

Abstract
Chronic heart failure (CHF) is a multi-factorial disease process that is characterized by over activation of the renin-angiotensin-aldosterone system (RAAS) and the sympathetic nervous system. Both of these systems are chronically activated in CHF. The RAAS consists of an excitatory arm involving Angiotensin II (Ang II), Angiotensin Converting Enzyme (ACE), and the Ang II type 1 Receptor (AT1R). The RAAS also consists of a protective arm consisting of Angiotensin-1-7 (Ang -1-7), the Ang II type 2 receptor (AT2R), ACE2 and the mas receptor. Sympathoexcitation in CHF is driven, in large part, by an imbalance of these two arms, with an increase in the Ang II-AT1R-ACE arm and a decrease in the AT2R-ACE2 arm. This imbalance is manifested in cardiovascular-control regions of the brain such as the rostral ventrolateral medulla and paraventricular nucleus in the hypothalamus. This review focuses on current literature that describes the components of these two arms of the RAAS, and their imbalance in the CHF state. Moreover, this review provides additional evidence for the relevance of ACE2 and Ang-1-7 as key players in the regulation of central sympathetic outflow in CHF. Finally we also examine the effects of exercise training as a therapeutic strategy and the molecular mechanisms at play in CHF, in part, because of the ability of exercise training to restore the balance of the RAAS axis and sympathetic outflow.
Introduction
Chronic heart failure (CHF) is the end result of various insults to the myocardium, the most common of which is ischemic heart disease [1]. Whenever cardiac output falls, more than momentarily, compensatory mechanisms are called into play in an attempt to maintain blood pressure and organ perfusion. Primary to this compensation is activation of the sympathetic nervous system and the renin angiotensin aldosterone system (RAAS). Increases in circulating norepinephrine (NE) and Angiotensin II (Ang II) evoke peripheral vasoconstriction and activate aldosterone secretion largely through α-adrenergic and Ang II Type 1 (AT1) receptors, respectively. An increase in renal renin release is undoubtedly caused by renal sympatho-excitation as well as a decrease in renal perfusion pressurein the CHF state[2-4]. While these compensatory changes are initially beneficial they become counterproductive if sustained for prolonged periods of time. Chronic increases in both plasma and tissue NE not only result in a down-regulation of β-adrenergic receptors in the heart[5, 6] but also contribute to further myocyte death[7]. Activation of the RAAS contributes in a major way to salt and water retention and to further sympatho-excitation[8, 9].
Central AT1R in Heart Failure
A large body of evidence points to the central RAAS in mediating sympatho-excitation in the setting of CHF[3, 10-14]. Areas in the rostral ventrolateral medulla (RVLM) and in the hypothalamus, (e.g. the paraventricular nucleus (PVN)) have been shown to mediate an increase in sympathetic outflow in response to microinjection of Ang II[15-25]. In studies carried out in rabbits with pacing-induced CHF Liu et al.[26] showed that central infusion of the AT1R blocker losartan reduced renal sympathetic nerve activity (RSNA). Furthermore, Zucker and co-workers showed an increase in Ang II in the cerebral spinal fluid of dogs with pacing-induced CHF[27] compared to sham animals. In an attempt to further understand the role of central AT1R in the sympathoexcitatory process, Gao et al. [28]measured AT1R protein and message in the RVLM of rabbits with pacing-induced CHF, both of which were increased (figure 1). Interestingly, in this and other studies[29] it was shown that this increase was associated with increased oxidative stress and RSNA in these animals. Similar observations have been made in rats with coronary artery ligation-induced CHF[30, 31]. In the rat model, Zhu et al.[32] carried out a novel study in which rats were infused with the antisense oligonucleotide to the AT1R while recording RSNA and blood pressure (figure 2). Compared to animals infused with scrambled antisense inhibition of the AT1R resulted in a decrease in RSNA. Upregulation of central AT1R is not limited to the RVLM and PVN in CHF; the subfornical organ (SFO), nucleus of the solitary tract (NTS), and area postrema (AP) are other areas of the brain have also shown an upregulation of AT1R in the setting of CHF[33, 34].
Figure 1.

A, RT-PCR analysis for mRNA expression of the AT1 receptor in the RVLM of sham and CHF rabbits. Top, A representative RT-PCR image showing the upregulated AT1 receptor mRNA expression in the RVLM of a CHF rabbit. β-Actin was used as internal control. Bottom, Results of densitometric analysis representing means±SE. ***P<0.001 compared with sham; n=9 each group. B, Western blot analysis for protein expression of the AT1 receptor in the RVLM of sham and CHF rabbits. Top, Representative Western blots showing the upregulation of AT1 receptor protein expression in RVLM of CHF rabbit. Bottom, Results of densitometric analysis representing means±SE. **P<0.0 compared with sham; n=8 each group. From Gao et al[28].
Figure 2.

Effects of intracerebroventricular (ICV) administration of antisense oligodeoxynucleotides (AS-ODN) and scrambled oligodeoxynucleotides (Scr-ODN) on baseline renal sympathetic nerve activity (RSNA; A), mean arterial pressure (MAP; B), and heart rate (HR; C) in sham-operated (Sham) and chronic heart failure (CHF) rats. AS-ODN significantly decreased baseline RSNA, MAP, and HR (n = 7 for each group) over time. Values are means ± SE. *P < 0.05 compared with administration of Scr-ODN. From Zhu et al[32]. Reprinted with Permission from the American Journal of Physiology.
The ability of the central RAAS to be a key player in the reverberating circuit of heart failure is not limited to the central nervous system. Indeed, intracerebroventricular (icv) blockade of the angiotensin receptor improved baroreflex sensitivity and decreased efferent renal sympathetic nerve activity in CHF rats[35, 36]. Central blockade of angiotensin converting enzyme (ACE) similarly decreased renal sympathetic nerve activity, improved the blunted baroreflex sensitivity, and normalized sodium consumption, urine sodium and urine volume in CHF rats[11]. Further, in a myocardial infarction rat model with a transgenic deletion of angiotensinogen, left ventricular enddiastolic pressure did not increase to the same extent as control CHF rats and LV dp/dt max did not decrease to the same extent as control CHF rats[37]. Taken together, the hyperactive central RAAS is a contributor to the global physiological changes as well as the cardiovascular dysfunction seen in CHF.
This apparent increase in AT1R signaling in the CNS appears to be mediated by a positive feedback of AT1R on the transcriptional regulation of the protein. The AT1R is upregulated in response to Ang II and in the CHF state[17, 29, 38, 39]. In a neuronal cell line (CATH.a) that express AT1Rs Mitra et al.[40] showed that in response to Ang II (100 nM), an NFkB-dependent increase in AT1R transcription ensued. This increase was also dependent on the downstream activation of two additional transcription factors, namely Ets-like kinase 1 (Elk-1) and activator protein 1 (AP-1). This pathway was confirmed in intact rabbits with CHF in which c-jun (one of the two transcription factors that dimerizes to form AP-1) and Jun N-terminal kinase (JNK) was increased in the RVLM[38]. The NFkB pathway has been shown to mediate an increase in sympathetic nerve activity in CHF since its blockade reduces sympathetic outflow, AT1R expression and oxidative stress in rats with CHF and hypertension[30, 41, 42]. This sympatho-excitation in response to central Ang II is most likely mediated by a decrease in the outward potassium current[43-45]. Recently, Gao et al.[46] showed that a potassium channel protein, Kv4.3 was downregulated in the brainstem of rats with CHF suggesting that this may contribute to enhanced membrane depolarization and action potential generation. Kv4.3 contributes to the transient outward current and is most prominently reduced in cardiac myocytes in CHF[47, 48]. The mechanism by which Ang II decreased K+ current is not clear but may result from interaction of a Kv4.3-AT1R complex[49].
