

Abstract
Background
We have used a sensitized ENU mutagenesis screen to produce mouse lines that carry mutations in genes required for epigenetic regulation. We call these lines Modifiers of murine metastable epialleles (Mommes).
Results
We report a basic molecular and phenotypic characterization for twenty of the Momme mouse lines, and in each case we also identify the causative mutation. Three of the lines carry a mutation in a novel epigenetic modifier, Rearranged L-myc fusion (Rlf), and one gene, Rap-interacting factor 1 (Rif1), has not previously been reported to be involved in transcriptional regulation in mammals. Many of the other lines are novel alleles of known epigenetic regulators. For two genes, Rlf and Widely-interspaced zinc finger (Wiz), we describe the first mouse mutants. All of the Momme mutants show some degree of homozygous embryonic lethality, emphasizing the importance of epigenetic processes. The penetrance of lethality is incomplete in a number of cases. Similarly, abnormalities in phenotype seen in the heterozygous individuals of some lines occur with incomplete penetrance.
Conclusions
Recent advances in sequencing enhance the power of sensitized mutagenesis screens to identify the function of previously uncharacterized factors and to discover additional functions for previously characterized proteins. The observation of incomplete penetrance of phenotypes in these inbred mutant mice, at various stages of development, is of interest. Overall, the Momme collection of mouse mutants provides a valuable resource for researchers across many disciplines.
Background
Mutagenesis screens for modifiers of position effect variegation in Drosophila have played a defining role in the development of the field of epigenetics [1]. The screens used a fly strain that showed variegated expression of the white (w) locus, resulting in red and white patches in the eye, as a result of the stochastic establishment of epigenetic state. The genes identified by these screens turn out to have pivotal roles in gene silencing [2-4]. We have designed a similar screen in the mouse, using a green fluorescent protein (GFP) transgene that shows variegated expression in red blood cells. Offspring of N-ethyl-N-nitrosourea (ENU)-treated males are screened for changes in the percentage of erythrocytes expressing GFP; this is a screen for dominant effects. Each mutant line is named a Modifier of murine metastable epiallele Dominant, MommeD[5]. The underlying mutations have been identified and published for nine lines and the mutations occur in DNA methyltransferases (Dnmt1 and Dnmt3b), chromatin remodelers (Smarca5 and Baz1b), a histone deacetylase (Hdac1), a transcriptional co-repressor (Trim28), a eukaryotic translation initiation factor (eIF3h) [6-10] and a previously unknown gene, Smchd1, now shown to be required for X-inactivation in the mouse [11,12]. Recently, Smchd1 has been shown to act as a tumor suppressor [13] and mutations in SMCHD1 have been shown to be tightly associated with the human disease facioscapulohumeral dystrophy type 2 (FSHD2) [14].
Results and discussion
Identification of Momme mutants
We have now screened a total of approximately 5,000 G1 offspring, recovered 42 MommeD lines and identified the underlying mutations in 29 cases. These lines carry mutations in 18 unique genes (Table 1). A number of the genes have been hit more than once. Based on their effect on GFP expression, 14 lines were classified as suppressors of variegation, that is, the mutation increased the percentage of erythrocytes expressing GFP, and 15 lines were classified as enhancers of variegation, that is, the mutation decreased the percentage of erythrocytes expressing GFP (Table 1). Here we report, for the first time, the underlying mutations in 20 MommeD lines, which carry mutations in 10 unique genes.
Table 1.
MommeD mutants, causative mutations and disease association
| Name | Effect on variegation | Gene | Nature of mutation | Homozygous phenotype | References | Human homolog | Disease association |
|---|---|---|---|---|---|---|---|
|
MommeD1 |
Suppressor |
Smchd1 |
C->T exon 23; introduces Stop |
Null |
[5,11] |
SMCHD1 |
FSHD2 [14] |
|
MommeD2 |
Suppressor |
Dnmt1 |
C->A exon 25; T812K |
Null |
[6] |
DNMT1 |
Schizophrenia, breast and prostate cancer [15-19] |
|
MommeD4 |
Enhancer |
Smarca5 |
T->A exon 12; W520R |
Hypomorphic? |
[5,6] |
SMARCA5 |
Acute myeloid leukemia [20] |
|
MommeD5 |
Enhancer |
Hdac1 |
7 bp deletion in exon 13; Frameshift |
Null |
[5,7] |
HDAC1 |
Schizophrenia, neural development [2] |
|
MommeD8 |
Enhancer |
Rlf |
G->T exon 8; C1558F |
Hypomorphic |
This study |
RLF |
|
|
MommeD9 |
Enhancer |
Trim28 |
T->C splice donor site of intron 13 |
Null |
[8] |
TRIM28 |
|
|
MommeD10 |
Enhancer |
Baz1b |
T->G exon 7; L733R |
Hypomorphic |
[7] |
BAZ1B |
Williams-Beuren syndrome [21] |
|
MommeD12 |
Enhancer |
eIF3h |
T->A - 10 bp before splice acceptor site of intron 4 |
Null |
[9] |
eIF3H |
|
|
MommeD13 |
Suppressor |
Setdb1 |
A->G exon 20; results in splicing defect |
Null |
This study |
SETDB1 |
Melanoma [22] |
|
MommeD14 |
Suppressor |
Dnmt3b |
T->C splice acceptor site of intron 12 |
Hypomorphic |
[10] |
DNMT3B |
ICF syndrome [23] |
|
MommeD16 |
Enhancer |
Baz1b |
T->C exon 2; L75P |
Hypomorphic? |
This study |
BAZ1B |
Williams-Beuren syndrome [21] |
|
MommeD17 |
Suppressor |
Setdb1 |
T->C exon 21; V1248A |
Hypomorphic |
This study |
SETDB1 |
Melanoma [22] |
|
MommeD18 |
Suppressor |
Rif1 |
C->T exon 29; Q1669 Stop |
Null |
This study |
RIF1 |
Breast cancer [24] |
|
MommeD19 |
Suppressor |
Smarcc1 |
T->G splice acceptor site of intron 10 |
Null |
This study |
SMARCC1 |
Colorectal cancer [25,26] |
|
MommeD23 |
Suppressor |
Smchd1 |
A->T exon 12; R498 Stop |
Null? |
This study |
SMCHD1 |
FSHD2 [14] |
|
MommeD27 |
Suppressor |
Pbrm1 |
A->G exon 17; Y733C |
Hypomorphic? |
This study |
PBRM1 |
Renal cancer [27] |
|
MommeD28 |
Enhancer |
Rlf |
A->G splice acceptor site of intron 4 |
Null |
This study |
RLF |
|
|
MommeD30 |
Enhancer |
Wiz |
1 bp deletion in exon 5; Frameshift at amino acid 553 |
Null |
This study |
WIZ |
|
|
MommeD31 |
Enhancer |
Trim 28 |
T->A exon 3; C178S |
Null? |
This study |
TRIM28 |
|
|
MommeD32 |
Suppressor |
Dnmt1 |
T->C exon 29; L1045P |
Null? |
This study |
DNMT1 |
Schizophrenia, breast and prostate cancer [15-19] |
|
MommeD33 |
Suppressor |
Suv39h1 |
A->G splice donor site of intron 1 |
Null |
This study |
SUV39H1 |
Lupus, retinoblastoma [2] |
|
MommeD34 |
Enhancer |
Rlf |
C->A exon 7; C355 Stop |
Null |
This study |
RLF |
|
|
MommeD35 |
Enhancer |
Smarca5 |
A-> G exon 9; N341S |
Hypomorphic? |
This study |
SMARCA5 |
Acute myeloid leukemia [20] |
|
MommeD36 |
Suppressor |
Smchd1 |
C->T exon 42; Q1732 Stop |
Null? |
This study |
SMCHD1 |
FSHD2 [14] |
|
MommeD37 |
Enhancer |
Smarca5 |
T->C exon 13; L565P |
Null? |
This study |
SMARCA5 |
Acute myeloid leukemia [20] |
|
MommeD38 |
Enhancer |
eIF3h |
G->A exon 7; R291 Stop |
Null |
[9] |
eIF3H |
|
|
MommeD39 |
Suppressor |
Smarca4 |
G->A splice donor site of intron 20 |
Null |
This study |
SMARCA4 |
Coffin-Siris syndrome [28] |
|
MommeD40 |
Suppressor |
Uhrf1 |
T->A exon17; Y778 Stop |
Null |
This study |
UHRF1 |
|
| MommeD42 | Enhancer | Brd1 | T->A exon 11; C411 Stop | Null | This study | BRD1 | Schizophrenia, bipolar affective disorder [29] |
In some cases the mutation can be defined as hypomorphic or null and in other cases this designation is less certain and we have added a question mark.
The experimental pipeline for the screen is shown in Figure 1. In brief, ENU mutagenesis was carried out as described previously [5] and mapping was carried out following a G2 backcross to Line3C (a C57BL/6J strain carrying the same GFP transgene array at the same location) using traditional microsatellite and SNP genotyping or an Illumina GoldenGate SNP genotyping assay. Mapping intervals for the latter group are shown as Manhattan plots in Additional file 1.
Figure 1.
