

Abstract
UDP-glucuronosyltransferases (UGTs) are phase II drug-metabolizing enzymes that catalyze glucuronidation of various drugs. Although experimental rodents are used in preclinical studies to predict glucuronidation and toxicity of drugs in humans, species differences in glucuronidation and drug-induced toxicity have been reported. Humanized UGT1 mice in which the original Ugt1 locus was disrupted and replaced with the human UGT1 locus (hUGT1 mice) were recently developed. In this study, acyl-glucuronidations of etodolac, diclofenac, and ibuprofen in liver microsomes of hUGT1 mice were examined and compared with those of humans and regular mice. The kinetics of etodolac, diclofenac, and ibuprofen acyl-glucuronidation in hUGT1 mice were almost comparable to those in humans, rather than in mice. We further investigated the hepatotoxicity of ibuprofen in hUGT1 mice and regular mice by measuring serum alanine amino transferase (ALT) levels. Because ALT levels were increased at 6 hours after dosing in hUGT1 mice and at 24 hours after dosing in regular mice, the onset pattern of ibuprofen-induced liver toxicity in hUGT1 mice was different from that in regular mice. These data suggest that hUGT1 mice can be valuable tools for understanding glucuronidations of drugs and drug-induced toxicity in humans.
Introduction
UDP-glucuronosyltransferases (UGTs) that catalyze glucuronidation of compounds by transferring glucuronic acid from a cosubstrate (UDP-glucuronic acid) to substrates are important in phase II metabolism reactions (Dutton, 1980). UGTs are superfamily enzymes that are mainly classified into two subfamilies (UGT1 and UGT2) on the basis of evolutionary divergence (Mackenzie et al., 2005). The human UGT1 gene is located on chromosome 2q37 and encodes multiple unique exons 1 and common exons 2–5, producing nine functional UGT1A isoforms (UGT1A1 and UGT1A3–UGT1A10) (Ritter et al., 1992). The UGT2B genes are located on chromosome 4q13, and seven functional UGT2B isoforms are encoded by individual genes (Mackenzie et al., 2005). Each UGT enzyme expresses in a tissue-specific manner and exhibits substrate specificity (Tukey and Strassburg, 2000). UGT1 family enzymes are responsible for more than 50% of the glucuronidation of most prescribed drugs (Williams et al., 2004).
The UGT gene is conserved in mammals such as humans, mice, and rats (Mackenzie et al., 2005). Therefore, to predict glucuronidation of drugs in humans, preclinical studies use not only in vitro systems such as human UGTs-expressing cells (Katoh et al., 2007) but also experimental rodents (Deguchi et al., 2011). Although most drugs are similarly glucuronidated in humans and rodents, species differences have been reported for some drugs (Magdalou et al., 1990; Brocks and Jamali, 1991; King et al., 2001; Tougou et al., 2004). Naproxen, diclofenac, and ibuprofen are glucuronidated differently because the Km values and Vmax values are different in liver microsomes of human and rodents.
Adverse drug reactions (ADRs) are a major complication of drug therapy (Lazarou et al., 1998; Pirmohamed et al., 1998). ADRs may preclude effective drug therapy. If ADRs are quite serious, they may occasionally lead to drug withdrawal (Jefferys et al., 1998). For instance, zomepirac was withdrawn on March 4, 1983, relatively soon after its November 1980 market release, due to unexplained fatal anaphylactic reactions (Ross-Degnan et al., 1993). Almost all body systems may be adversely affected by drugs, but the most common serious reactions are those that involve the liver, skin, hemopoietic system, and more generalized toxicities such as systemic anaphylaxis (Park et al., 2001). Nonsteroidal anti-inflammatory drugs (NSAIDs) are well known as drugs that induce ADRs. NSAIDs that have a carboxylic group can be glucuronidated by multiple UGT isoforms (Table 1). It has been postulated that acyl-glucuronides, which are generated by glucuronidation of the carboxylic group of NSAIDs, can bind to proteins and cause idiosyncratic drug toxicity (Pumford et al., 1993), although the conclusive evidence that acyl-glucuronides cause toxicities has not been obtained. In fact, the most serious ADRs of NSAIDs usually occur in the liver and kidney, organs associated with high exposure to acyl-glucuronides. To date, both direct toxicity and/or immune-mediated indirect toxicity of acyl-glucuronides have been suggested as possible mechanisms of idiosyncratic liver injury. With direct toxicity, covalent protein binding via acyl-glucuronide may disrupt the normal physiologic function of a critical protein or some critical regulatory pathway that leads to cellular necrosis (Pirmohamed et al., 1996). Inflammation-mediated toxicity was investigated in a recent study, which showed that acyl-glucuronides induce inflammatory toxicity and cytotoxicity against CD14+ cells via the p38 mitogen-activated protein kinase pathway (Miyashita et al., 2014). In the case of diclofenac, it was previously demonstrated that the acyl-glucuronide can either bind directly to protein with displacement of the glucuronide group, or can rearrange to form a reactive imine intermediate that binds to proteins (Kretz-Rommel and Boelsterli, 1994). It is assumed that these chemical pathways of activation initiate either direct or immune-mediated indirect hepatotoxicity (Spahn-Langguth and Benet, 1992).