A Role for Angiotensin Converting Enzyme (ACE) 2 in the Sympatho-Excitatory Process
The discovery of ACE2 and generation of Ang 1-7[50, 51] as important components of the RAAS has resulted in an explosion of studies on the biological and therapeutic effects of this arm of the RAAS. ACE2 activation has been shown to have beneficial effects in a variety of disease states[52]. Furthermore, Ang 1-7 has been shown to be beneficial in the setting of systemic hypertension[53, 54], pulmonary hypertension[55], renal disease[56] and cancer[57-59]. Since ACE2 has been found in the brain[60, 61] it seems reasonable that this enzyme and its product, Ang 1-7, would modulate the generation of central sympathetic outflow in CHF. Utilizing a unique transgenic mouse model that overexpresses human ACE2 selectively in neurons[53]Xiao et al.[62] examined the sympathetic nervous effects in transgenic and wild type mice subjected to a chronic myocardial infarction and the subsequent development of CHF. While there were no major differences in cardiac function in both groups of mice, transgenic mice exhibited a lower RSNA and an improvement in arterial baroreflex function (figure 3). Mice that overexpressed central ACE2 were able to suppress RSNA to zero during increases in blood pressure in contrast to wild type mice with CHF who could not lower RSNA in response to an increase in blood pressure. Examination of the spontaneous baroreflex control of heart rate also indicated an enhanced sympatho-inhibitory process in these mice. In a recent study by Zheng et al.[63] it was shown that viral overexpression of ACE2 reduced RSNA in a rat CHF model. This effect was apparently mediated by an increase in nitric oxide.
Figure 3.
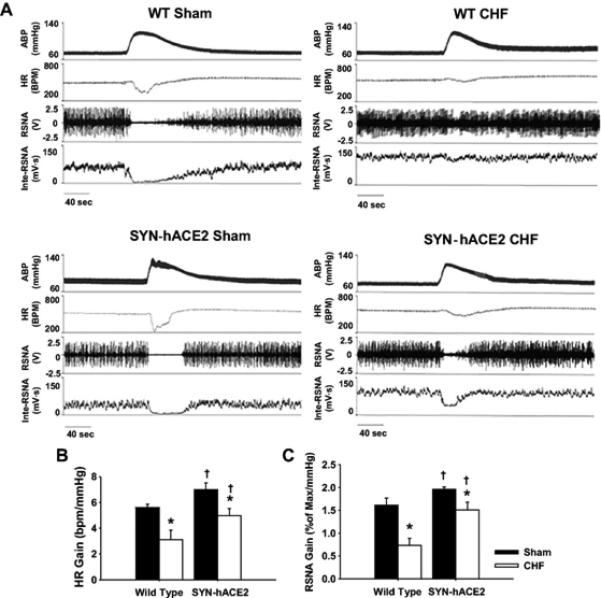
Baroreflex response to elevation in blood pressure induced by phenylephrine. A, Representative recordings for arterial blood pressure (BP), heart rate (HR), raw renal sympathetic nerve activity (RSNA), and integrated RSNA from anesthetized wild-type (WT) and SYN-hACE2 mice with either sham surgery or chronic heart failure (CHF). Mean values of the gain for HR and RSNA in each group are shown in B and C. *P<0.05 vs the corresponding group in sham mice; †P<0.05 vs the WT-CHF group. n=4 to 5 per group. From Xiao et al[62]
In an attempt to understand the cellular mechanisms by which over expression of ACE2 in neurons regulates AT1R expression we carried out in vitro studies where the neuronal cell line, CATH.a was transfected with a lentivirus that resulted in overexpression of human ACE2. As can be seen in figure 4, Ang II stimulated an increase in AT1R expression which was blocked by the AT1R antagonist, losartan. Overexpression of ACE2 completely prevented the increase in AT1R in response to Ang II. Interestingly, this suppression was not reversed by the mas receptor antagonist A-779. These results suggest that the efficacy of ACE2 to prevent the upregulation of AT1R may be related to its ability to degrade Ang II rather than to the generation of Ang 1-7. Because over expression of human ACE2 may be quite nonphysiologic and expression may be increased by several fold over endogenous mouse ACE2, the effects of exogenous Ang 1-7 was tested in order to determine if this peptide could alter AT1R expression in vitro. Figure 5 shows the results from this experiment. Again losartan prevented the upregulation of the AT1R in response to Ang II. Ang 1-7 also increased AT1R expression. Both the response to Ang II and the response to Ang 1-7 were blocked by losartan and neither was blocked by A-779. Taken together these data strongly suggest, at least in vitro, that Ang 1-7 does not prevent the up regulation of the AT1R and that the efficacy of ACE2 in this response is mediated by an AT1R dependent reduction in Ang II. Therefore, increases in Ang 1-7 may be secondary to a reduction in AT1R activation as a therapy in the setting of CHF. However, therapies that both stimulate the Ang 1-7 pathway and decrease AT1R would be of additive benefit. It is also possible that Ang 1-7 signaling through the mas receptor augments nitric oxide production which has been shown to decrease AT1R expression[64] .
Figure 4.
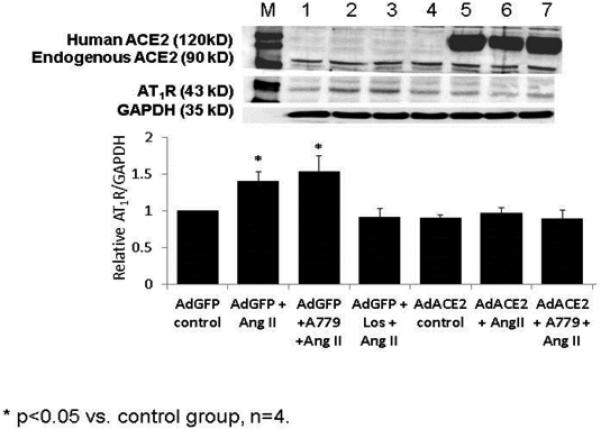
AT1R protein expression in Cath.a neurons which were incubated with Ang II alone or in combination with either a mas receptor antagonist, A-779 or an AT1R receptor antagonist, losartan. Experiments were conducted with either a GFP adenovirus or human ACE2 adenovirus. ACE2 overexpression inhibited the upregulation of the AT1R in response to Ang II however this was not reversed by A-779. Data generated by Dr. Liang Xiao.
Figure 5.
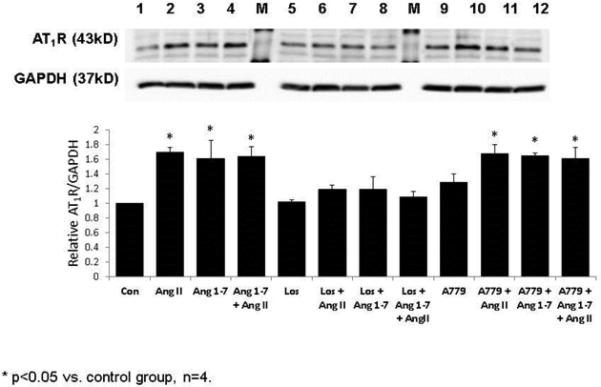
AT1R expression is upregulated by both Ang II and Ang (1-7) in CATH.a cultured neurons. Losartan but not A-779 blocked the response to both peptides. Data generated by Dr. Liang Xiao.
