doi: 10.1186/1471-2105-9-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier N, Doan S, Kawazoe A, Goodwin RM, Conway M, Tateno Y, Ngo QH, Dien D, Kawtrakul A, Takeuchi K, Shigematsu M, Taniguchi K. BioCaster: detecting public health rumors with a Web-based text mining system. Bioinformatics. 2008;14:2940–2941. doi: 10.1093/bioinformatics/btn534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JD, Pyysalo S, Ohta T, Bossy R, Nguyen N, Tsujii JI. Proceedings of the BioNLP Shared Task 2011 Workshop, Association for Computational Linguistics: June 24, 2011; Portland. New York: Curran Associates; 2012. Overview of BioNLP shared task 2011; pp. 1–6. [Google Scholar]
- Morgan AA, Lu Z, Wang X, Cohen AM, Fluck J, Ruch P, Divoli A, Fundel K, Leaman R, Hakenberg J, Sun C, Hui Liu H, Torres R, Krauthammer M, Lau WW, Liu H, Hsu CN, Schuemie M, Cohen KB, Hirschman L. Overview of BioCreative II gene normalization. Genome Biol. 2008;14:S3. doi: 10.1186/gb-2008-9-s2-s3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SN, Weber G, Mendis M, Gainer V, Chueh HC, Churchill S, Kohane I. Serving the enterprise and beyond with informatics for integrating biology and the bedside (i2b2) J Am Med Inform Assoc. 2010;14:124–130. doi: 10.1136/jamia.2009.000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorisson GA, Muilu J, Brookes AJ. Genotype-phenotype databases: challenges and solutions for the post-genomic era. Nat Rev Genet. 2009;14:9–18. doi: 10.1038/nrg2483. [DOI] [PubMed] [Google Scholar]
- Spasic I, Ananiadou S, McNaught J, Kumar A. Text mining and ontologies in biomedicine: Making sense of raw text. Brief Bioinform. 2005;14:239–251. doi: 10.1093/bib/6.3.239. [DOI] [PubMed] [Google Scholar]
- Morgan AA, Hirschman L, Colosimo M, Yeh AS, Colombe JB. Gene name identification and normalization using a model organism database. J Biomed Inform. 2004;14:396–410. doi: 10.1016/j.jbi.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Cohen KB, Ogren PV, Fox L, Hunter L. Proceedings of the ACL-ISMB Workshop on Linking Biological Literature, Ontologies and Databases: Mining Biological Semantics, Association for Computational Linguistics: June 24, 2005; Detroit. Pennsylvania: Association for Computational Linguistics; 2005. Corpus design for biomedical natural language processing; pp. 38–45. [Google Scholar]
- Aronson AR. Proceedings of the American Medical Informatics Association Symposium 2001. Bethesda: American Medical Informatics Association; 2001. Effective mapping of biomedical text to the UMLS Metathesaurus: the MetaMap program; pp. 17–21. [PMC free article] [PubMed] [Google Scholar]
- Bodenreider O. The Unified Medical Language System (UMLS): integrating biomedical terminology. Nucleic Acids Res. 2004;14:D267–D270. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonquet C, Shah NH, Musen MA. Proceedings of the Summit on Translational Bioinformatics: March 15–17, 2009; San Francisco. Bethesda: American Medical Informatics Association; 2009. The Open Biomedical Annotator; pp. 56–60. [PMC free article] [PubMed] [Google Scholar]
- Groza T, Hunter J, Zankl A. Mining skeletal phenotype descriptions from scientific literature. PLoS One. 2013;14:e55656. doi: 10.1371/journal.pone.0055656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balhoff JP, Dahdul WM, Kothari CR, Lapp H, Lundberg JG, Mabee P, Midford PE, Westerfield M, Vision TJ. Phenex: Ontological annotation of phenotypic diversity. PLoS One. 2010;14:e10500. doi: 10.1371/journal.pone.0010500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midford P, Mabee P, Vision T, Lapp H, Balhoff J, Dahdul W, Kothari C, Lundberg J, Westerfield M. Phenoscape: Ontologies for large multi-species phenotype datasets. Nat Precedings. 2009.
- Uzuner O, Luo Y, Szolovits P. Evaluating the state-of-the-art in automatic de-identification. J Am Med Inform Assoc. 2007;14:550–563. doi: 10.1197/jamia.M2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belleau F, Nolin MA, Tourigny N, Rigault P, Morissette J. Bio2RDF: towards a mashup to build bioinformatics knowledge systems. J Biomed Inform. 2008;14:706–716. doi: 10.1016/j.jbi.2008.03.004. [DOI] [PubMed] [Google Scholar]