TABLE 1.
The recombinant UGT isoforms responsible for acyl-glucuronidations of NSAIDs in humans
| NSAID | Concentration | Glucuronidation Activity of Recombinant UGT Isoforms | Reference |
|---|---|---|---|
| μM | |||
| Diclofenac | 500 | 2B7 > 1A3 > 2B17 > 1A9 > 2B15 > 1A6 | Kuehl et al., 2005 |
| Edotolac | 50 | 1A9 > 1A10 ≥ 2B7 | Tougou et al., 2004 |
| Ibuprofen | 500 | 2B7 > 1A3 > 1A9 > 2B4 | Kuehl et al., 2005 |
| Ketoprofen | 500 | 2B7 > 2B4 > 1A3 > 1A9 | Kuehl et al., 2005 |
| Indomethacin | 500 | 1A9 > 2B7 > 1A3 > 1A1 | Kuehl et al., 2005 |
| Naproxen | 500 | 2B7 > 1A3 > 1A9 > 2B4 | Kuehl et al., 2005 |
Species differences in toxicity based on metabolism were previously reported (O'Brien et al., 1983; Eberhart et al., 1991). To predict and avoid human-specific drug- and metabolite-induced toxicities based on metabolism, the species difference in glucuronidation, especially of carboxylic acid drugs, needs to be carefully evaluated in preclinical studies. To overcome the species difference in drug glucuronidation, humanized UGT1 mice in which the original Ugt1 locus was disrupted and replaced with the human UGT1 locus (hUGT1 mice) were recently developed (Fujiwara et al., 2010, 2012). In this study, glucuronidations of etodolac, naproxen, and ibuprofen in hUGT1 mice were examined and compared with those in human and regular mice. Furthermore, we investigated the hepatotoxicity of ibuprofen in hUGT1 mice and regular mice.
Materials and Methods
Chemicals and Reagents.
UDP-glucuronic acid (UDPGA) and alamethicin were purchased from Sigma-Aldrich (St. Louis, MO). Etodolac and diclofenac sodium were purchased from Wako Pure Chemical (Osaka, Japan). S-Ibuprofen was purchased from LKT Laboratories (Saint Paul, MN). Diclofenac acyl-glucuronide, ibuprofen acyl-glucuronide, R-ibuprofen, and racemic (rac) ibuprofen were purchased from Toronto Research Chemicals (Toronto, ON, Canada). Human and mouse liver microsomes were obtained from BD Gentest (Woburn, MA). All other chemicals and solvents were of analytical grade or the highest grade commercially available.
Animals and Preparation of Liver Microsomes.