While manipulations of ACE2 may provide insight into the autonomic effects of this enzyme they do not directly address the question of whether Ang 1-7 is capable of modulating sympathetic nerve activity in a beneficial direction, especially in cardiovascular disease states. There has been controversy over the effects of Ang 1-7 on sympathetic outflow and baroreflex function. In early experiments Potts et al.[65] and Da Silva et al.[66] provided evidence for a sympatho-excitatory effect of Ang 1-7. On the other hand, Xia et al.[67] and Diz et al.[68] have provided evidence that Ang 1-7 exerts sympatho-inhibitory effects. In a recent study by Kar et al.[69] conscious rabbits with and without CHF were infused by the intracerebroventricular (icv) route with Ang 1-7 for several days. Autonomic balance to the heart was assessed by evaluating the heart rate response to an acute bolus injection of either atropine to assess vagal tone or metoprolol to evaluate cardiac sympathetic outflow. Ang 1-7 increased vagal tone in CHF rabbits (i.e. a greater increase in heart rate in response to atropine) and decreased sympathetic tone (a smaller decrease in heart rate in response to metoprolol). There was no effect of Ang 1-7 in sham rabbits. In addition, chronic icv infusion of Ang 1-7 profoundly reduced RSNA in conscious CHF rabbits while having no effect in normal rabbits. Importantly, this effect was mediated by the mas receptor since it was reversed when Ang 1-7 was co-infused with A-779 (figure 6). Consistent with these results baroreflex gain for both heart rate and RSNA was increased in rabbits with CHF.
Figure 6.

AT1R (A), GRK5 (B), p65 NF-κB (C) are increased in the PVN (solid bars) and RVLM (open bars) of CHF animals and normalized by ExT. GRK2 (D), another kinase implicated in regulating AT1R expression, is unchanged in the PVN during both CHF and ExT. *P<0.05 versus Sham-Sed. †P<0.05 versus CHF-Sed; n=5 to 7. From Haack et al[79].
Modulation of Central RAS Components by Exercise Training in CHF
There is a growing trend to consider the use of non-pharmacological therapy in the treatment of CHF. One such intervention is exercise training (ExT). In 2003 a position statement by The American Heart Association concluded that ExT of patients with CHF is safe and likely to be an effective treatment paradigm[70].There is now clear evidence that ExT of patients with CHF increases quality of life and improves survival from all cardiac events[71-74] even if cardiac function per se is not improved. However, in elderly patients the benefits may not be as great[75, 76]. In experiments carried out in rabbits with pacing-induced CHF, Mousa et al.[77] and Liu et al.[78] showed that ExT reduced AT1R expression in the RVLM and reduced plasma Ang II. Similar results have been reported in rats with MI-induced CHF[79]. In humans, studies by Roveda et al.[80] and by Negrão and colleagues[81-83] have clearly shown a decrease in muscle sympathetic nerve activity (MSNA) in CHF patients following ExT. These studies raise important questions concerning the role of the central RAAS in mediating the sympatho-inhibitory effects of ExT in the setting of CHF. What does ExT do to central Ang II signaling and to the components of the RAAS system in the central nervous system? Exercise training in experimental CHF has been shown to reduce central AT1R[77, 84] and oxidative stress[85-87]while at the same time increasing the sympatho-inhibitory effects of nitric oxide[84, 88] and improving baroreflex function[77, 78, 86, 89]. There is much less known about ACE and ACE2 in the central nervous system following ExT in the CHF state. In a study carried out in rabbits with pacing-induced CHF Kar et al.[90] examined the relationship between ExT and the expression of ACE and ACE2 in the RVLM and PVN. In both regions ACE was elevated in the CHF state and ACE2 was decreased. Following ExT the expression of these two proteins were reversed and looked very similar to sham animals. While there are several studies showing that ExT reduces ACE in the myocardium[91, 92] the above is the first study showing a reversal of the ACE/ACE2 ratio in the brain of animals with CHF. Because Ang 1-7 is thought to be sympatho-inhibitory and Ang II sympatho-excitatory, a decrease in the ratio of ACE to ACE2 should be beneficial in reducing sympathetic outflow in CHF and hypertension. This in turn would also reduce oxidative stress and increase nitric oxide at the local level. Theoretically, the ACE/ACE2 balance would also mediate the relative concentrations of Ang II and Ang 1-7 in the brain. In vitro evidence has shown that Ang 1-7 can mediate an increase in neuronal potassium current by a nitric oxide and mas receptor-dependent mechanism[93].
The expression of AT1Rs in the brain is critically tied to signaling through the AT1R in animals with CHF. This positive feedback nature was shown in CHF rabbits that underwent an ExT regimen while simultaneously receiving a chronic infusion of Ang II so that plasma Ang II would not be normalized[77]. Under these conditions AT1R message and protein in the RVLM remained elevated (compared to non-ExT CHF rabbits). Furthermore, resting RSNA and arterial baroreflex function remained elevated and depressed, respectively. These data, along with those from chronic central losartan infusion, strongly suggest that both high levels of Ang II and increased AT1R signaling are necessary for sympatho-excitation and that the normalization of these parameters by ExT are mediated, at least in part, by a reduction in both.
Regulation of Central RAS Components
Because plasma Ang II is reduced following ExT in CHF animals[39] (although it is unclear if this is also true for Ang II in the brain) it is of some interest to determine the influence of Ang II on ACE and ACE2 expression. In a recent in vitro study, Xiao et al.[94] clearly showed that Ang II treatment of neurons resulted in an increase in ACE and a decrease in ACE2 in a dose-dependent manner. At the message level the changes for both proteins in response to Ang II could be inhibited by blocking p38 MAPK or ERK1/2. This seems to be selectively mediated by the AT1R as it was also blocked by losartan but not by the AT2R blocker PD123319. This reciprocal relationship appears to hold in other tissues and in other diseases states such as hypertension[95- 98]. These data suggest that Ang II can set into motion a series of transcriptional events through common cell signaling pathways to regulate the balance between ACE and ACE2. However, the transcription factors that drive the regulation of both ACE and ACE2 have not yet been identified.
While the transcription of new AT1R protein may be an important contributor to central Ang II signaling and another mechanism of potential importance relates to the way the AT1R is turned over. The AT1R is a G-protein coupled receptor and signals through a Gq/11 and other G protein mechanisms[99]. As such, its phosphorylation following agonist binding is mediated by G-protein receptor kinases (GRK)[100]. Following phosphorylation the protein is targeted for internalization by β-arrestin after which it is degraded in lysosomes[101]. Recent experiments carried out in rats with CHF have shown that GRK5 is upregulated in the RVLM and PVN (figure 6) and binds to the AT1R[79]. On the other hand, GRK2, the more classical β-adrenergic receptor kinase that can also regulate AT1R is not changed. Interestingly, the increase in GRK5 occurs at the same time as the AT1R is upregulated. Following ExT both GRK5 and AT1R were decreased. This parallel change in both proteins suggests that the increase in GRK5 is a compensatory response to the increased AT1R expression. The increase in GRK5 may not be effective in decreasing AT1R expression due to intense stimuli that increase the transcription of this receptor (e.g. NFkB) in CHF.