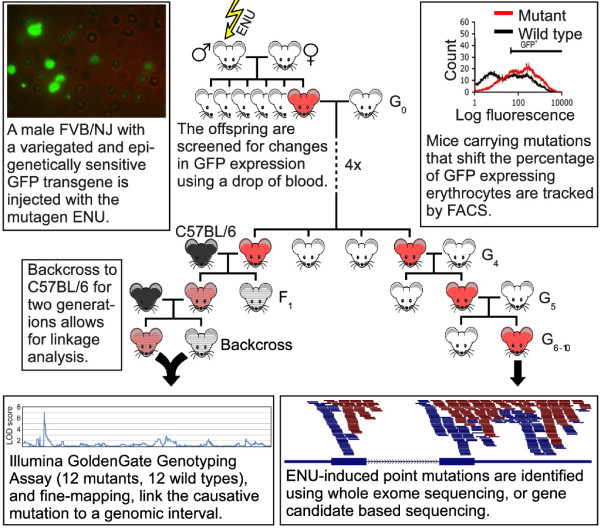
An ENU mutagenesis/whole exome deep sequencing pipeline enables rapid identification of causative mutations in mice with defects in epigenetic gene silencing. A schematic overview of the ENU mutagenesis gene discovery pipeline is presented. The major components of the screen are described in the figure. Briefly, male FVB/NJ mice carrying an epigenetically sensitive GFP transgene (Line3) were treated with ENU and mated with female Line3 mice. Offspring were screened for a shift in the percentage of GFP expressing erythrocytes using flow cytometry. Putative mutants were mated with Line3 mice for four generations to test for heritability and reproducibility of the GFP expression, and to reduce the number of non-causative ENU mutations within the genomes. Linkage analysis was carried out on the offspring from two generations of backcrosses between putative mutants and C57BL/6J mice, which also carry the GFP transgene (Line3C). Causative mutations were identified by whole exome deep sequencing, or gene candidate sequencing on individuals that had been maintained on the Line3 background for at least seven generations. FACS, fluorescence-activated cell sorting.
To identify the underlying mutations, several different strategies have been used. In ten cases, the mutation was identified following whole exome deep sequencing of DNA from one mutant mouse from the MommeD inbred colony. For these, variants were called within the intervals linked to the causative mutation. For some lines, variant calling was extended to exome-wide, to assess for passenger mutations but only a small number was found (<10). These did not reflect the typical ENU sequence bias (T/A) [30] and probably represent normal background mutations.
In two instances, we have used a custom capture array to specifically enrich for DNA from a 4.2 Mbp interval previously identified by linkage analysis [7] followed by deep sequencing to identify the underlying mutations. In the remaining eight cases, Sanger sequencing of the exons (including splice sites) of candidate genes in the linked intervals was carried out. Details on identification of the mutations are provided in Materials and methods. In all cases, putative mutations were verified by PCR and Sanger sequencing in larger cohorts (at least 100 mice per mutant line).
In the two cases in which a custom capture array was used, we had an opportunity to obtain an estimate of the ENU mutation rate. In one case four mutations were found in the 4.2 Mbp captured interval and in the other case six mutations were found. Based on these results we estimated the ENU mutation rate at approximately 1 per Mbp. This is consistent with reports by others [31].
The heritability of the mutations was tested over at least six generations. Flow cytometric expression profiles and the percentage of erythrocytes expressing the transgene for each MommeD (presented here for the first time) are shown in Additional file 2.
MommeD30 mice are haploinsufficient for Widely interspaced zinc finger (Wiz)
MommeD30 was classified as an enhancer of variegation. We mapped the MommeD30 mutation to a 1.9 Mbp interval on chromosome 17 encompassing 48 genes (Additional file 3). Exome deep sequencing identified a single base deletion in exon 5 of the Wiz gene, causing a frame shift mutation that is predicted to introduce a premature stop codon (Figure 2a and Table 1). No other putative ENU variants were identified in the linked region.
Figure 2.
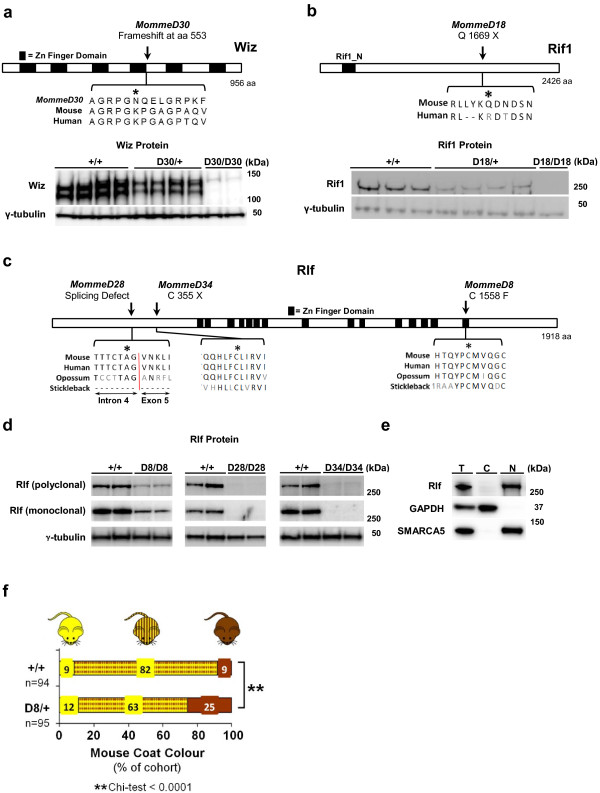
Causative mutations in MommeD30, MommeD18, MommeD8, MommeD28 and MommeD34. (a)MommeD30 carries a 1 bp deletion in Wiz that leads to a frame-shift at amino acid 553. Western blot analysis of embryo heads at 12.5 days post-coitum (dpc) shows reduced levels of Wiz protein in WizMommeD30 heterozygotes and no Wiz protein in homozygotes. Wiz protein was detected at approximately 120 and approximately 130 kDa. Each track represents a different animal. Tubulin was used as a loading control. (b)MommeD18 harbors a point mutation in Rif1 that introduces a premature stop codon at amino acid 1,669. Western analysis of testes tissue shows that RifMommeD18 heterozygotes have a reduced dosage of Rif1. No Rif1 protein was detected in a homozygote. Each track represents a different animal. Rif1 protein was detected at approximately 260 kDa and Tubulin was used as a loading control. (c) ENU point mutations in MommeD8, MommeD28 and MommeD34 occur in conserved regions of the Rlf protein. Asterisks indicate mutation. (d) Western blotting of Rlf in embryo head lysates from 14.5 dpc Rlf+/+, RlfMommeD8/D8, RlfD28/D28 and RlfD34/D34 revealed greatly reduced amounts of Rlf protein. Each track represents a different animal. Rlf protein was detected at approximately 280 kDa. Tubulin was used as a loading control. (e) Total, cytoplasmic and nuclear fractions of HeLa cells were isolated and protein concentration quantified. Equal amounts of each fraction were immunoblotted with anti-Rlf, GAPDH (cytoplasmic marker) and SMARCA5 (nuclear marker) antibodies, revealing nuclear localization of RLF. Rlf protein was detected at approximately 280 kDa, GAPDH protein at 37 kDa and SMARCA5 at approximately 120 kDa. (f) Coat color of offspring carrying the Avy allele produced from a RlfMommeD8 heterozygous female crossed to a pseudoagouti Avy male. RlfMommeD8 heterozygous offspring showed a shift in coat color towards pseudoagouti compared to wild-type littermates.
Wiz contains six Kruppel-type zinc finger motifs in a widely interspaced manner and has been reported to associate with the histone H3 lysine 9 (H3K9) methyltransferases G9a and GLP in cell lines [32,33]. No mouse mutants for Wiz have so far been described. Western blot analysis using an anti-Wiz antibody detected two bands (about 120 and 130 kDa), as expected, in wild-type embryos (Figure 2a). Embryos heterozygous for the MommeD30 mutation showed approximately half the amount of Wiz protein and no Wiz protein was detected in homozygotes (Figure 2a). We conclude that MommeD30 mice are haploinsufficient for Wiz. We refer to this allele as WizMommeD30.
MommeD18 carries a mutation in Rap interacting factor 1 (Rif1)
MommeD18 was classified as a suppressor of variegation and candidate gene sequencing revealed that MommeD18 mice carry a nonsense mutation in exon 29 of the Rif1 gene (Figure 2b and Table 1). Two other candidate genes (Epc2 and Mbd5) were sequenced and no mutations were identified in these genes. Western blot analysis showed approximately half the amount of full-length Rif1 protein in MommeD18 heterozygotes and no full-length Rif1 protein in a homozygote (Figure 2b). Furthermore, a Rif1 gene trap allele (Materials and methods) had a similar effect on transgene expression as that observed with the MommeD18 mutation, increasing the percentage of expressing cells in mice heterozygous for the gene-trap allele (Additional file 4).
We conclude that the MommeD18 allele is the result of a mutation in Rif1 that we refer to as Rif1MommeD18. Rif1 has recently been shown to be involved in DNA double strand break repair [34-37] and replication timing in mammals [38,39], both processes involving significant chromatin reorganization. Our findings suggest a role for Rif1 in transcription.
MommeD8, MommeD28 and MommeD34 carry mutations in Rearranged L-myc fusion (Rlf), a gene about which little is known
Three enhancers of variegation, MommeD8, MommeD28 and MommeD34, were found to carry mutations in Rlf. Whole interval capture revealed that MommeD8 and MommeD34 mice have mutations in exon 8 and exon 7, respectively. MommeD8 is a missense mutation and MommeD34 is a nonsense mutation (Figure 2c and Table 1). The MommeD28 mutation was identified by candidate sequencing and affects the splice acceptor site of intron 4 (Figure 2c and Table 1). Western blot analysis using two independent anti-Rlf antibodies (one polyclonal and one monoclonal) detected an approximately 280 kDa band in protein lysates made from wild-type embryos (Figure 2d). Although the band detected on the western blots is substantially higher than the predicted molecular weight of 218 kDa (NCBI Mouse Build 37), this could be due to post-translational modifications to the Rlf protein. We concluded that it is likely that this band is Rlf. Reduced amounts (MommeD8) or no (MommeD28 and MommeD34) Rlf protein was detected in homozygous embryos (Figure 2d). Together, these results suggest that MommeD8 is a hypomorphic allele and that MommeD28 and MommeD34 are null alleles.
Rlf encodes a protein predicted to contain 16 widely spaced zinc finger domains, which has led to the suggestion that it has a role in transcriptional regulation [40]. Rlf is conserved across vertebrates. To determine the localization of Rlf in the cell, we carried out cell fractionation experiments. Rlf protein was specifically detected in the nuclear fraction of HeLa cells (Figure 2e). To our knowledge, we have identified the first mouse mutants for Rlf and hereafter refer to these alleles as RlfMommeD8, RlfMommeD28 and RlfMommeD34.