Tg(UGT1A1*28)Ugt1−/− (hUGT1) mice were developed previously in a C57BL/6 background (Fujiwara et al., 2010). All animals received food and water ad libitum, and mouse handling and experimental procedures were conducted in accordance with the animal care protocol approved by Kitasato University (Tokyo, Japan). For tissue collections, mice were anesthetized by diethyl ether inhalation, and the liver was perfused with ice-cold 1.15% KCl. The liver was rinsed in cold 1.15% KCl and stored at −80°C. Liver microsomes were prepared using the following procedure. Perfused liver with 1.15% KCl was homogenized in three volumes of homogenization buffer (1.15% KCl/10 mM potassium phosphate buffer, pH 7.4). The homogenate was centrifuged at 10,000 × g for 30 minutes at 4°C, and the supernatant was collected. The supernatant was centrifuged at 105,000 × g for 60 minutes at 4°C, and the pellet was suspended in the same buffer and used as the microsomal fraction. Protein concentrations of microsomal fractions were measured by the Bradford (1976) method using bovine serum albumin.
Enzyme Assays.
Etodolac and diclofenac acyl-glucuronide formation was determined according to the method of Fujiwara et al. (2007) with slight modifications. Briefly, a typical incubation mixture (200 μl of total volume) contained 100 mM phosphate buffer (pH 7.4), 4 mM MgCl2, 5 mM UDPGA, 50 μg/ml alamethicin, 0.1 mg/ml liver microsomes, and 50 μM to 4 mM etodolac or 10 µM to 1 mM diclofenac. The reaction was initiated by the addition of UDPGA after a 3-minute preincubation at 37°C. After incubation at 37°C for 30 minutes, the reaction was terminated by the addition of 200 μl cold acetonitrile. After removal of the protein by centrifugation at 12,000 × g for 5 minutes, a 50-μl portion of the sample was subjected to high-performance liquid chromatography (HPLC).
S-Ibuprofen, R-ibuprofen, and rac-ibuprofen acyl-glucuronide formation was determined according to the method of Fujiwara et al. (2007) with slight modifications. Briefly, a typical incubation mixture (200 μl of total volume) contained 100 mM phosphate buffer (pH 7.4), 4 mM MgCl2, 5 mM UDPGA, 50 μg/ml alamethicin, 0.2 mg/ml, and 50 μM to 3 mM S-ibuprofen, 50 μM to 3 mM R-ibuprofen, or 25 μM to 2 mM rac-ibuprofen. The reaction was initiated by the addition of UDPGA after a 3-minute preincubation at 37°C. After incubation at 37°C for 60 minutes, the reaction was terminated by the addition of 200 μl cold methanol. After removal of the protein by centrifugation at 12,000 × g for 5 minutes, a 50-μl portion of the sample was subjected to HPLC.
HPLC Conditions.
Glucuronides were determined by the HPLC system with a LC-10AD pump (Shimadzu, Kyoto, Japan), a FP-2020 fluorescence detector (JASCO, Tokyo Japan), a SIL-10A autosampler (Shimadzu), a SLC-10A system controller (Shimadzu), and a Mightysil RP-18 GP column (4.6 × 150 mm, 5 μm; Kanto Chemical, Tokyo, Japan). The mobile phases were 50% acetonitrile containing 1% aqueous acetic acid for the etodolac glucuronide, 65% methanol containing 0.05 M KH2PO4 for diclofenac glucuronide, and 65% methanol containing pH 3.0 phosphoric acid aqueous solution for ibuprofen glucuronides. The flow rate was 1.0 ml/min. Glucuronides were detected with a fluorescence detector at 276 nm excitation and 667 nm emission for the etodolac glucuronide, at 282 nm excitation and 365 nm emission for the diclofenac glucuronide, and at 263 nm excitation and 288 nm emission for the ibuprofen glucuronides. Quantification of diclofenac and ibuprofen glucuronides was carried out by comparing the HPLC peak area with that of the authentic standard. Quantification of etodolac glucuronides was conducted by the following procedure. The incubation mixture including 5 µM etodolac and 1 mg/ml human liver microsomes, 100 mM phosphate buffer (pH 7.4), 4 mM MgCl2, 5 mM UDPGA, and 50 μg/ml alamethicin was incubated at 37°C for 0 hours and 3 hours. The increase of HPLC peak area of the etodolac glucuronide was compared with the decrease of the peak area of the parent compound. As the relationship between the peak area of etodolac glucuronide and the amount of etodolac glucuronide was determined, the etodolac glucuronide in the reaction mixture was quantified by measuring the peak area of glucuronide. The retention times of etodolac glucuronide, diclofenac glucuronide, and ibuprofen glucuronides were 4.0, 6.2, and 5.9 minutes, respectively.