In vitro experiments carried out in CATH.a neurons confirmed that substantial upregulation of GRK5 results in a decrease in AT1R protein. Under these conditions, the Ang II – mediated increase in AT1R expression was completely blocked (Figure 7). On the other hand, GRK5 knock down with an siRNA caused an increase in AT1R expression in response to Ang II. Taken together these studies suggest that a balance exists between the transcriptional regulators of AT1R and the pathways responsible for degrading the AT1R. In the setting of CHF the former apparently predominate, thus promoting an Ang II-dependent neuronal depolarization and increase in sympathetic outflow.
Figure 7.

GRK5 overexpression normalizes AT1R and p65 NF-κB protein levels following stimulation with Ang II. Values are expressed as a ratio of protein to GAPDH and normalized to no ligand. A, AT1R. B, p65 NF-κB. C, GRK5. *P<0.05 versus no ligand; n=5 to 7. From Haack et al[79].
Despite our increasing knowledge on the regulatory pathways of AT1R and central RAS components, the induction of the central RAS, especially in regions with an intact blood-brain barrier like the RVLM, remain unclear. While angiotensinogen is found in brain extracellular fluid and cerebrospinal fluid, astrocytes and more recently in the neurons of many brain regions including the PVN, NTS, RVLM and SFO, the cell types in which renin, ACE, aminopeptidase A and aminopeptidase N are found in the brain are still controversial. To date only low levels of Ang I, Ang II and Ang 1-7 have been identified in brain tissue[22]. Therefore, one possibility is that the Ang II from the periphery detected in circumventricular organs (CVOs) would induce a signaling cascade in non-CVO nuclei. Conversely, given that much of the RAS is expressed centrally between neurons, astrocytes and endothelial cells, angiotensins are not only present in the brain, but may function as neurotransmitters[102] .
Data from this laboratory and others strongly suggests that circulating Ang II is a primary driver of the imbalance for AT1R-ACE and AT2R-ACE2. Both in vitro and in vivo studies have demonstrated that Ang II mediated increase in AT1R and its pathway components are dependent on AT1R signaling; pretreatment with Losartan blunts this imbalance[29, 38].
Summary
Clearly, the regulation of sympathetic nerve activity is a complicated and multifactorial process. This review only highlights one mechanism that plays a role in this process. Ang II along with other peptide and non-peptide mediators can alter neuronal membrane potential, in part, by reducing outward potassium currents. Most central pre-sympathetic neurons express all the receptors involved in signaling through the RAS. Therefore, the balance between Ang II and other peptides such as Ang 1-7 and the balance between AT1R, AT2R and mas receptors may be critical to establishing the level of neuronal activation. Furthermore, the synthesis of Ang II and Ang 1-7 due to ACE and ACE2 also appears to contribute to sympatho-excitation in CHF. Figure 8 outlines the major mechanisms in the RAS system that has thus far been defined to regulate sympatho-excitation in CHF. Increases in central Ang II initiate a positive feedback process whereby the AT1R is upregulated by an Ang II-AT1R-dependent mechanism. Intracellular activation of NFkB and downstream transcription factors (Elk-1 and AP1) increase the transcription of the AT1R. This process is accompanied by a compensatory increase in GRK5 in an attempt to limit AT1R upregulation. However, the apparent intensity of the stimuli to up regulate this protein far outweighs the ability of GRK5 alone to reduce the AT1R protein.
Figure 8.
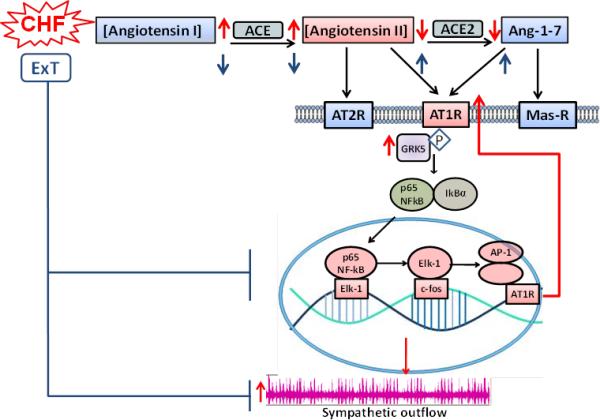
Summary schematic showing the relationship between RAS metabolites, the AT1R signalling cascade, and sympathetic outflow in CHF and following ExT. CHF, chronic heart failure; ExT, exercise training; AP1, Activator Protein 1; NFkB, Nuclear Factor kappa B; ELK1, ETS Like Kinase; ACE, Angiotensin Converting Enzyme. In neurons Ang II stimulates the up regulation of the AT1R by an NFkB initiating process. ACE is increased and ACE2 decreased resulting in an imbalance between Ang II and Ang 1-7. ExT restores this imbalance and reduces AT1R signalling by increasing ACE2 and reducing Ang II.
The relative paucity of new pharmacological agents in the treatment of CHF has stimulated a search for non-pharmacological therapies. In addition to novel device therapy (e.g. baroreflex stimulation, renal denervation, vagal stimulation), ExT has been promoted as a way of reducing mortality and increasing the quality of life for patients with CHF. The mechanisms by which ExT is efficacious in this regard are not well understood. While ExT is known to affect virtually every organ system, the focus on central sympathetic remodeling is starting to define some of the pathways impacted by this intervention[103]. ExT impacts the central RAS in a major way. Importantly, it reduces oxidative stress in the RVLM and causes an upregulation of both SOD1 and SOD2[86]. Since Ang II signals, in part, through the NADPH oxidase – dependent production of superoxide, ExT is likely to have a major impact on Ang II signaling. Indeed ExT lowers plasma Ang II and reduces AT1R expression which is dependent on activation of the AT1R (thus, positive feedback). Furthemore, ExT reduces ACE and increases ACE2 in the setting of CHF. Therefore, the role of ExT in modulating the central RAS would seem a fruitful area of continued investigation. This simple and inexpensive intervention may provide some of the benefits of currently used pharmacological therapies and can also be used as adjunctive therapy.
Future Directions
Many questions still remain in the regulation of central RAS in the setting of CHF. It will be important to determine the precise location and mechanism(s) by which central Ang II is generated, and what initiating signal drives the feed-forward activation of the RAS in the setting of CHF. Conversely, the precise central and peripheral signals generated by ExT that trigger the downstream effects outlined in this review that lead to protection in the setting of CHF also remain unclear. Additionally, the mechanism(s) by which nitric oxide can negatively regulate AT1R are still unknown. Finally, because many existing therapies that target the RAS do not improve cardiac parameters, it will be important to develop novel therapies that both improve autonomic imbalance and hemodynamic parameters.