We have identified Rlf from a screen that is based on the expression of a multi-copy variegating transgene. To test if Rlf is required for the establishment of epigenetic state at an endogenous locus, we studied the effect of RlfMommeD8 on expression of agouti viable yellow (Avy). Avy is a single copy gene that, like the transgene, is known to be sensitive to epigenetic state [41]. The Avy allele is the result of an intracisternal-A-particle (IAP) retrotransposon insertion upstream of the agouti gene and isogenic mice carrying the Avy allele can be yellow, mottled or pseudoagouti (dark brown). Coat color has been shown to correlate with the level of DNA methylation at the long-terminal repeat (LTR) promoter [42]. It has been shown that haploinsufficiency for modifiers of epigenetic reprogramming can alter the ratio of the different coat colors [5,6,43,44]. We crossed females heterozygous for RlfMommeD8 with pseudoagouti Avy/a mice and scored the offspring for coat color. Offspring that inherited the RlfMommeD8 allele were more likely to be pseudoagouti than their wild-type littermates, that is, haploinsufficiency for Rlf increased the probability of silencing at the Avy locus (Figure 2f). This is consistent with Rlf being an enhancer of variegation and suggests that Rlf has a general role in epigenetic regulation.
Novel alleles of known epigenetic regulators
In addition to the genes described above, we have produced 15 MommeD lines that carry mutations in genes already known to be involved in epigenetic regulation in the mouse. Exome deep sequencing of three suppressors of variegation, MommeD19, MommeD27 and MommeD39, identified mutations in the chromatin remodelers SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily c, member 1 (Smarcc1), Polybromo 1 (Pbrm1) and SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4 (Smarca4), respectively (Table 1). Smarcc1MommeD19 carries a mutation in the splice acceptor site of intron 10 predicted to lead to an in-frame premature stop codon and likely nonsense-mediated mRNA decay. Quantitative real-time RT-PCR and western blot analysis showed that Smarcc1 mRNA and Smarcc1 protein levels were reduced in heterozygous animals (Figure 3a). The Pbrm1MommeD27 mutation is a missense mutation in exon 17. Western blot analysis revealed reduced levels of Pbrm1 protein in homozygotes (Figure 3b). Smarca4MommeD39 has a mutation in the splice donor site of intron 20. cDNA sequencing revealed an extended exonic sequence (data not shown) that is predicted to lead to an in-frame premature stop codon and nonsense-mediated mRNA decay. Quantitative real-time RT-PCR analysis showed reduced Smarca4 mRNA in heterozygotes (Figure 3c).
Figure 3.
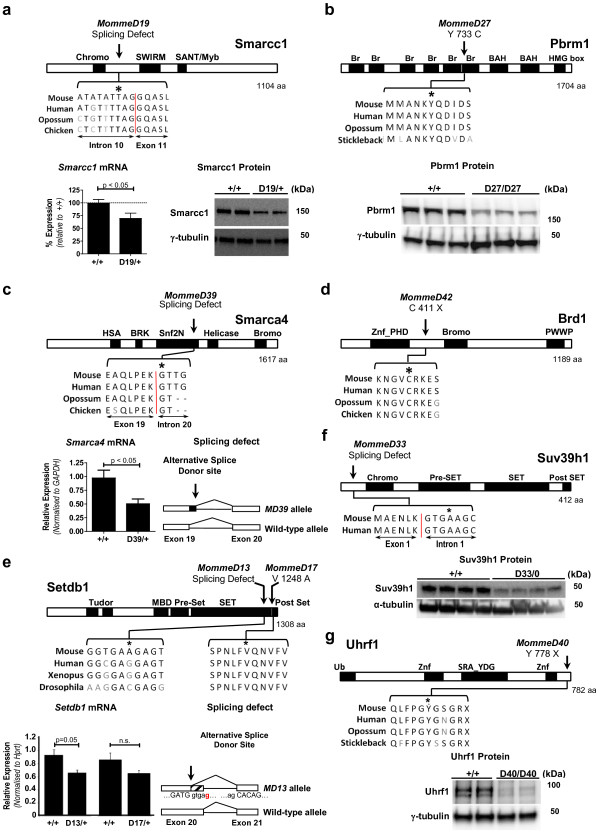
Causative mutations in MommeD19, MommeD27, MommeD39, MommeD42, MommeD13, MommeD33 and MommeD40. (a)MommeD19 carries a mutation at a splice site of Smarcc1. Real-time RT-PCR and immunoblotting of embryos at 12.5 days post-coitum (dpc) showed reduced levels of Smarcc1 mRNA and Smarcc1 protein in heterozygotes (n ≥ 5 mice). Each track represents a different animal. Smarcc1 protein was detected at approximately 160 kDa. (b)MommeD27 harbors a mutation in a bromodomain of Pbrm1. Western blot analysis of 14.5 dpc embryo heads showed reduced levels of Pbrm1 in homozygotes. Each track represents a different animal. Pbrm1 was detected at approximately 190 kDa. (c)MommeD39 carries a mutation at a splice site of Smarca4. Real-time RT-PCR showed reduced Smarca4 mRNA in testes of heterozygotes (n ≥ 4 mice). cDNA sequencing revealed that the mutation in Smarca4MommeD39 results in use of an alternative splice donor site. (d) The mutation in MommeD42 introduces a premature stop codon at amino acid 411 of the Brd1 protein. (e) Mutations in MommeD13 and MommeD17 occur in the conserved SET domain of Setdb1. Real-time RT-PCR of Setdb1 mRNA from testes of heterozygotes and age-matched wild types (n = 4 mice). cDNA analysis revealed that the Setdb1MommeD13 allele is associated with the use of an alternative splice donor site in exon 20, leading to a 62 bp truncation. (f)MommeD33 carries a mutation at a splice site in Suv39h1. Western blot analysis of Suv39h1 in adult thymus showed reduced Suv39h1 in hemizygous mutant males. Each track represents a different animal. Suv39h1 protein was detected at 48 kDa. (g) The mutation in MommeD40 introduces a premature stop codon at amino acid 778 of Uhrf1. Western blot analysis of Uhrf1 revealed greatly reduced levels in 9.5 dpc embryos homozygous for the Uhrf1MommeD40 mutation. Each track represents a different animal. Uhrf1 protein was detected at approximately 90 kDa. Error bars indicate ± standard error of the mean. N.s., not significant. Asterisks indicate mutation.
MommeD42 was identified as an enhancer of variegation and carries a nonsense mutation in exon 11 of Bromodomain containing 1 (Brd1; Figure 3d and Table 1). Brd1 has recently been reported to form a complex with the histone acetyltransferase HBO1 and is required for the transcriptional activation of the erythroid-specific regulator genes in fetal liver [45].
Three suppressors of variegation were found to carry mutations in H3K9 methyltransferases. MommeD13 and MommeD17 carry mutations in Setdb1 and MommeD33 has a mutation in Suv39h1 (Table 1). The Setdb1MommeD13 mutation introduces a novel splice donor site leading to an in-frame premature stop codon in exon 20 (Figure 3e). cDNA analysis confirmed the presence of the predicted transcript (data not shown). Setdb1MommeD17 is a missense mutation in exon 21 of Setdb1 (Figure 3e). RNA analysis indicates reduced levels of Setdb1 mRNA in the MommeD13 mutants whereas Setdb1 mRNA levels were not significantly different in the MommeD17 mutants (Figure 3e). The Suv39h1MommeD33 mutation is at the splice donor side of intron 20. This mutation is predicted to lead to a premature stop codon. Consistent with this, western blot analysis showed that the level of Suv39h1 protein was greatly reduced in hemizygous mutant males (Suv39h1 is on the X chromosome; Figure 3f).
MommeD40, a suppressor of variegation, carries a nonsense mutation in exon 17 of the Uhrf1 gene (Figure 3g and Table 1). Western blot analysis revealed greatly reduced levels of Uhrf1 protein in homozygous embryos (Figure 3g). Uhrf1 is known to associate with the maintenance DNA methyltransferase Dnmt1 [46] and has recently been shown to link DNA methylation and H3K9 methylation in human cells [47].
Seven MommeD lines are new mutant alleles of genes already identified in the screen and we have designated these lines Baz1bMommeD16, Smchd1MommeD23, Trim28MommeD31, Dnmt1MommeD32, Smarca5MommeD35, Smchd1MommeD36 and Smarca5MommeD37 (Table 1; Additional file 5).
Approximately 60 Suppressor of variegation (Su(var)) and 25 Enhancer of variegation (E(var)) genes were identified in the Drosophila screens [2]. We have now documented 12 Su(var) and 8 E(var) genes in the mouse and these encode some of the key factors for DNA methylation and H3K9 methylation, as well as a large group of chromatin remodelers (Figure 4). Some of these genes, such as Dnmt1, Suv39h and Trim28, have been identified as core heterochromatin factors, whereas others, such as Smarca5 and Hdac1, have been associated with gene silencing at euchromatic positions [2]. This is consistent with the variegating phenotype of the transgene. Interestingly, we have not yet recovered any members of the RNA interference/piwi-interacting RNA (piRNA) pathway or Polycomb and trithorax groups of proteins but the screen has not reached saturation.
Figure 4.
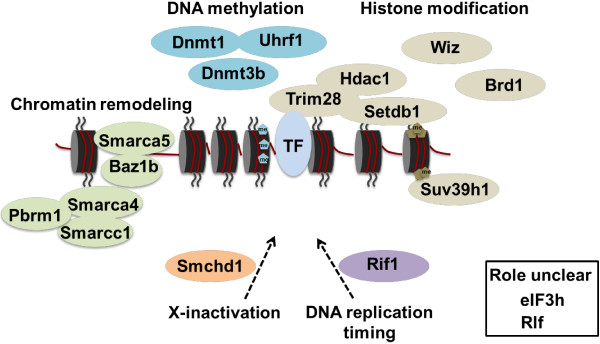
MommeD genes involved in transgene silencing in the mouse. Using an ENU mutagenesis screen, 18 unique genes were identified to be involved in transgene silencing in the mouse. Factors are grouped by their broad mechanistic role in epigenetic regulation. TF, transcription factor; Me, methyl. Figure adapted from [2].