Data Analysis.
When kinetics of drug metabolism follows a simple Michaelis–Menten equation, the relationship between substrate concentration and velocity can be described by the following equation (eq. 1):
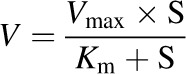 |
(1) |
where V is the initial velocity of the metabolic reaction, S is the substrate concentration, Vmax is the maximum rate of metabolism, and Km is the Michaelis constant, which is defined as the substrate concentration at one-half the maximum velocity. Although the clearance is substrate concentration dependent, the rate is constant when the substrate concentration is much smaller than Km, providing the intrinsic clearance (CLint) parameter (eq. 2):
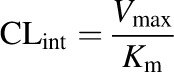 |
(2) |
When substrate inhibition was observed, the data were analyzed by eq. 3:
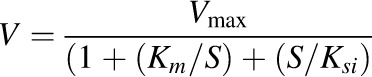 |
(3) |
where Ksi is the constant describing the substrate inhibition interaction.
For sigmoidal kinetics, kinetic parameters were obtained by the Hill equation (eq. 4):
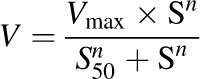 |
(4) |
where S50 is the substrate concentration showing the one-half Vmax and n is the Hill coefficient.
Kinetic data were also analyzed using Eadie–Hofstee plots. Furthermore, goodness of fit to kinetic models was assessed by calculation of the Akaike information criterion (AIC) values. AIC values of each model were compared and the best-fit model was determined.
Ibuprofen-Induced Liver Injury.
Rac-Ibuprofen suspended in canola oil containing 10% dimethylsulfoxide was administered to hUGT1 mice and regular mice (750 mg/kg, p.o.). Canola oil containing 10% dimethylsulfoxide without rac-ibuprofen was also administered to hUGT1 mice for a control. Blood was obtained from the submandibular vein immediately before dosing and at 6 hours and 24 hours after the dosing. Blood was centrifuged at 9200 × g for 10 minutes after incubation at 37°C for 10 minutes to facilitate the clotting reaction. The supernatant was collected to obtain serum. Liver damage was assessed by measuring the serum activity of alanine amino transaminase (ALT), which is a sensitive diagnostic indicator of hepatotoxicity (Ozer et al., 2008), using the IDTox ALT Color Endpoint Assay Kit (ID Laboratories Inc., London, ON, Canada).
Pharmacokinetics Study of Ibuprofen.
Rac-ibuprofen suspended in canola oil was administered to hUGT1 mice and regular mice (200 mg/kg, p.o.). Blood was collected from the submandibular vein into heparinized tubes at 15, 30, 60, 120, 240, and 480 minutes after the administration and immediately centrifuged at 9200 × g for 10 minutes to obtain plasma. For removal of protein, plasma was mixed with 2-fold volume of acetonitrile and centrifuged at 12,000 × g for 5 minutes and the supernatant was filtered through a 0.45-μm cellulose acetate membrane filter (Cosmonice Filter S; Nakalai Tesque, Kyoto, Japan). The plasma concentrations of ibuprofen and ibuprofen acyl-glucuronides were analyzed by HPLC as described above. The peak plasma concentration (Cmax) and the time to peak concentration (Tmax) were obtained from experimental observation. The area under the plasma concentration-time curve from 0 hours to 8 hours (AUC0–8 h) was calculated using the trapezoidal integration without extrapolation to infinity.
Statistical Analysis.
All data were presented as means ± S.D. The t test was used to analyze data from the pharmacokinetics study of ibuprofen. Analysis of variance and Dunnett’s procedure for multiple comparisons were used to analyze data on ibuprofen-induced liver injury. P < 0.05 was considered significant.
Results
Etodolac Glucuronidation in Liver Microsomes from hUGT1 Mice, Humans, and Regular Mice.