Acknowledgements
Some of the data shown in this review was supported by a grant from the National Heart, Lung and Blood Institute, HL PO1 62222. Dr. Haack was supported by HL F32-116172.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2011;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dzau VJ, Colucci WS, Hollenberg NK, Williams GH. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation. 1981;63:645–651. doi: 10.1161/01.cir.63.3.645. [DOI] [PubMed] [Google Scholar]
- 3.Francis GS. The relationship of the sympathetic nervous system and the renin-angiotensin system in congestive heart failure. Am Heart J. 1989;118:642–648. doi: 10.1016/0002-8703(89)90291-3. [DOI] [PubMed] [Google Scholar]
- 4.Holtz J. Pathophysiology of heart failure and the renin-angiotensin-system. Basic Res Cardiol. 88 Suppl. 1993;1:183–201. doi: 10.1007/978-3-642-72497-8_13. [DOI] [PubMed] [Google Scholar]
- 5.Bristow MR, Minobe W, Rasmussen R, Larrabee P, Skerl L, Klein JW, Anderson FL, Murray J, Mestroni L, Karwande SV, et al. Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest. 1992;89:803–815. doi: 10.1172/JCI115659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P, Jamieson S, et al. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res. 1986;59:297–309. doi: 10.1161/01.res.59.3.297. [DOI] [PubMed] [Google Scholar]
- 7.Freeman K, Lerman I, Kranias EG, Bohlmeyer T, Bristow MR, Lefkowitz RJ, Iaccarino G, Koch WJ, Leinwand LA. Alterations in cardiac adrenergic signaling and calcium cycling differentially affect the progression of cardiomyopathy. J Clin Invest. 2001;107:967–974. doi: 10.1172/JCI12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ljungman S, Laragh JH, Cody RJ. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs. 39 Suppl. 1990;4:10–21. doi: 10.2165/00003495-199000394-00004. discussion 22-14. [DOI] [PubMed] [Google Scholar]
- 9.Sobotka PA, Krum H, Bohm M, Francis DP, Schlaich MP. The role of renal denervation in the treatment of heart failure. Curr Cardiol Rep. 2012;14:285–292. doi: 10.1007/s11886-012-0258-x. [DOI] [PubMed] [Google Scholar]
- 10.Felder RB, Francis J, Zhang ZH, Wei SG, Weiss RM, Johnson AK. Heart failure and the brain: new perspectives. Am J Physiol Regul Integr Comp Physiol. 2003;284:R259–276. doi: 10.1152/ajpregu.00317.2002. [DOI] [PubMed] [Google Scholar]
- 11.Francis J, Wei SG, Weiss RM, Felder RB. Brain angiotensin-converting enzyme activity and autonomic regulation in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H2138–2146. doi: 10.1152/ajpheart.00112.2004. [DOI] [PubMed] [Google Scholar]
- 12.Hegarty AA, Hayward LF, Felder RB. Influence of circulating angiotensin II and vasopressin on neurons of the nucleus of the solitary tract. Am J Physiol. 1996;270:R675–681. doi: 10.1152/ajpregu.1996.270.3.R675. [DOI] [PubMed] [Google Scholar]
- 13.Huang BS, Leenen FH. The brain renin-angiotensinaldosterone system: a major mechanism for sympathetic hyperactivity and left ventricular remodeling and dysfunction after myocardial infarction. Curr Heart Fail Rep. 2009;6:81–88. doi: 10.1007/s11897-009-0013-9. [DOI] [PubMed] [Google Scholar]
- 14.Zucker IH, Schultz HD, Li YF, Wang Y, Wang W, Patel KP. The origin of sympathetic outflow in heart failure: the roles of angiotensin II and nitric oxide. Prog Biophys Mol Biol. 2004;84:217–232. doi: 10.1016/j.pbiomolbio.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 15.Bains JS, Potyok A, Ferguson AV. Angiotensin II actions in paraventricular nucleus: functional evidence for neurotransmitter role in efferents originating in subfornical organ. Brain Res. 1992;599:223–229. doi: 10.1016/0006-8993(92)90395-p. [DOI] [PubMed] [Google Scholar]
- 16.Chen F, Liu F, Badoer E. AT1 receptors in the paraventricular nucleus mediate the hyperthermia-induced reflex reduction of renal blood flow in rats. Am J Physiol Regul Integr Comp Physiol. 2011;300:R479–485. doi: 10.1152/ajpregu.00604.2010. [DOI] [PubMed] [Google Scholar]
- 17.Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Sympathoexcitation by central ANG II: roles for AT1 receptor upregulation and NAD(P)H oxidase in RVLM. Am J Physiol Heart Circ Physiol. 2005;288:H2271–2279. doi: 10.1152/ajpheart.00949.2004. [DOI] [PubMed] [Google Scholar]
- 18.Gao L, Wang WZ, Wang W, Zucker IH. Imbalance of angiotensin type 1 receptor and angiotensin II type 2 receptor in the rostral ventrolateral medulla: potential mechanism for sympathetic overactivity in heart failure. Hypertension. 2008;52:708–714. doi: 10.1161/HYPERTENSIONAHA.108.116228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Head GA. Role of AT1 receptors in the central control of sympathetic vasomotor function. Clin Exp Pharmacol Physiol. 1996;(Suppl 3):S93–98. [PubMed] [Google Scholar]
- 20.Ito S, Komatsu K, Tsukamoto K, Kanmatsuse K, Sved AF. Ventrolateral medulla AT1 receptors support blood pressure in hypertensive rats. Hypertension. 2002;40:552–559. doi: 10.1161/01.hyp.0000033812.99089.92. [DOI] [PubMed] [Google Scholar]
- 21.Kleiber AC, Zheng H, Sharma NM, Patel KP. Chronic AT1 receptor blockade normalizes NMDA-mediated changes in renal sympathetic nerve activity and NR1 expression within the PVN in rats with heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H1546–1555. doi: 10.1152/ajpheart.01006.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, Oldfield BJ, Mendelsohn FA, Chai SY. The brain renin-angiotensin system: location and physiological roles. Int J Biochem Cell Biol. 2003;35:901–918. doi: 10.1016/s1357-2725(02)00306-0. [DOI] [PubMed] [Google Scholar]
- 23.Patel KP, Mayhan WG, Bidasee KR, Zheng H. Enhanced angiotensin II-mediated central sympathoexcitation in streptozotocin-induced diabetes: role of superoxide anion. Am J Physiol Regul Integr Comp Physiol. 2011;300:R311–320. doi: 10.1152/ajpregu.00246.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang G, Milner TA, Speth RC, Gore AC, Wu D, Iadecola C, Pierce JP. Sex differences in angiotensin signaling in bulbospinal neurons in the rat rostral ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1149–1157. doi: 10.1152/ajpregu.90485.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu GQ, Gao L, Patel KP, Zucker IH, Wang W. ANG II in the paraventricular nucleus potentiates the cardiac sympathetic afferent reflex in rats with heart failure. J Appl Physiol. 2004;97:1746–1754. doi: 10.1152/japplphysiol.00573.2004. [DOI] [PubMed] [Google Scholar]
- 26.Liu JL, Zucker IH. Regulation of sympathetic nerve activity in heart failure: a role for nitric oxide and angiotensin II. Circ Res. 1999;84:417–423. doi: 10.1161/01.res.84.4.417. [DOI] [PubMed] [Google Scholar]
- 27.Zucker IH, Wang W, Pliquett RU, Liu J-L, Patel KP. The regulation of sympathetic outflow in heart failure. In: Chapleau MW, Abboud FM, editors. Neuro-Cardiovascular Regulation. New York Academy of Sciences; New York: 2001. pp. 431–443. [PubMed] [Google Scholar]