Momme genes are required for normal embryonic development
In most of the MommeD lines, heterozygous mice were observed at expected ratios at weaning and did not show any obvious phenotypes. To determine the viability of homozygotes, we performed embryonic dissections following intercrosses. We have focused on those lines carrying mutations in genes about which there is no or few data available in the literature.
Following WizMommeD30 intercrosses, no homozygous animals were recovered at weaning out of 97 progeny (Figure 5a). To determine when WizMommeD30 homozygotes died, embryos from intercross matings were obtained at different stages of development and genotyped. Viable homozygotes were recovered at the expected ratios at 10.5 days post-coitum (dpc). However, these embryos appeared smaller than their wild-type and heterozygous littermates. At 12.5 dpc homozygotes were present in numbers less than expected (15% as opposed to 25%) and at 14.5 dpc no viable homozygous embryos were obtained (Figure 5a). We conclude from these data that embryonic death in mice homozygous for WizMommeD30 occurs between 10.5 and 12.5 dpc.
Figure 5.
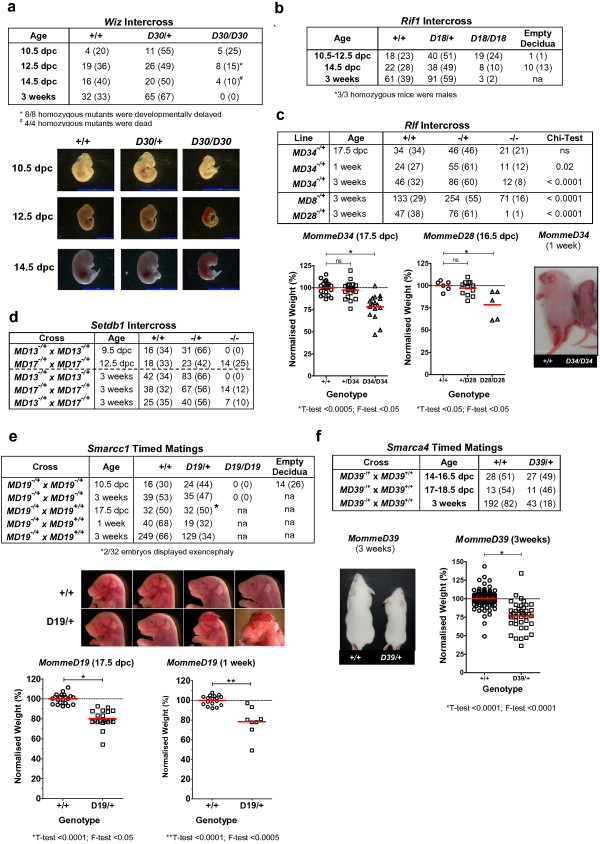
MommeD mutants show abnormal embryonic development. Timed matings and intercrosses of MommeD mutants. (a)WizMommeD30 mice; data show the number of mice observed (and in brackets the percentage) at 10.5 days post-coitum (dpc), 12.5 dpc, 14.5 dpc and at weaning. Representative embryos are shown. (b)Rif1MommeD18 mice; data show the number of mice observed (and in brackets the percentage) at 10.5 to 12.5 dpc, 14.5 dpc and at weaning. na, not applicable. (c)Rlf mutants; data show the number of mice observed (and in brackets the percentage). Embryonic weights were measured from intercrosses of RlfMommeD34 and RlfMommeD28 mice. Homozygous embryos from both had a significant increase in weight variation at 17.5 dpc or 16.5 dpc, respectively. Weights for each litter were normalized to the average weight of wild-type embryos in that litter. Each data point represents an individual. Homozygotes were smaller than wild-type littermates at one week after birth. ns, not significant. (d)Setdb1MommeD13 and Setdb1MommeD17 mice; data show the number of mice observed (and in brackets the percentage). (e)Smarcc1MommeD19 and Smarcc1MommeD19 mated to wild-type mice revealed incomplete penetrance of heterozygous lethality after birth. Data show the number of mice (and in brackets the percentage). Weights of 17.5 dpc Smarcc1MommeD19 embryos and pups were, on average, less than that of wild-type littermates (T-test, P < 0.0001) and showed greater variation (F-test, P < 0.05). Weights in each litter were normalized to the average weight of wild-type embryos in that litter. Each data point represents an individual. (f)Smarca4MommeD39 heterozygotes showed reduced viability. Data show the number of mice and in brackets the percentage. Heterozygotes were smaller than their wild-type littermates. Smarc4MommeD39/+ weighed less (T-test, P < 0.0001) and weights were more variable (F-test, P<0.0001) than wild types. Weights in each litter were normalized to the average weight of wild-type embryos in that litter. Each data point represents an individual.
Rif1MommeD18 intercrosses produced wild-type and heterozygous offspring at the expected ratios and occasional homozygous individuals (3 out of 155 offspring) at weaning (Figure 5b). All surviving homozygotes were males and two, out of three tested, were infertile. Timed matings revealed that the majority of the homozygotes died during the second half of gestation (Figure 5b). This is consistent with the literature [48].
Rlf intercrosses produced homozygous individuals that survived to weaning in less than expected numbers for RlfMommeD8, RlfMommeD28 and RlfMommeD34 (Figure 5c). RlfMommeD34 homozygotes died in the first week after birth, for unknown reasons (Figure 5c). A similar finding was made for RlfMommeD28 homozygotes; most died in the first week after birth (data not shown). However, in both lines some homozygotes did survive to adulthood and were fertile (data not shown). Embryonic dissections indicated that, for the RlfMommeD28 and RlfMommeD34 lines, late gestation homozygous embryos weighed significantly less than their heterozygous or wild-type littermates (T-test, P < 0.05; Figure 5c). RlfMommeD8 behaves like a hypomorphic allele, with approximately half the expected number of homozygotes present at weaning (Figure 5c). Those surviving were smaller and were fertile (data not shown).
One advantage of ENU mutagenesis is that it produces point mutations, increasing the likelihood of hypomorphic mutations. We have reported hypomorphic alleles for Smarca5MommeD4 and Dnmt3bMommeD14 previously [6,10]. In addition to the RlfMommeD8 allele described above, SetdbMommeD17 behaves like a hypomorphic allele for the following reasons. Following intercrosses, some Setdb1MommeD17 homozygotes were present at weaning (Figure 5d) and some survived to adulthood but were smaller than their wild-type and heterozygous littermates and showed decreased fertility (data not shown). Mice homozygous for a null allele of Setdb1 have been reported to die at the early post-implantation stage [49]. Consistent with this, timed matings with Setdb1MommeD13 heterozygous mice produced no viable homozygous offspring at 9.5 dpc (Figure 5d). To our knowledge, Setdb1MommeD17 is the first hypomorphic allele for Setdb1.
In two cases, Smarcc1MommeD19 and Smarca4MommeD39, heterozygous mutants showed signs of stochastic death prior to weaning. Following Smarcc1MommeD19 intercrosses, no viable homozygotes were obtained at weaning and timed matings revealed that homozygous lethality occurred prior to mid-gestation, as expected based on the literature [50] (Figure 5e). At weaning the percentage of heterozygotes to wild types (47% and 53%, respectively) was less than that expected (66% and 33%, respectively). Maintenance of the colony, which involved heterozygous to wild type crosses, also produced fewer mutants than expected at weaning (34% as opposed to 50%) (Figure 5e). Further investigation suggested that this stochastic death of heterozygotes was occurring after 17.5 dpc and before 1 week postnatal (Figure 5e). At 17.5 dpc, some heterozygotes had exencephaly, although this could not account for all the perinatal lethality observed (Figure 5e). The average weight of heterozygous mice at E17.5 and at 1 week postnatal was reduced (T-test, P < 0.0001) and greater variance (F-test, P < 0.05) was observed among heterozygotes than among wild-type littermates (Figure 5e).
Maintenance of the Smarca4MommeD39 colony (heterozygote to wild type crosses) also revealed non-Mendelian ratios; heterozygotes were present in less than expected numbers at weaning (18% as opposed to 50%) and were smaller than their wild-type littermates (Figure 5f). Timed matings showed that the ratios were close to Mendelian prior to birth (Figure 5f). Body weights of heterozygotes were lower than those of wild-type littermates at weaning (T-test, P < 0.0001) and, again, the variance was greater in the heterozygotes (F-test, P < 0.0001) (Figure 5f). Similar stochastic death of heterozygotes has been described by others for mice with knock-out alleles for Smarcc1 and Smarca4[50,51]. In these cases, however, the genetic heterogeneity among the mice could have been an underlying factor.
We have previously reported increased variance in body weight and behavioral responses in adult mice heterozygous for Trim28[8] and others have made similar findings with respect to Trim28 during the oocyte to embryo transition [52]. We have also reported that reduced levels of Dnmt3a are associated with an increase in body weight variance in adults [8]. These observations, in the context of inbred strains reared in controlled environments, are consistent with a role for the genes identified in this screen in canalization, a term coined by Waddington to describe phenotypic robustness during development. Exactly how important probabilistic developmental events are in determining phenotype is not yet clear [53]. We have so far reported only three measures; the penetrance of homozygous or heterozygous death in the colony, the body weight of heterozygotes or homozygotes (at various ages) in the colony and behavioral responses. More extensive phenotyping of these inbred MommeD lines should enable us to gain a better understanding of this phenomenon.
In the remaining MommeD lines examined, Baz1bMommeD16, Smchd1MommeD23, Pbrm1MommeD27, Trim28MommeD31, Dnmt1MommeD32, Suv39h1MommeD33, Smarca5MommeD35, Smchd1MommeD36, Smarca5MommeD37, Uhrf1MommeD40 and Brd1MommeD41, we observed homozygous embryonic death that is similar to that reported for knock-out alleles of these genes (Additional file 6).