Etodolac is one of the drugs that are subject to species-different glucuronidation because the Km values and Vmax values are different between liver microsomes of humans and rats (Brocks and Jamali, 1991; Tougou et al., 2004). To examine whether etodolac glucuronidation in hUGT1 mice is similar to that in humans, liver microsomes were prepared from adult hUGT1 mice and etodolac glucuronidation was determined. The etodolac acyl-glucuronide formation by the liver microsomes from hUGT1 mice followed a simple Michaelis–Menten equation (Fig. 1A) because Eadie–Hofstee plots were linear (Supplemental Fig. 1A). Obtained parameter values were as follows: Km = 483 μM, Vmax = 379 pmol/min per mg, and CLint = 0.79 μl/min per mg (Table 2). In human liver microsomes, Eadie–Hofstee plots were almost liner (Supplemental Fig. 1A). AIC values for a simple Michaelis–Menten equation and the Hill equation were compared and the Michaelis–Menten equation gave smaller AIC values (Fig. 1A). The kinetic parameters of etodolac acyl-glucuronide in human liver microsomes were as follows: Km = 483 μM, Vmax = 246 pmol/min per mg, and CLint = 0.51 μl/min per mg (Table 2). The etodolac acyl-glucuronide formation by the mouse liver microsomes followed a simple Michaelis–Menten equation because Eadie–Hofstee plots were liner (Fig. 1A). Obtained parameter values were as follows: Km = 1170 μM, Vmax = 201 pmol/min per mg, and CLint = 0.17 μl/min per mg (Table 2). These data showed that the kinetic parameters obtained in liver microsomes from hUGT1 mice were closer to those in human liver microsomes than those in mice liver microsomes.
Fig. 1.

Kinetic analyses of etodolac, diclofenac, rac-ibuprofen, S-ibuprofen, and R-ibuprofen acyl-glucuronide formations in liver microsomes. The substrate concentration-velocity curves of the etodolac (A), diclofenac (B), rac-ibuprofen (C), S-ibuprofen (D), and R-ibuprofen (E) glucuronide formations are shown. Pooled liver microsomes of hUGT1 mice, humans, and regular mice were incubated with each substrate and 5 mM UDP-glucuronic acid at 37°C for 30 minutes. Data are the means ± S.D. of three independent determinations.
TABLE 2.
Kinetic parameters of diclofenac, etodolac, and ibuprofen glucuronidation in liver microsomes
| Compound | Liver Microsome | Kinetic Parameter |
|||
|---|---|---|---|---|---|
|
Km |
Vmax |
Ki |
CLint |
||
| μM | nmol/min per mg | mM | µl/min per mg | ||
| Etodolac | Humanized UGT1 micea | 483 ± 40 | 0.379 ± 0.003 | ― | 0.79 ± 0.06 |
| Humansa | 483 ± 50 | 0.246 ± 0.012 | ― | 0.51 ± 0.03 | |
| Micea | 1170 ± 287 | 0.201 ± 0.029 | ― | 0.17 ± 0.01 | |
| Diclofenac | Humanized UGT1 micea | 31.8 ± 1.91 | 7.05 ± 0.04 | ― | 222 ± 12.2 |
| Humansb | 33.1 ± 1.70 | 10.4 ± 0.28 | 22.5 ± 1.00 | 313 ± 8.88 | |
| Micea | 55.6 ± 1.52 | 11.8 ± 0.10 | ― | 211 ± 5.62 | |
| rac-Ibuprofen | Humanized UGT1 micea | 319 ± 26 | 4.27 ± 0.12 | ― | 13.4 ± 0.71 |
| Humansa | 651 ± 8.1 | 6.44 ± 0.05 | ― | 9.9 ± 0.05 | |
| Micea | 364 ± 12 | 11.2 ± 0.19 | ― | 30.7 ± 0.54 | |
| S-Ibuprofen | Humanized UGT1 micea | 381 ± 26 | 7.54 ± 0.16 | ― | 19.9 ± 1.00 |
| Humansa | 699 ± 34 | 8.71 ± 0.17 | ― | 12.5 ± 0.38 | |
| Micea | 364 ± 5.0 | 16.5 ± 0.19 | ― | 45.1 ± 0.50 | |
| R-Ibuprofen | Humanized UGT1 micea | 320 ± 38 | 2.18 ± 0.07 | ― | 6.87 ± 0.56 |
| Humansa | 428 ± 11 | 3.37 ± 0.03 | ― | 7.86 ± 0.13 | |
| Micea | 424 ± 19 | 8.00 ± 0.09 | ― | 18.9 ± 0.70 | |
Calculated using the Michaelis–Menten equation.