- 28.Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Superoxide mediates sympathoexcitation in heart failure: roles of angiotensin II and NAD(P)H oxidase. Circ Res. 2004;95:937–944. doi: 10.1161/01.RES.0000146676.04359.64. [DOI] [PubMed] [Google Scholar]
- 29.Liu D, Gao L, Roy SK, Cornish KG, Zucker IH. Role of oxidant stress on AT1 receptor expression in neurons of rabbits with heart failure and in cultured neurons. Circ Res. 2008;103:186–193. doi: 10.1161/CIRCRESAHA.108.179408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guggilam A, Cardinale JP, Mariappan N, Sriramula S, Haque M, Francis J. Central TNF inhibition results in attenuated neurohumoral excitation in heart failure: a role for superoxide and nitric oxide. Basic Res Cardiol. 2011;106:273–286. doi: 10.1007/s00395-010-0146-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang BS, Zheng H, Tan J, Patel KP, Leenen FH. Regulation of hypothalamic renin-angiotensin system and oxidative stress by aldosterone. Exp Physiol. 2011;96:1028–1038. doi: 10.1113/expphysiol.2011.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu GQ, Gao L, Li Y, Patel KP, Zucker IH, Wang W. AT1 receptor mRNA antisense normalizes enhanced cardiac sympathetic afferent reflex in rats with chronic heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1828–1835. doi: 10.1152/ajpheart.01245.2003. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Seto SW, Golledge J. Angiotensin II, sympathetic nerve activity and chronic heart failure. Heart Fail Rev. 2012 doi: 10.1007/s10741-012-9368-1. [DOI] [PubMed] [Google Scholar]
- 34.Yoshimura R, Sato T, Kawada T, Shishido T, Inagaki M, Miyano H, Nakahara T, Miyashita H, Takaki H, Tatewaki T, Yanagiya Y, Sugimachi M, Sunagawa K. Increased brain angiotensin receptor in rats with chronic high-output heart failure. J Card Fail. 2000;6:66–72. doi: 10.1016/s1071-9164(00)00013-0. [DOI] [PubMed] [Google Scholar]
- 35.DiBona GF, Jones SY, Brooks VL. ANG II receptor blockade and arterial baroreflex regulation of renal nerve activity in cardiac failure. Am J Physiol. 1995;269:R1189–1196. doi: 10.1152/ajpregu.1995.269.5.R1189. [DOI] [PubMed] [Google Scholar]
- 36.Zhang W, Huang BS, Leenen FH. Brain renin-angiotensin system and sympathetic hyperactivity in rats after myocardial infarction. Am J Physiol. 1999;276:H1608–1615. doi: 10.1152/ajpheart.1999.276.5.H1608. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, Huang BS, Ganten D, Leenen FH. Prevention of sympathetic and cardiac dysfunction after myocardial infarction in transgenic rats deficient in brain angiotensinogen. Circ Res. 2004;94:843. doi: 10.1161/01.res.0000120864.21172.5a. [DOI] [PubMed] [Google Scholar]
- 38.Liu D, Gao L, Roy SK, Cornish KG, Zucker IH. Neuronal angiotensin II type 1 receptor upregulation in heart failure: activation of activator protein 1 and Jun N-terminal kinase. Circ Res. 2006;99:1004–1011. doi: 10.1161/01.RES.0000247066.19878.93. [DOI] [PubMed] [Google Scholar]
- 39.Liu JL, Irvine S, Reid IA, Patel KP, Zucker IH. Chronic exercise reduces sympathetic nerve activity in rabbits with pacing-induced heart failure: A role for angiotensin II. Circulation. 2000;102:1854–1862. doi: 10.1161/01.cir.102.15.1854. [DOI] [PubMed] [Google Scholar]
- 40.Mitra AK, Zucker IH. Convergence of p38MAPK pathway and nuclear signals involving NFkB/CREB following Ang II stimulation in neurons. In: FASEB J, editor. FASEB J; Washington D.C.: 2011. [Google Scholar]
- 41.Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S, Yang ZM, Francis J. Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension. Cardiovasc Res. 2009;82:503–512. doi: 10.1093/cvr/cvp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elks CM, Mariappan N, Haque M, Guggilam A, Majid DS, Francis J. Chronic NF-{kappa}B blockade reduces cytosolic and mitochondrial oxidative stress and attenuates renal injury and hypertension in SHR. Am J Physiol Renal Physiol. 2009;296:F298–305. doi: 10.1152/ajprenal.90628.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sumners C, Gelband CH. Neuronal ion channel signalling pathways: modulation by angiotensin II. Cell Signal. 1998;10:303–311. doi: 10.1016/s0898-6568(97)00133-2. [DOI] [PubMed] [Google Scholar]
- 44.Sumners C, Zhu M, Gelband CH, Posner P. Angiotensin II type 1 receptor modulation of neuronal K+ and Ca2+ currents: intracellular mechanisms. Am J Physiol. 1996;271:C154–163. doi: 10.1152/ajpcell.1996.271.1.C154. [DOI] [PubMed] [Google Scholar]
- 45.Zhu M, Neubig RR, Wade SM, Posner P, Gelband CH, Sumners C. Modulation of K+ and Ca2+ currents in cultured neurons by an angiotensin II type 1a receptor peptide. Am J Physiol. 1997;273:C1040–1048. doi: 10.1152/ajpcell.1997.273.3.C1040. [DOI] [PubMed] [Google Scholar]
- 46.Gao L, Li Y, Schultz HD, Wang WZ, Wang W, Finch M, Smith LM, Zucker IH. Downregulated Kv4.3 expression in the RVLM as a potential mechanism for sympathoexcitation in rats with chronic heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H945–955. doi: 10.1152/ajpheart.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin H, Hadri L, Palomeque J, Morel C, Karakikes I, Kaprielian R, Hajjar R, Lebeche D. KChIP2 attenuates cardiac hypertrophy through regulation of Ito and intracellular calcium signaling. J Mol Cell Cardiol. 2010;48:1169–1179. doi: 10.1016/j.yjmcc.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Tang K, Xie B, Li S, Rozanski GJ. Regulation of Kv4 channel expression in failing rat heart by the thioredoxin system. Am J Physiol Heart Circ Physiol. 2008;295:H416–424. doi: 10.1152/ajpheart.91446.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozgen N, Lu Z, Boink GJ, Lau DH, Shlapakova IN, Bobkov Y, Danilo P,, Jr., Cohen IS, Rosen MR. Microtubules and angiotensin II receptors contribute to modulation of repolarization induced by ventricular pacing. Heart Rhythm. 2012;9:1865–1872. doi: 10.1016/j.hrthm.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 51.Ferrario CM, Santos RA, Brosnihan KB, Block CH, Schiavone MT, Khosla MC, Greene LJ. A hypothesis regarding the function of angiotensin peptides in the brain. Clin Exp Hypertens A. 1988;10(Suppl 1):107–121. doi: 10.3109/10641968809075966. [DOI] [PubMed] [Google Scholar]