DNA methylation levels at the transgene are increased in Rlf mutants
Changes in transcription can correlate with changes in DNA methylation at transgenes and metastable epialleles [42,54-56]. We have shown previously that changes in the percentage of red blood cells expressing GFP can be accompanied by changes in DNA methylation at the transgene [7,10]. Using bisulfite sequencing, we investigated DNA methylation at the HS-40 enhancer region of the transgene in MommeD lines. We used adult spleens from wild-type mice and mice heterozygous for a MommeD mutation. Consistent with our previous reports, in Line3 wild-type mice around 60% of the CpGs in the HS-40 element were methylated (Figure 6a). Mice heterozygous for Dnmt1MommeD32 suggested a decrease in CpG methylation (around 50%; Figure 6a). This is consistent with an increase in expression of the GFP transgene in these mutants and with its role as the maintenance DNA methyltransferase. In mice heterozygous for the Wiz and Rif1 mutations DNA methylation patterns at the HS-40 region were unaffected (Figure 6b).
Figure 6.

Bisulfite sequencing of the HS-40 enhancer region of the GFP transgene. (a) DNA methylation levels were reduced in Dnmt1MommeD32 heterozygous mice compared to wild-type mice in adult spleens (n = 4 wild-type mice and n = 2 Dnmt1MommeD32 mutant mice). Filled circles represent methylated CpG sites. (b) DNA methylation levels in adult spleens were unaffected in mice heterozygous for WizMommeD30 and RifMommeD18 (n = 4 for WizMommeD30 and n = 2 for Rif1MommeD18). (c) DNA methylation levels in adult spleens of RlfMommeD8, RlfMommeD28 and RlfMommeD34 heterozygotes were increased compared to wild-type mice (n = 2 mice for all genotypes). (d)Rlf homozygotes showed significantly increased DNA methylation (T-test, P < 0.05) in adult male spleen (RlfMommeD8 and RlfMommeD34) and 16.5 dpc embryo (RlfMommeD28) compared to wild types (n = 3 for all genotypes).
In the case of the Rlf alleles, we detected higher levels of DNA methylation at the HS-40 element in adult spleens in RlfMommeD8 (82%, T-test <0.05) and RlfMommeD34 (81%, T-test <0.05) homozygotes compared to wild types (67%) (Figure 6d). A similar trend (not statistically significant) was observed in the RlfMommeD8 and the RlfMommeD34 heterozygotes compared with that seen in Line3 wild-type (Figure 6a,c). In the case of RlfMommeD28, where adult homozygotes are extremely rare, we analyzed DNA methylation patterns in 16.5 dpc embryos and made a similar finding. The HS-40 element was more methylated in RlfMommeD28 homozygotes (79%, T-test <0.05) than it was in wild-type controls (51%) (Figure 6d). This is the first time that we have observed hypermethylation of the transgene in a MommeD line. An increase in DNA methylation at the transgene locus in the Rlf mutants is consistent with the decreased expression of the transgene in these mice.
Conclusions
Our results suggest that transgene silencing in the mouse acts through a mechanism common to transposon silencing, X-inactivation and imprinting. We provide evidence that a novel gene, Rlf, is involved in this process. Depletion of Rlf leads to DNA hypermethylation at the transgene. How this is achieved is currently unknown and requires further investigation. We have isolated the first mouse mutants for Rlf and Wiz and shown that the genes are required for normal embryonic development. Notably, the human homologs of many of the genes recovered from our screen have been found to be associated with human diseases, in some cases identified in family studies and in other cases suggested by genome-wide association studies (Table 1).
Since the mouse is often used as a disease model, we anticipate that our collection of Momme mutants will provide a valuable resource for researchers across many disciplines.
Material and methods
Generation and screening of mutant mice
Procedures were approved by the Animal Ethics Committee of the QIMR Berghofer Medical Research Institute. The ENU screen was carried out in the FVB/NJ inbred transgenic line, Line3, which is homozygous for a multicopy GFP transgene, as described previously [5]. All MommeD lines were maintained in this background and are homozygous for the GFP transgene. All experimental data: crosses to other lines, intercrosses, and so on, were carried out using heterozygous MommeD mice five generations or more removed from the MommeD founder.
Other mouse strains
The congenic strain, Line3C, used for linkage studies, was produced by crossing Line3 to C57BL/6J for 10 generations, selecting for mice carrying the transgene by flow cytometry. Inbred C57BL/6J mice were purchased from ARC Perth (Perth, WA, Australia). Rif1GT mice were generated by the Australian Phenomics Network from an embryonic stem cell clone carrying a trapped Rif1 allele (A045A01; German Gene Trap Consortium), crossed to Line3C (C57BL/6J) and maintained on this background.
Flow cytometry
Mice were analyzed by flow cytometry at 3 weeks of age. A drop of blood was collected in Osmosol buffer and analyzed on a Guava easyCyte HT (Merck/Millipore, Darmstadt, Germany). The data were analyzed by using Guava InCyte software with a GFP-positive gate set to exclude 99.9% of wild-type erythrocytes. Histograms shown depict only the GFP fluorescence channel.
Linkage analysis
Heterozygous mutant MommeD mice, at least four generations down from the founder, were backcrossed twice to Line3C (see above) and phenotyped for GFP expression by flow cytometry. DNA from tail tips was used to perform linkage analysis. The Illumina GoldenGate genotyping assay (Mouse Medium Density Linkage Panel) was used for MommeD13, MommeD16 to MommeD19, MommeD27, MommeD30, MommeD32, MommeD34, MommeD35, MommeD37, MommeD39, MommeD40 and MommeD42 on at least 12 wild-type and 12 heterozygous mice from each line. The Mouse Medium Density Linkage panel contains 766 measurable SNPs between C57BL/6J and FVB/NJ. Samples were genotyped following the Illumina protocol and genotype calls were made using the Genotyping module of the GenomeStudio v1.1 software. Only samples with a call rate >95 were accepted. Linked intervals were identified based on LOD scores of 3 or higher (Additional file 7).
For MommeD8, MommeD23, MommeD28, MommeD32, MommeD33 and MommeD36 the linked chromosomes were identified using microsatellite or SNP markers. Additional markers were used to reduce the linked intervals using wild-type and heterozygous mice. Mice wild-type for the mutation should only have C57BL/6J chromosomes in the linked region, while mice heterozygous for the mutation should carry both C57BL/6J and FVB/NJ chromosomes.
For MommeD13, MommeD16, MommeD18, MommeD23, MommeD28, MommeD32, MommeD33 and MommeD35, mutations were found using Sanger sequencing. For the remaining MommeD lines deep sequencing approaches were used.
Whole exome deep sequencing and interval capture
Mouse exomes were captured and sequenced using exome capture reagents from either Agilent (SureselectXT Mouse All Exon v.1, Santa Clara, CA, USA) or Roche NimbleGen (SeqCap EZ Mouse Exome, version Beta 2, 110603_MM9_exome_rebal_2EZ_HX1, Madison, WI, USA). The Agilent captures were carried out as detailed in the manufacturer’s protocol (version 1.1.1), with the following exceptions: a Bioruptor (Diagenode, Liège, Belgium) was used for DNA shearing, producing fragment sizes of approximately 200 to 300 bp, and sample pooling/multiplexing was omitted. The Bioruptor sonication settings were 3 × 10 minutes on the low setting with cycles of 30 seconds on and 30 seconds off. The Roche NimbleGen captures were carried out as outlined in the Illumina optimized Roche NimbleGen SeqCap User’s Guide (version 1.0), also using the Bioruptor for DNA fragmentation.
For two mutant lines, MommeD8 and MommeD34, a 4.2 Mbp linked interval was sequenced in full using a custom capture array designed and manufactured by Roche NimbleGen. MommeD8 was captured and sequenced by Roche NimbleGen using Roche 454 sequencing (110317_MM9_mommeD8_Rocky_cap_HX3). Three mice were sequenced; a wild type, a heterozygote and a homozygote. For MommeD34, a heterozygous mutant was captured and sequenced in-house using the same array capture design on 2.1M arrays with HX3 mixers, according to the Roche NimbleGen Arrays User’s Guide, Illumina Optimized protocol (version 1.0).
SNP calling
The sequencing reads from the targeted sequence capture experiments were aligned to the mouse genome (build 37, mm9) using the program bwa version 0.6.1 [57]. Datasets generated by Roche on the 454 platform were mapped using bwa bwasw [58] while the remaining datasets, generated on the Illumina HiSeq or GAIIx platforms, were mapped using bwa aln, with default settings, followed by bwa sampe with the default settings. The resulting sam files were converted to bam files and coordinate-sorted using SAMtools version 0.1.17 [59] and PCR duplicates were subsequently eliminated using the program Picard MarkDuplicates, version 1.48 [60].
For each sample, nucleotide variants were identified within the intervals for which linkage had previously been determined. This was achieved by creating a pileup file of the linked region using SAMtools mpileup, using the option -q 20, followed by variant calling using the program Varscan version v2.2.8 [61] using the 'somatic' feature and the settings -min-coverage 15 and -min-var-freq 0.3. Varscan somatic calls sequence differences between a case and a control sample; as control the sequenced exome from a different ENU mutant was used. The case and control exomes were constructed using the same library preparation methods and sequenced in the same deep sequencing run, but had different linked intervals. All exomes were mapped and processed in parallel, using identical settings, to minimize post-sequencing artifacts. The output from Varscan was manually screened for likely ENU mutations, appearing as heterozygous SNPs in the mutant and wild type in the control. These SNPs were in turn validated using the Sanger method.
The custom capture and sequencing of the MommeD34 linked region was carried out in-house without a matched wild-type control; instead, a merge of the three MommeD8 deep sequencing samples previously sequenced by Roche was used as control. A merge of these 454 datasets was used in order to achieve greater read depth across the region. SNP calling was subsequently carried out as described above. Exome sequencing datasets generated in this study are accessible via European Nucleotide Archive (ENA) under accession ERP003831.