Calculated using substrate inhibition.
Diclofenac Glucuronidation in Liver Microsomes from hUGT1 Mice, Humans, and Regular Mice.
Previous reports indicate that diclofenac is glucuronidated differently in humans and rodents because the Km values and Vmax are different between liver microsomes of humans and rats (King et al., 2001). Diclofenac glucuronidation in hUGT1 mice, humans, and mice liver microsomes was determined and the kinetic data were analyzed. Although the diclofenac acyl-glucuronide formations by the liver microsomes from hUGT1 mice and regular mice followed a simple Michaelis–Menten equation (Fig. 1B) because Eadie–Hofstee plots were linear (Supplemental Fig. 1B), those by liver microsomes from humans followed the Michaelis–Menten equation with substrate inhibition. In human liver microsomes, the Eadie–Hofstee plots were not linear at the higher substrate concentration (Supplemental Fig. 1B). In the human liver microsomes, the AIC values for a simple Michaelis–Menten equation and a Michaelis–Menten equation with a substrate inhibition were compared and the Michaelis–Menten with substrate inhibition model gave a smaller AIC value. The kinetic parameters were as follows: Km = 31.8 μM, Vmax = 7.05 nmol/min per mg, and CLint = 222 μl/min per mg in hUGT1 mice liver microsomes; Km = 55.6 μM, Vmax = 11.8 nmol/min per mg, and CLint = 211 μl/min per mg in mouse liver microsomes; and Km = 33.1 μM, Vmax = 10.4 nmol/min per mg, Ki = 22.5 mM, and CLint = 313 μl/min per mg in human liver microsomes (Table 2). Although the Vmax and CLint values in hUGT1 mice were not very close to those in humans compared with in regular mice, the Km value in hUGT1 mice was closer to that in humans than those in regular mice.
Rac-Ibuprofen Glucuronidation in Liver Microsomes from hUGT1 Mice, Humans, and Regular Mice.
Ibuprofen possesses a chiral center in the propionic acid moiety. The pharmacological activity resides mainly in the S-ibuprofen. Because much of R-ibuprofen is converted to the active S-form (Hutt and Caldwell, 1983), the racemate has clinically been used. A previous report indicated that ibuprofen was glucuronidated differently in humans and rodents because the Km values and Vmax were different between liver microsomes of humans, rats, and mice (Magdalou et al., 1990). Rac-Ibuprofen glucuronide formation in hUGT1 mice, humans, and mouse liver microsomes was determined and the kinetic data were analyzed. Rac-Ibuprofen glucuronidation in liver microsomes of hUGT1 mice, humans, and regular mice best fit to a simple Michaelis–Menten equation (Fig. 1C) because the Eadie–Hofstee plots were linear (Supplemental Fig. 1C). The kinetic parameters were as follows: Km = 319 μM, Vmax = 4.27 nmol/min per mg, and CLint = 13.4 μl/min per mg in hUGT1 mice liver microsomes; Km = 651 μM, Vmax = 6.44 nmol/min per mg, and CLint = 9.9 μl/min per mg in human liver microsomes; and Km = 364 μM, Vmax = 11.2 nmol/min per mg, and CLint = 30.7 μl/min per mg in regular mice liver microsomes (Table 2). In racemate glucuronidations, Vmax and intrinsic clearance in liver microsomes from hUGT1 mice were similar to those in human liver microsomes than those in mouse liver microsomes. The intrinsic clearance in hUGT1 mice and humans was similar and approximately 2- and 3- fold lower than that in regular mice.
S-Ibuprofen Glucuronidation in Liver Microsomes from hUGT1 Mice, Humans, and Regular Mice.