- 52.Ferreira AJ, Murca TM, Fraga-Silva RA, Castro CH, Raizada MK, Santos RA. New cardiovascular and pulmonary therapeutic strategies based on the Angiotensin-converting enzyme 2/angiotensin-(1-7)/mas receptor axis. Int J Hypertens. 2012;2012:147825. doi: 10.1155/2012/147825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feng Y, Xia H, Cai Y, Halabi CM, Becker LK, Santos RA, Speth RC, Sigmund CD, Lazartigues E. Brain-selective overexpression of human Angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res. 2009;106:373–382. doi: 10.1161/CIRCRESAHA.109.208645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xia H, Feng Y, Obr TD, Hickman PJ, Lazartigues E. Angiotensin II type 1 receptor-mediated reduction of angiotensin-converting enzyme 2 activity in the brain impairs baroreflex function in hypertensive mice. Hypertension. 2009;53:210–216. doi: 10.1161/HYPERTENSIONAHA.108.123844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shenoy V, Qi Y, Katovich MJ, Raizada MK. ACE2, a promising therapeutic target for pulmonary hypertension. Curr Opin Pharmacol. 2011;11:150–155. doi: 10.1016/j.coph.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zimmerman D, Burns KD. Angiotensin-(1-7) in kidney disease: a review of the controversies. Clin Sci (Lond) 2012;123:333–346. doi: 10.1042/CS20120111. [DOI] [PubMed] [Google Scholar]
- 57.Qian YR, Guo Y, Wan HY, Fan L, Feng Y, Ni L, Xiang Y, Li QY. Angiotensin-converting enzyme 2 attenuates the metastasis of non-small cell lung cancer through inhibition of epithelial-mesenchymal transition. Oncol Rep. 2013;29:2408–2414. doi: 10.3892/or.2013.2370. [DOI] [PubMed] [Google Scholar]
- 58.Gallagher PE, Tallant EA. Inhibition of human lung cancer cell growth by angiotensin-(1-7). Carcinogenesis. 2004;25:2045–2052. doi: 10.1093/carcin/bgh236. [DOI] [PubMed] [Google Scholar]
- 59.Krishnan B, Smith TL, Dubey P, Zapadka ME, Torti FM, Willingham MC, Tallant EA, Gallagher PE. Angiotensin-(1-7) attenuates metastatic prostate cancer and reduces osteoclastogenesis. Prostate. 73:71–82. doi: 10.1002/pros.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2007;292:R373–381. doi: 10.1152/ajpregu.00292.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiao L, Gao L, Lazartigues E, Zucker IH. Brain-Selective Overexpression of Angiotensin-Converting Enzyme 2 Attenuates Sympathetic Nerve Activity and Enhances Baroreflex Function in Chronic Heart Failure. Hypertension. 2011;58:8. doi: 10.1161/HYPERTENSIONAHA.111.176636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng H, Liu X, Patel KP. Angiotensin-converting enzyme 2 overexpression improves central nitric oxide-mediated sympathetic outflow in chronic heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2402–2412. doi: 10.1152/ajpheart.00330.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma NM, Zheng H, Li YF, Patel KP. Nitric oxide inhibits the expression of AT1 receptors in neurons. Am J Physiol Cell Physiol. 2012;302:C1162–1173. doi: 10.1152/ajpcell.00258.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Potts PD, Horiuchi J, Coleman MJ, Dampney RA. The cardiovascular effects of angiotensin-(1-7) in the rostral and caudal ventrolateral medulla of the rabbit. Brain Res. 2000;877:58–64. doi: 10.1016/s0006-8993(00)02626-3. [DOI] [PubMed] [Google Scholar]
- 66.Silva AQ, Santos RA, Fontes MA. Blockade of endogenous angiotensin-(1-7) in the hypothalamic paraventricular nucleus reduces renal sympathetic tone. Hypertension. 2005;46:341–348. doi: 10.1161/01.HYP.0000179216.04357.49. [DOI] [PubMed] [Google Scholar]
- 67.Xia H, Suda S, Bindom S, Feng Y, Gurley SB, Seth D, Navar LG, Lazartigues E. ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS One. 2011;6:e22682. doi: 10.1371/journal.pone.0022682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Diz DI, Garcia-Espinosa MA, Gallagher PE, Ganten D, Ferrario CM, Averill DB. Angiotensin-(1-7) and baroreflex function in nucleus tractus solitarii of (mRen2)27 transgenic rats. J Cardiovasc Pharmacol. 2008;51:542–548. doi: 10.1097/FJC.0b013e3181734a54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kar S, Gao L, Belatti DA, Curry PL, Zucker IH. Central angiotensin (1-7) enhances baroreflex gain in conscious rabbits with heart failure. Hypertension. 2011;58:627–634. doi: 10.1161/HYPERTENSIONAHA.111.177600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pina IL, Apstein CS, Balady GJ, Belardinelli R, Chaitman BR, Duscha BD, Fletcher BJ, Fleg JL, Myers JN, Sullivan MJ. Exercise and heart failure: A statement from the American Heart Association Committee on exercise, rehabilitation, and prevention. Circulation. 2003;107:1210–1225. doi: 10.1161/01.cir.0000055013.92097.40. [DOI] [PubMed] [Google Scholar]
- 71.Belardinelli R, Georgiou D, Ginzton L, Cianci G, Purcaro A. Effects of moderate exercise training on thallium uptake and contractile response to low-dose dobutamine of dysfunctional myocardium in patients with ischemic cardiomyopathy. Circulation. 1998;97:553–561. doi: 10.1161/01.cir.97.6.553. [DOI] [PubMed] [Google Scholar]
- 72.Belardinelli R, Georgiou D, Cianci G, Purcaro A. Randomized, controlled trial of long-term moderate exercise training in chronic heart failure: effects on functional capacity, quality of life, and clinical outcome. Circulation. 1999;99:1173–1182. doi: 10.1161/01.cir.99.9.1173. [DOI] [PubMed] [Google Scholar]
- 73.Georgiou D, Chen Y, Appadoo S, Belardinelli R, Greene R, Parides MK, Glied S. Cost-effectiveness analysis of long-term moderate exercise training in chronic heart failure. Am J Cardiol. 2001;87:984–988. A984. doi: 10.1016/s0002-9149(01)01434-5. [DOI] [PubMed] [Google Scholar]
- 74.Belardinelli R, Paolini I, Cianci G, Piva R, Georgiou D, Purcaro A. Exercise training intervention after coronary angioplasty: the ETICA trial. J Am Coll Cardiol. 2001;37:1891–1900. doi: 10.1016/s0735-1097(01)01236-0. [DOI] [PubMed] [Google Scholar]
- 75.Kitzman DW, Brubaker PH, Morgan TM, Stewart KP, Little WC. Exercise training in older patients with heart failure and preserved ejection fraction: a randomized, controlled, single-blind trial. Circ Heart Fail. 2010;3:659–667. doi: 10.1161/CIRCHEARTFAILURE.110.958785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brubaker PH, Moore JB, Stewart KP, Wesley DJ, Kitzman DW. Endurance exercise training in older patients with heart failure: results from a randomized, controlled, single-blind trial. J Am Geriatr Soc. 2009;57:1982–1989. doi: 10.1111/j.1532-5415.2009.02499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mousa TM, Liu D, Cornish KG, Zucker IH. Exercise training enhances baroreflex sensitivity by an angiotensin II-dependent mechanism in chronic heart failure. J Appl Physiol. 2008;104:616–624. doi: 10.1152/japplphysiol.00601.2007. [DOI] [PubMed] [Google Scholar]
- 78.Liu JL, Kulakofsky J, Zucker IH. Exercise training enhances baroreflex control of heart rate by a vagal mechanism in rabbits with heart failure. J Appl Physiol. 2002;92:2403–2408. doi: 10.1152/japplphysiol.00039.2002. [DOI] [PubMed] [Google Scholar]
- 79.Haack KK, Engler CW, Papoutsi E, Pipinos II, Patel KP, Zucker IH. Parallel changes in neuronal AT1R and GRK5 expression following exercise training in heart failure. Hypertension. 2012;60:354–361. doi: 10.1161/HYPERTENSIONAHA.112.195693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roveda F, Middlekauff HR, Rondon MU, Reis SF, Souza M, Nastari L, Barretto AC, Krieger EM, Negrao CE. The effects of exercise training on sympathetic neural activation in advanced heart failure: a randomized controlled trial. J Am Coll Cardiol. 2003;42:854–860. doi: 10.1016/s0735-1097(03)00831-3. [DOI] [PubMed] [Google Scholar]