Genotyping
Once the mutations had been identified, genotyping was carried out by either sequencing (MommeD13, MommeD16, MommeD17, MommeD18, MommeD23, MommeD27, MommeD28, MommeD30, MommeD32, MommeD34, MommeD35, MommeD36, MommeD37, MommeD39, MommeD40 and MommeD42) or, if the mutation had changed a restriction enzyme recognition site, by PCR and digestion and gel electrophoresis (MommeD8 and MommeD19). Genotyping primers are provided in Additional file 8 and Sanger sequencing traces in Additional file 9.
Embryo dissections
All embryos were produced by natural matings and detection of a vaginal plug was counted as 0.5 dpc. Except where otherwise stated, embryos were produced by intercrosses.
Introduction of the mutant lines into mice carrying the Avy allele
FVB/NJ mice heterozygous for the MommeD8 mutation and homozygous for the GFP transgene were mated with C57BL/6J mice heterozygous for the Avy allele. The coat color phenotype was classified at weaning by a trained observer as either yellow, mottled, or agouti. GFP expression was determined by flow cytometry and used to classify the mice into mutants or wild-type for the MommeD8 mutation. FVB/NJ mice carry the A locus and C57BL/6J mice carry the a locus. Yellow and mottled offspring carry the Avy allele. All agouti-colored offspring were genotyped by PCR, to assess whether they were Avy/A and pseudoagouti, or A/a, as reported [56].
Bisulfite sequencing of the transgene HS-40 enhancer
Bisulfite conversion of DNA was carried out using the EpiTect Bisulfite Kit (Qiagen, Doncaster, VIC, Australia) according to the manufacturer’s instructions. At least two male adult spleens were used for each MommeD line. The bisulfite conversion rate was at least 97% and sequences were analyzed using the BiQ Analyser software [62]. Oligonucleotides to the bisulfite converted HS-40 enhancer region were as follows (5’-3’): GFPbisF1: AAAATAAAATTTTTGGATTGTTATTATTATAA; GFPbisF2: ATATTTGTAATTTTAGTATTTTGGGAGGTT and GFPbisR: AATCTCTACTCACTACAAACTCCATCTC. Cycling conditions were as follows: 94°C for 2 minutes for 1 cycle; 94°C for 30 seconds, 60°C for 30 seconds, 72°C for 45 seconds for 35 cycles and 72°C for 6 minutes for 1 cycle.
Protein analysis
Whole-cell extracts were prepared from various tissues of adult mice. Protein concentration was measured by BCA (Thermo Scientific, Waltham, MA, USA) and protein lysates were separated using polyacrylamide gels (Invitrogen, Carlsbad, CA, USA, BioRad, Hercules, CA, USA or ECL, Rydalmere, NSW, Australia). Antibodies used were as follows: anti-γ-tubulin (T5192, Sigma-Aldrich, St. Louis, MO, USA), anti-Rif1 (ab13422, Abcam, Cambridge, ENG, UK), anti-Wiz (gift from Yoichi Shinkai), anti-Smarcc1 (9053S, Cell Signaling, Danvers, MA, USA), anti-Rlf (ab115011, Abcam - rabbit polyclonal) and (M05, clone2G2, Abnova - mouse monoclonal, Neihu District, Taipei City, Taiwan), anti-Pbrm1 (Rabbit polyclonal ABE70, Millipore), anti-Suv39h1 (clone D11B5, Cell Signaling), anti-Uhrf1 (ab151187 Abcam), anti-GAPDH (D16H11 XP, Cell Signaling) and anti-Snf2H (ab3749, Abcam).
Cell fractionation
HeLa cells were trypsinized, pelleted and washed twice with phosphate-buffered saline before re-suspending in ice-cold cytoplasmic extraction buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40). A small aliquot of this suspension, the total cell lysate, was transferred to a separated tube. The remaining suspension was then centrifuged (500 rpm, 10 minutes, 4°C) to pellet the nuclei. The supernatant (cytoplasmic fraction) was aliquoted into a separate tube. Nuclei were re-suspended in an 8 M urea lysis buffer (8 M urea, 1/10 vol. glycerol, 1/20 vol. 20% SDS, 1/2,000 vol. 1 M dithiothreitol, 1/100 vol. 1 M Tris, pH 6.8).
RNA isolation, cDNA analysis and quantitative real-time RT-PCR
Total RNA was extracted from various tissues using TRI reagent (Invitrogen). cDNA was synthesized from total RNA using SuperScriptIII reverse transcriptase (Invitrogen) or AMV reverse transcriptase (Roche) and random hexamer primers. Quantitative real-time PCR was performed with the Platinum SYBR Green qPCR Super Mix -UDG (Invitrogen) with primers designed to span exon/intron boundaries. All reactions were performed in triplicates and normalized to Hprt or Gapdh. PCRs were run on a Viia7 (Applied Biosystems, Mulgrave, VIC, Australia) or on a Corbett Research Rotor-Gene (Qiagen). Cryp-Skip [63] was used for splice site prediction. cDNA from mutant alleles was sequenced using Sanger sequencing. Primer sequences are provided in Additional file 8.
Statistical analysis
Statistical significance of quantitative data was determined by two-tailed Student’s T-test. F-test was used to test whether variance was significantly different between wild-type and mutant groups. The proportions of genotypes were compared to expected Mendelian ratios using a χ2 test. For all datasets a minimum of three biological replicates were analyzed.
Abbreviations
bp: base pair; dpc: days post-coitum; ENU: N-ethyl-N-nitrosourea; FSHD2: facioscapulohumeral dystrophy type 2; GFP: green fluorescent protein; H3K9: histone H3 lysine 9; Momme: Modifier of murine metastable epiallele; PCR: polymerase chain reaction; SNP: single-nucleotide polymorphism.
Competing interests
The authors declare no competing financial interests.
Authors’ contributions
LD, SKH, and HO contributed to study design, carried out experiments, interpreted results and helped to draft the manuscript. TE, LI, EH, NW, AA, AS, JY, VB, JS, AA, ZP, NW, DG, MEB and JAJ carried out experiments and interpreted results. EW conceived the study and project design, performed ENU mutagenesis, interpreted results and helped to draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Linked intervals. Manhattan plots showing linked intervals identified by Illumina GoldenGate SNP genotyping analysis. The x-axis represents the chromosomes and the y-axis is the LOD score. Peaks with a LOD score of 3 or higher are considered significant.
Flow cytometric profiles of the MommeDs. (a) Representative flow cytometry profiles show the percentage of GFP-expressing erythrocytes from wild-type and heterozygous mutant littermates (n = 3). The x-axis represents the erythrocyte fluorescence on a logarithmic scale and the y-axis is the number of cells detected at each fluorescence level. (b) Percentage of GFP-expressing cells in wild-type, heterozygous mutant and homozygous mutant (where viable) mice at three weeks of age (mean ± standard error of the mean).
Mapping interval for MommeD30. (a)MommeD30 was produced in the FVB/NJ strain of mice and mapped by crossing twice onto Line3C in a C57BL/6J background. The results of the genotype for seven SNP markers and one microsatellite marker surrounding the linked interval are shown. The number of mice classified into each haplotype is shown on top. Our estimate of the linked interval is between rs29539305 and rs33446195 on chromosome 17 (highlighted). (b) List of genes in the MommeD30 linked interval on chromosome 17. The Mus musculus Ensembl database (release 37) was used to export a list of transcripts (protein coding and non-coding RNAs) within the 1.9 Mbp MommeD30 interval.
GFP expression in offspring of a Rif1GTheterozygote crossed to Line3C. A Rif1 gene trap allele (Rif1GT) had a similar effect on transgene expression as that observed with the MommeD18 mutation, increasing the percentage of expressing cells in mice heterozygous for the gene-trap allele.
Novel mutant alleles of Baz1b, Smchd1, Trim28, Dnmt1 and Smarca5. (a)MommeD16 carries a point mutation resulting in a non-conservative amino acid change in the Wstf domain of Baz1b. (b)MommeD23 and MommeD36 carry point mutations in Smchd1. Both mutations introduce premature stop codons in the Smchd1 protein. (c)MommeD31 carries a point mutation in Trim28 that results in an amino acid change in a highly conserved zinc finger domain. (d)MommeD32 carries a point mutation that results in an amino acid change in the BAH domain of Dnmt1. (e)MommeD35 and MommeD37 carry mutations that result in amino acid changes in highly conserved domains of the Smarca5 protein.
Embryonic development in MommeD mutants. Tabulated data shows the number of observed mice and in brackets the percentage of total. (a) Intercrosses of Pbrm1MommeD27 mice produce no homozygous offspring at three weeks. (b)Brd1MommeD42 homozygous mice are embryonic lethal around 10.5 dpc. (c) Mice carrying mutations in Suv39h1MommeD33 are viable at three weeks. (d)Uhrf1MommeD40 homozygotes are embryonic lethal around 10.5 dpc. (e) Some homozygous Baz1bMommeD16 mice were obtained at three weeks but less than expected. (f) No (Smchd1MommeD36) or few (Smchd1MommeD23) homozygous individuals were recovered from intercrosses. The Smchd1MommeD23 homozygotes that survived were males. (g)Trim28MommeD31 homozygous mice are embryonic lethal prior to 10.5 dpc. (h) Mice homozygous for the Dnmt1MommeD32 mutation die around 10.5 dpc. (i) Intercrosses of Smarca5MommeD35 and Smarca5MommeD37 produced no homozygous offspring at three weeks. Timed matings show that survival of homozygous mutants at 12.5 to 14.5 dpc was rare in Smarca5MommeD35 mutants. In Smarca5MommeD37 mutants homozygous embryonic death occurred prior to 14.5 dpc.
Linkage analysis using Illumina GoldenGate genotyping assay.
List of primer sequences.
Sanger traces.
Contributor Information
Lucia Daxinger, Email: l.clemens-daxinger@latrobe.edu.au.