S-Ibuprofen glucuronide formation in hUGT1 mice, humans, and mouse liver microsomes was determined and the kinetic data were analyzed. S-Ibuprofen glucuronidation in liver microsomes of hUGT1 mice, humans, and regular mice best fi to a simple Michaelis–Menten equation (Fig. 1D) because the Eadie–Hofstee plots were linear (Supplemental Fig. 1D). The kinetic parameters were as follows: Km = 381 μM, Vmax = 7.54 nmol/min per mg, and CLint = 19.9 μl/min per mg in hUGT1 mice liver microsomes; Km = 699 μM, Vmax = 8.71 nmol/min per mg, and CLint = 12.5 μl/min per mg in human liver microsomes; and Km = 364 μM, Vmax = 16.5 nmol/min per mg, and CLint = 45.1 μl/min per mg in mouse liver microsomes (Table 2). In S-ibuprofen glucuronidations, Vmax values and intrinsic clearance in liver microsomes from hUGT1 mice were similar to those in human liver microsomes than those in mouse liver microsomes, whereas Km values in hUGT1 mice were not similar to that in humans.
R-Ibuprofen Glucuronidation in Liver Microsomes from hUGT1 Mice, Humans, and Regular Mice.
R-Ibuprofen glucuronide formation in hUGT1 mice, humans, and mouse liver microsomes was determined and the kinetic data were analyzed. R-Ibuprofen glucuronidation in liver microsomes of hUGT1 mice, humans, and regular mice best fit to a simple Michaelis–Menten equation (Fig. 1E) because the Eadie–Hofstee plots were linear (Supplemental Fig. 1E). The kinetic parameters were as follows: Km = 320 μM, Vmax = 2.18 nmol/min per mg, and CLint = 6.87 μl/min per mg in hUGT1 mice liver microsomes; Km = 428 μM, Vmax = 3.37 nmol/min per mg, and CLint = 7.86 μl/min per mg in human liver microsomes; and Km = 424 μM, Vmax = 8.00 nmol/min per mg, and CLint = 18.9 μl/min per mg in mouse liver microsomes (Table 2). In R-ibuprofen glucuronidations, Vmax values and intrinsic clearance in liver microsomes from hUGT1 mice were similar to those in human liver microsomes than those in mouse liver microsomes, whereas Km values in hUGT1 mice were not similar to that in humans.
Ibuprofen-Induced Liver Injury.
Ibuprofen is one of the most commonly used NSAIDs acting as a cyclooxygenase inhibitor and is most frequently administered orally as a racemic mixture. Although ibuprofen is considered to have a lower risk of liver injury, several cases of serious liver disease have been reported with ibuprofen (Rubenstein and Laine, 2004; Lapeyre-Mestre et al., 2006). In this study, rac-ibuprofen–induced liver injury was investigated in hUGT1 mice and regular mice to examine the differences in the onset pattern of toxicity between hUGT1 and regular mice. In the control group, there were no statistical differences in ALT levels before dosing and at 6 hours and 24 hours after dosing (data not shown). In hUGT1 mice, the serum ALT levels at 6 hours after 750 mg/kg rac-ibuprofen administration significantly increased by approximately 2- fold compared with those before dosing (P < 0.01). ALT levels at 24 hours after dosing slightly increased compared with those before dosing (Fig. 2A). By contrast, the ALT levels at 6 hours after rac-ibuprofen dosing slightly increased compared with those before dosing in regular mice. The ALT levels at 24 hours after dosing significantly increased by approximately 3-fold compared with those before dosing in regular mice (P < 0.01) (Fig. 2B). These results suggested that there were differences in the onset pattern of ibuprofen-induced liver injury between hUGT1 mice and regular mice.
Fig. 2.

Serum ALT activity in mice treated with rac-ibuprofen. ALT activities of hUGT1 mice (A) and those of regular mice (B) treated with 750 mg/kg rac-ibuprofen are shown. Blood was collected before dosing and 6 h and 24 h after oral dosing. The serum was analyzed for ALT activity, and the results are shown as means ± S.D. of the ALT activity (units per liter) (n ≥ 3). **P < 0.01, compared with the group before dosing.