- 81.Antunes-Correa LM, Melo RC, Nobre TS, Ueno LM, Franco FG, Braga AM, Rondon MU, Brum PC, Barretto AC, Middlekauff HR, Negrao CE. Impact of gender on benefits of exercise training on sympathetic nerve activity and muscle blood flow in heart failure. Eur J Heart Fail. 2010;12:58–65. doi: 10.1093/eurjhf/hfp168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Mello Franco FG, Santos AC, Rondon MU, Trombetta IC, Strunz C, Braga AM, Middlekauff H, Negrao CE, Pereira Barretto AC. Effects of home-based exercise training on neurovascular control in patients with heart failure. Eur J Heart Fail. 2006;8:851–855. doi: 10.1016/j.ejheart.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 83.Fraga R, Franco FG, Roveda F, de Matos LN, Braga AM, Rondon MU, Rotta DR, Brum PC, Barretto AC, Middlekauff HR, Negrao CE. Exercise training reduces sympathetic nerve activity in heart failure patients treated with carvedilol. Eur J Heart Fail. 2007;9:630–636. doi: 10.1016/j.ejheart.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 84.Zheng H, Sharma NM, Liu X, Patel KP. Exercise training normalizes enhanced sympathetic activation from the paraventricular nucleus in chronic heart failure: role of angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2012;303:R387–394. doi: 10.1152/ajpregu.00046.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bertagnolli M, Schenkel PC, Campos C, Mostarda CT, Casarini DE, Bello-Klein A, Irigoyen MC, Rigatto K. Exercise training reduces sympathetic modulation on cardiovascular system and cardiac oxidative stress in spontaneously hypertensive rats. Am J Hypertens. 2008;21:1188–1193. doi: 10.1038/ajh.2008.270. [DOI] [PubMed] [Google Scholar]
- 86.Gao L, Wang W, Liu D, Zucker IH. Exercise training normalizes sympathetic outflow by central antioxidant mechanisms in rabbits with pacing-induced chronic heart failure. Circulation. 2007;115:3095–3102. doi: 10.1161/CIRCULATIONAHA.106.677989. [DOI] [PubMed] [Google Scholar]
- 87.Kishi T, Hirooka Y, Katsuki M, Ogawa K, Shinohara K, Isegawa K, Sunagawa K. Exercise training causes sympathoinhibition through antioxidant effect in the rostral ventrolateral medulla of hypertensive rats. Clin Exp Hypertens. 2012;34:278–283. doi: 10.3109/10641963.2012.681084. [DOI] [PubMed] [Google Scholar]
- 88.Wang Y, Golledge J. Neuronal nitric oxide synthase and sympathetic nerve activity in neurovascular and metabolic systems. Curr Neurovasc Res. 2013;10:81–89. doi: 10.2174/156720213804805963. [DOI] [PubMed] [Google Scholar]
- 89.Coats AJ, Adamopoulos S, Radaelli A, McCance A, Meyer TE, Bernardi L, Solda PL, Davey P, Ormerod O, Forfar C, et al. Controlled trial of physical training in chronic heart failure. Exercise performance, hemodynamics, ventilation, and autonomic function. Circulation. 1992;85:2119–2131. doi: 10.1161/01.cir.85.6.2119. [DOI] [PubMed] [Google Scholar]
- 90.Kar S, Gao L, Zucker IH. Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing-induced heart failure. J Appl Physiol. 2010;108:923–932. doi: 10.1152/japplphysiol.00840.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Evangelista FS, Brum PC, Krieger JE. Duration-controlled swimming exercise training induces cardiac hypertrophy in mice. Braz J Med Biol Res. 2003;36:1751–1759. doi: 10.1590/s0100-879x2003001200018. [DOI] [PubMed] [Google Scholar]
- 92.Fernandes T, Hashimoto NY, Magalhaes FC, Fernandes FB, Casarini DE, Carmona AK, Krieger JE, Phillips MI, Oliveira EM. Aerobic exercise training-induced left ventricular hypertrophy involves regulatory MicroRNAs, decreased angiotensin-converting enzyme-angiotensin ii, and synergistic regulation of angiotensin-converting enzyme 2-angiotensin (1-7). Hypertension. 2011;58:182–189. doi: 10.1161/HYPERTENSIONAHA.110.168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang RF, Yin JX, Li YL, Zimmerman MC, Schultz HD. Angiotensin-(1-7) increases neuronal potassium current via a nitric oxide-dependent mechanism. Am J Physiol Cell Physiol. 2011;300:C58–64. doi: 10.1152/ajpcell.00369.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xiao L, Haack KK, Zucker IH. Angiotensin II regulates ACE and ACE2 in neurons through p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling. Am J Physiol Cell Physiol. 2013;304:C1073–1079. doi: 10.1152/ajpcell.00364.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burrell LM, Burchill L, Dean RG, Griggs K, Patel SK, Velkoska E. Chronic kidney disease: cardiac and renal angiotensin-converting enzyme (ACE) 2 expression in rats after subtotal nephrectomy and the effect of ACE inhibition. Exp Physiol. 97:477–485. doi: 10.1113/expphysiol.2011.063156. [DOI] [PubMed] [Google Scholar]
- 96.Sriramula S, Cardinale JP, Francis J. Inhibition of TNF in the brain reverses alterations in RAS components and attenuates angiotensin II-induced hypertension. PLoS One. 2013;8:e63847. doi: 10.1371/journal.pone.0063847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wakahara S, Konoshita T, Mizuno S, Motomura M, Aoyama C, Makino Y, Kato N, Koni I, Miyamori I. Synergistic expression of angiotensin-converting enzyme (ACE) and ACE2 in human renal tissue and confounding effects of hypertension on the ACE to ACE2 ratio. Endocrinology. 2007;148:2453–2457. doi: 10.1210/en.2006-1287. [DOI] [PubMed] [Google Scholar]
- 98.Zhang R, Wu Y, Zhao M, Liu C, Zhou L, Shen S, Liao S, Yang K, Li Q, Wan H. Role of HIF-1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L631–640. doi: 10.1152/ajplung.90415.2008. [DOI] [PubMed] [Google Scholar]
- 99.Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 2007;112:417–428. doi: 10.1042/CS20060342. [DOI] [PubMed] [Google Scholar]
- 100.Oppermann M, Freedman NJ, Alexander RW, Lefkowitz RJ. Phosphorylation of the type 1A angiotensin II receptor by G protein-coupled receptor kinases and protein kinase C. J Biol Chem. 1996;271:13266–13272. doi: 10.1074/jbc.271.22.13266. [DOI] [PubMed] [Google Scholar]
- 101.Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, Lefkowitz RJ. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci U S A. 2005;102:1442–1447. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grobe JL, Xu D, Sigmund CD. An intracellular reninangiotensin system in neurons: fact, hypothesis, or fantasy. Physiology (Bethesda) 2008;23:187–193. doi: 10.1152/physiol.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mueller PJ. Exercise training and sympathetic nervous system activity: evidence for physical activity dependent neural plasticity. Clin Exp Pharmacol Physiol. 2007;34:377–384. doi: 10.1111/j.1440-1681.2007.04590.x. [DOI] [PubMed] [Google Scholar]