Sarah K Harten, Email: Sarah.Harten@qimrberghofer.edu.au.
Harald Oey, Email: h.oey@latrobe.edu.au.
Trevor Epp, Email: eppt@img.cas.cz.
Luke Isbel, Email: l.isbel@latrobe.edu.au.
Edward Huang, Email: edward.huang@qimrberghofer.edu.au.
Nadia Whitelaw, Email: n.hamilton@griffith.edu.au.
Anwyn Apedaile, Email: chapman.anwyn.e@edumail.vic.gov.au.
Anabel Sorolla, Email: anabel.sorolla@qimrberghofer.edu.au.
Joan Yong, Email: wolfjay89@gmail.com.
Vandhana Bharti, Email: vandhana.bharti@qimrberghofer.edu.au.
Joanne Sutton, Email: joanne.sutton@qimrberghofer.edu.au.
Alyson Ashe, Email: alysonashe@gmail.com.
Zhenyi Pang, Email: zhenyi.pang@qimrberghofer.edu.au.
Nathan Wallace, Email: nathan.wallace@qimrberghofer.edu.au.
Daniel J Gerhardt, Email: daniel.gerhardt@mmri.mater.org.au.
Marnie E Blewitt, Email: blewitt@wehi.edu.au.
Jeffrey A Jeddeloh, Email: jeffrey.jeddeloh@roche.com.
Emma Whitelaw, Email: e.whitelaw@latrobe.edu.au.
Acknowledgements
This study was supported by National Health and Medical Research Council of Australia grants to SH and EW. We are grateful to Yoichi Shinkai for providing the anti-Wiz antibody.
References
- Henikoff S. Position-effect variegation after 60 years. Trends Genet. 1990;14:422–426. doi: 10.1016/0168-9525(90)90304-o. [DOI] [PubMed] [Google Scholar]
- Fodor BD, Shukeir N, Reuter G, Jenuwein T. Mammalian Su(var) genes in chromatin control. Annu Rev Cell Dev Biol. 2010;14:471–501. doi: 10.1146/annurev.cellbio.042308.113225. [DOI] [PubMed] [Google Scholar]
- Schotta G, Ebert A, Dorn R, Reuter G. Position-effect variegation and the genetic dissection of chromatin regulation in Drosophila. Semin Cell Dev Biol. 2003;14:67–75. doi: 10.1016/S1084-9521(02)00138-6. [DOI] [PubMed] [Google Scholar]
- Reuter G, Spierer P. Position effect variegation and chromatin proteins. Bioessays. 1992;14:605–612. doi: 10.1002/bies.950140907. [DOI] [PubMed] [Google Scholar]
- Blewitt ME, Vickaryous NK, Hemley SJ, Ashe A, Bruxner TJ, Preis JI, Arkell R, Whitelaw E. An N-ethyl-N-nitrosourea screen for genes involved in variegation in the mouse. Proc Natl Acad Sci U S A. 2005;14:7629–7634. doi: 10.1073/pnas.0409375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Vickaryous N, Ashe A, Zamudio N, Youngson N, Hemley S, Stopka T, Skoultchi A, Matthews J, Scott HS, de Kretser D, O'Bryan M, Blewitt M, Whitelaw E. Modifiers of epigenetic reprogramming show paternal effects in the mouse. Nat Genet. 2007;14:614–622. doi: 10.1038/ng2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe A, Morgan DK, Whitelaw NC, Bruxner TJ, Vickaryous NK, Cox LL, Butterfield NC, Wicking C, Blewitt ME, Wilkins SJ, Anderson GJ, Cox TC, Whitelaw E. A genome-wide screen for modifiers of transgene variegation identifies genes with critical roles in development. Genome Biol. 2008;14:R182. doi: 10.1186/gb-2008-9-12-r182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelaw NC, Chong S, Morgan DK, Nestor C, Bruxner TJ, Ashe A, Lambley E, Meehan R, Whitelaw E. Reduced levels of two modifiers of epigenetic gene silencing, Dnmt3a and Trim28, cause increased phenotypic noise. Genome Biol. 2010;14:R111. doi: 10.1186/gb-2010-11-11-r111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxinger L, Oey H, Apedaile A, Sutton J, Ashe A, Whitelaw E. A forward genetic screen identifies eukaryotic translation initiation factor 3, subunit H (eIF3h), as an enhancer of variegation in the mouse. G3 (Bethesda) 2012;14:1393–1396. doi: 10.1534/g3.112.004036. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngson NA, Epp T, Roberts AR, Daxinger L, Ashe A, Huang E, Lester KL, Harten SK, Kay GF, Cox T, Matthews JM, Chong S, Whitelaw E. No evidence for cumulative effects in a Dnmt3b hypomorph across multiple generations. Mamm Genome. 2013;14:206–217. doi: 10.1007/s00335-013-9451-5. [DOI] [PubMed] [Google Scholar]
- Blewitt ME, Gendrel AV, Pang Z, Sparrow DB, Whitelaw N, Craig JM, Apedaile A, Hilton DJ, Dunwoodie SL, Brockdorff N, Kay GF, Whitelaw E. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat Genet. 2008;14:663–669. doi: 10.1038/ng.142. [DOI] [PubMed] [Google Scholar]
- Gendrel AV, Apedaile A, Coker H, Termanis A, Zvetkova I, Godwin J, Tang YA, Huntley D, Montana G, Taylor S, Giannoulatou E, Heard E, Stancheva I, Brockdorff N. Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X chromosome. Dev Cell. 2012;14:265–279. doi: 10.1016/j.devcel.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong HS, Chen K, Hu Y, Lee S, Corbin J, Pakusch M, Murphy JM, Majewski IJ, Smyth GK, Alexander WS, Hilton DJ, Blewitt ME. Epigenetic regulator Smchd1 functions as a tumor suppressor. Cancer Res. 2013;14:1591–1599. doi: 10.1158/0008-5472.CAN-12-3019. [DOI] [PubMed] [Google Scholar]
- Lemmers RJ, Tawil R, Petek LM, Balog J, Block GJ, Santen GW, Amell AM, van der Vliet PJ, Almomani R, Straasheijm KR, Krom YD, Klooster R, Sun Y, den Dunnen JT, Helmer Q, Donlin-Smith CM, Padberg GW, van Engelen BG, de Greef JC, Aartsma-Rus AM, Frants RR, de Visser M, Desnuelle C, Sacconi S, Filippova GN, Bakker B, Bamshad MJ, Tapscott SJ, Miller DG, van der Maarel SM. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;14:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H, Karpf AR, Wallace DC, Simon M, Lander C, Boardman LA, Cunningham JM, Smith GE, Litchy WJ, Boes B, Atkinson EJ, Middha S, Dyck PJ B, Parisi JE, Mer G, Smith DI, Dyck PJ. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. 2011;14:595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelmann J, Lin L, Schormair B, Kornum BR, Faraco J, Plazzi G, Melberg A, Cornelio F, Urban AE, Pizza F, Poli F, Grubert F, Wieland T, Graf E, Hallmayer J, Strom TM, Mignot E. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet. 2012;14:2205–2210. doi: 10.1093/hmg/dds035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann K, Deryal M, Ong MF, Schmidt W, Mahlknecht U. DNMT1 genetic polymorphisms affect breast cancer risk in the central European Caucasian population. Clin Epigenetics. 2013;14:7. doi: 10.1186/1868-7083-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldic M, Guidotti A, Maloku E, Davis JM, Costa E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc Natl Acad Sci U S A. 2005;14:2152–2157. doi: 10.1073/pnas.0409665102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra SK, Patra A, Zhao H, Dahiya R. DNA methyltransferase and demethylase in human prostate cancer. Mol Carcinog. 2002;14:163–171. doi: 10.1002/mc.10033. [DOI] [PubMed] [Google Scholar]
- Stopka T, Zakova D, Fuchs O, Kubrova O, Blafkova J, Jelinek J, Necas E, Zivny J. Chromatin remodeling gene SMARCA5 is dysregulated in primitive hematopoietic cells of acute leukemia. Leukemia. 2000;14:1247–1252. doi: 10.1038/sj.leu.2401807. [DOI] [PubMed] [Google Scholar]
- Lu X, Meng X, Morris CA, Keating MT. A novel human gene, WSTF, is deleted in Williams syndrome. Genomics. 1998;14:241–249. doi: 10.1006/geno.1998.5578. [DOI] [PubMed] [Google Scholar]
- Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferré F, Bourque C, Burke CJ, Turner L, Uong A, Johnson LA, Beroukhim R, Mermel CH, Loda M, Ait-Si-Ali S, Garraway LA, Young RA, Zon LI. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;14:513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GL, Bestor TH, Bourc'his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Pequignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;14:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhao A, Chen L, Zhong X, Liao J, Gao M, Cai M, Lee DH, Li J, Chowdhury D, Yang YG, Pfeifer GP, Yen Y, Xu X. Human RIF1 encodes an anti-apoptotic factor required for DNA repair. Carcinogenesis. 2009;14:1314–1319. doi: 10.1093/carcin/bgp136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CL, Christensen LL, Thorsen K, Schepeler T, Sorensen FB, Verspaget HW, Simon R, Kruhoffer M, Aaltonen LA, Laurberg S, Orntoft TF. Dysregulation of the transcription factors SOX4, CBFB and SMARCC1 correlates with outcome of colorectal cancer. Br J Cancer. 2009;14:511–523. doi: 10.1038/sj.bjc.6604884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelBove J, Rosson G, Strobeck M, Chen J, Archer TK, Wang W, Knudsen ES, Weissman BE. Identification of a core member of the SWI/SNF complex, BAF155/SMARCC1, as a human tumor suppressor gene. Epigenetics. 2011;14:1444–1453. doi: 10.4161/epi.6.12.18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, Bignell G, Butler A, Cho J, Dalgliesh GL, Galappaththige D, Greenman C, Hardy C, Jia M, Latimer C, Lau KW, Marshall J, McLaren S, Menzies A, Mudie L, Stebbings L, Largaespada DA, Wessels LF, Richard S, Kahnoski RJ, Anema J, Tuveson DA. et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;14:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N, Matsumoto N. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;14:376–378. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
- Severinsen JE, Bjarkam CR, Kiaer-Larsen S, Olsen IM, Nielsen MM, Blechingberg J, Nielsen AL, Holm IE, Foldager L, Young BD, Muir WJ, Blackwood DH, Corydon TJ, Mors O, Børglum AD. Evidence implicating BRD1 with brain development and susceptibility to both schizophrenia and bipolar affective disorder. Mol Psychiatry. 2006;14:1126–1138. doi: 10.1038/sj.mp.4001885. [DOI] [PubMed] [Google Scholar]
- Arnold CN, Barnes MJ, Berger M, Blasius AL, Brandl K, Croker B, Crozat K, Du X, Eidenschenk C, Georgel P, Hoebe K, Huang H, Jiang Z, Krebs P, La Vine D, Li X, Lyon S, Moresco EM, Murray AR, Popkin DL, Rutschmann S, Siggs OM, Smart NG, Sun L, Tabeta K, Webster V, Tomisato W, Won S, Xia Y, Xiao N, Beutler B. ENU-induced phenovariance in mice: inferences from 587 mutations. BMC Res Notes. 2012;14:577. doi: 10.1186/1756-0500-5-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keays DA, Clark TG, Campbell TG, Broxholme J, Valdar W. Estimating the number of coding mutations in genotypic and phenotypic driven N-ethyl-N-nitrosourea (ENU) screens: revisited. Mamm Genome. 2007;14:123–124. doi: 10.1007/s00335-006-0065-z. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005;14:815–826. doi: 10.1101/gad.1284005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda J, Tachibana M, Ikura T, Shinkai Y. Zinc finger protein Wiz links G9a/GLP histone methyltransferases to the co-repressor molecule CtBP. J Biol Chem. 2006;14:20120–20128. doi: 10.1074/jbc.M603087200. [DOI] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;14:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, Nussenzweig A, Casellas R, Robbiani DF, Nussenzweig MC. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;14:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD, Xu D, Durocher D. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;14:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science. 2013;14:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella-Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SB. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012;14:3678–3690. doi: 10.1038/emboj.2012.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012;14:3667–3677. doi: 10.1038/emboj.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela TP, Hellsten E, Vesa J, Hirvonen H, Palotie A, Peltonen L, Alitalo K. The rearranged L-myc fusion gene (RLF) encodes a Zn-15 related zinc finger protein. Oncogene. 1995;14:2699–2704. [PubMed] [Google Scholar]
- Waterland RA. Assessing the effects of high methionine intake on DNA methylation. J Nutr. 2006;14:1706S–1710S. doi: 10.1093/jn/136.6.1706S. [DOI] [PubMed] [Google Scholar]
- Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;14:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- Blewitt ME, Vickaryous NK, Paldi A, Koseki H, Whitelaw E. Dynamic reprogramming of DNA methylation at an epigenetically sensitive allele in mice. PLoS Genet. 2006;14:e49. doi: 10.1371/journal.pgen.0020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet F, Rideout WM 3rd, Meissner A, Dausman J, Leonhardt H, Jaenisch R. Dnmt1 expression in pre- and postimplantation embryogenesis and the maintenance of IAP silencing. Mol Cell Biol. 2004;14:1640–1648. doi: 10.1128/MCB.24.4.1640-1648.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima Y, Miyagi S, Saraya A, Negishi M, Endoh M, Endo TA, Toyoda T, Shinga J, Katsumoto T, Chiba T, Yamaguchi N, Kitabayashi I, Koseki H, Iwama A. The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood. 2011;14:2443–2453. doi: 10.1182/blood-2011-01-331892. [DOI] [PubMed] [Google Scholar]
- Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, Tajima S, Mitsuya K, Okano M, Koseki H. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;14:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- Rothbart SB, Dickson BM, Ong MS, Krajewski K, Houliston S, Kireev DB, Arrowsmith CH, Strahl BD. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013;14:1288–1298. doi: 10.1101/gad.220467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonomo SB, Wu Y, Ferguson D, de Lange T. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J Cell Biol. 2009;14:385–398. doi: 10.1083/jcb.200902039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge JE, Kang YK, Beppu H, Lei H, Li E. Histone H3-K9 methyltransferase ESET is essential for early development. Mol Cell Biol. 2004;14:2478–2486. doi: 10.1128/MCB.24.6.2478-2486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Huh SO, Choi H, Lee KS, Shin D, Lee C, Nam JS, Kim H, Chung H, Lee HW, Park SD, Seong RH. Srg3, a mouse homolog of yeast SWI3, is essential for early embryogenesis and involved in brain development. Mol Cell Biol. 2001;14:7787–7795. doi: 10.1128/MCB.21.22.7787-7795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, Magnuson T. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;14:1287–1295. doi: 10.1016/S1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science. 2012;14:1499–1502. doi: 10.1126/science.1216154. [DOI] [PubMed] [Google Scholar]
- Pujadas E, Feinberg AP. Regulated noise in the epigenetic landscape of development and disease. Cell. 2012;14:1123–1131. doi: 10.1016/j.cell.2012.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen ND, Norris ML, Surani MA. Epigenetic control of transgene expression and imprinting by genotype-specific modifiers. Cell. 1990;14:853–861. doi: 10.1016/0092-8674(90)90195-K. [DOI] [PubMed] [Google Scholar]
- Sutherland HG, Kearns M, Morgan HD, Headley AP, Morris C, Martin DI, Whitelaw E. Reactivation of heritably silenced gene expression in mice. Mamm Genome. 2000;14:347–355. doi: 10.1007/s003350010066. [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Chong S, Champ ME, Cuthbert PC, Morgan HD, Luu KV, Whitelaw E. Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc Natl Acad Sci U S A. 2003;14:2538–2543. doi: 10.1073/pnas.0436776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;14:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;14:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome Project Data Processing S: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;14:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard. http://picard.sourceforge.net.
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;14:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;14:4067–4068. doi: 10.1093/bioinformatics/bti652. [DOI] [PubMed] [Google Scholar]
- Divina P, Kvitkovicova A, Buratti E, Vorechovsky I. Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur J Hum Genet. 2009;14:759–765. doi: 10.1038/ejhg.2008.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Linked intervals. Manhattan plots showing linked intervals identified by Illumina GoldenGate SNP genotyping analysis. The x-axis represents the chromosomes and the y-axis is the LOD score. Peaks with a LOD score of 3 or higher are considered significant.
Flow cytometric profiles of the MommeDs. (a) Representative flow cytometry profiles show the percentage of GFP-expressing erythrocytes from wild-type and heterozygous mutant littermates (n = 3). The x-axis represents the erythrocyte fluorescence on a logarithmic scale and the y-axis is the number of cells detected at each fluorescence level. (b) Percentage of GFP-expressing cells in wild-type, heterozygous mutant and homozygous mutant (where viable) mice at three weeks of age (mean ± standard error of the mean).
Mapping interval for MommeD30. (a)MommeD30 was produced in the FVB/NJ strain of mice and mapped by crossing twice onto Line3C in a C57BL/6J background. The results of the genotype for seven SNP markers and one microsatellite marker surrounding the linked interval are shown. The number of mice classified into each haplotype is shown on top. Our estimate of the linked interval is between rs29539305 and rs33446195 on chromosome 17 (highlighted). (b) List of genes in the MommeD30 linked interval on chromosome 17. The Mus musculus Ensembl database (release 37) was used to export a list of transcripts (protein coding and non-coding RNAs) within the 1.9 Mbp MommeD30 interval.
GFP expression in offspring of a Rif1GTheterozygote crossed to Line3C. A Rif1 gene trap allele (Rif1GT) had a similar effect on transgene expression as that observed with the MommeD18 mutation, increasing the percentage of expressing cells in mice heterozygous for the gene-trap allele.
Novel mutant alleles of Baz1b, Smchd1, Trim28, Dnmt1 and Smarca5. (a)MommeD16 carries a point mutation resulting in a non-conservative amino acid change in the Wstf domain of Baz1b. (b)MommeD23 and MommeD36 carry point mutations in Smchd1. Both mutations introduce premature stop codons in the Smchd1 protein. (c)MommeD31 carries a point mutation in Trim28 that results in an amino acid change in a highly conserved zinc finger domain. (d)MommeD32 carries a point mutation that results in an amino acid change in the BAH domain of Dnmt1. (e)MommeD35 and MommeD37 carry mutations that result in amino acid changes in highly conserved domains of the Smarca5 protein.
Embryonic development in MommeD mutants. Tabulated data shows the number of observed mice and in brackets the percentage of total. (a) Intercrosses of Pbrm1MommeD27 mice produce no homozygous offspring at three weeks. (b)Brd1MommeD42 homozygous mice are embryonic lethal around 10.5 dpc. (c) Mice carrying mutations in Suv39h1MommeD33 are viable at three weeks. (d)Uhrf1MommeD40 homozygotes are embryonic lethal around 10.5 dpc. (e) Some homozygous Baz1bMommeD16 mice were obtained at three weeks but less than expected. (f) No (Smchd1MommeD36) or few (Smchd1MommeD23) homozygous individuals were recovered from intercrosses. The Smchd1MommeD23 homozygotes that survived were males. (g)Trim28MommeD31 homozygous mice are embryonic lethal prior to 10.5 dpc. (h) Mice homozygous for the Dnmt1MommeD32 mutation die around 10.5 dpc. (i) Intercrosses of Smarca5MommeD35 and Smarca5MommeD37 produced no homozygous offspring at three weeks. Timed matings show that survival of homozygous mutants at 12.5 to 14.5 dpc was rare in Smarca5MommeD35 mutants. In Smarca5MommeD37 mutants homozygous embryonic death occurred prior to 14.5 dpc.
Linkage analysis using Illumina GoldenGate genotyping assay.
List of primer sequences.
Sanger traces.