Pharmacokinetics Study of Ibuprofen.
A pharmacokinetics study was conducted to investigate the concentration of rac-ibuprofen and its glucuronide in hUGT1 mice and wild-type mice. As shown in Fig. 3A, the plasma level of ibuprofen rapidly increased (Tmax = 30 minutes) in both hUGT1 mice and regular mice with Cmax values of 368 ± 177 μM and 270 ± 60.4 μM, respectively. Furthermore, the AUC0–8 h values of ibuprofen in hUGT1 mice was statistically higher than that of regular mice (P < 0.05), because the AUC0–8 h values of ibuprofen were 1285.3 ± 282.1 μM⋅h in hUGT1 mice and 721.1 ± 246.3 μM⋅h in regular mice. Whereas the plasma level of ibuprofen acyl-glucuronide rapidly increased in regular mice, the plasma level of ibuprofen acyl-glucuronides slowly increased in hUGT1 mice (Fig. 3B). The Tmax value of ibuprofen acyl-glucuronides was different between hUGT1 mice and regular mice because Tmax values were 120 minutes in hUGT1 mice and 30 minutes in regular mice, with Cmax values of 96.7 ± 17.7 μM and 114 ± 23.4 μM, respectively. AUC0–8 h values of ibuprofen acyl-glucuronides in hUGT1 mice (634.8 ± 115.4 μM⋅h) was statistically higher than that in regular mice (452.1 ± 39.6 μM⋅h). Although ibuprofen acyl-glucuronides were stable in protein-free plasma, they were slightly unstable in mouse blood (data not shown). Therefore, the plasma concentrations of ibuprofen acyl-glucuronides shown in Fig. 3 might be slightly lower than actual concentrations.
Fig. 3.

Pharmacokinetics of ibuprofen and its glucuronides in hUGT1 mice and regular mice. Pharmacokinetics of ibuprofen (A) and ibuprofen acyl-glucuronides (B) in regular mice (closed circles) and hUGT1 mice (open circles) treated with ibuprofen (200 mg/kg, p.o.) are shown. Results are shown as means ± S.D. of the plasma concentration of ibuprofen or ibuprofen acyl-glucuronides (in micromoles) (n ≥ 3).
Discussion
There are marked species differences in metabolism (Bogaards et al., 2000). This is especially notable between humans and rodents such as rats and mice, the most commonly used experimental models for studies in pharmacology. It is assumed that metabolic enzymes of humans and experimental animals could have differences in affinity to substrate, expression levels, and gene expression profiles. For example, because human UGT1A4 is functional but rat UGT1A4 and mouse Ugt1a4 are pseudogenes, specific probes of UGT1A4 such as trifluoperazine and imipramine are not glucuronidated in mice or rats (Uchaipichat et al., 2006; Shiratani et al., 2008). The species differences of metabolic enzymes are typically assessed by comparing kinetic parameters such as Km, Vmax, and CLint. In this study, the method of preparation of liver microsomes was different because the microsomes of humans and regular mice were purchased and microsomes of hUGT1 mice were prepared by our above-described method. The difference in the method of microsome preparation may influence glucuronidation activities. However, in past studies, species differences of glucuronidation activities have been assessed with microsomes prepared by different methods (Xu et al., 2006; Cai et al., 2010).
One approach to overcome the gap of species difference is to generate humanized mice by introducing a human gene into the mouse genome (Gonzalez, 2003; Henderson and Wolf, 2003). Thus, humanized mice offer a better animal model to predict the human drug metabolism and understand the underlying mechanisms. In our recent study, hUGT1 mice showed glucuronidation activity of furosemide, naproxen, imipramine, and trifluoperazine that was comparable to that of humans (Kutsuno et al., 2013). In this study, we investigated the glucuronidation activity of etodolac, diclofenac, and ibuprofen metabolized by UGTs. Because glucuronidations of a chiral compound can be enantioselective like the several other chiral drugs reported (Silber et al., 1982; Zhang et al., 2000), there are enantiomer-enantiomer interactions between one enantiomer and the other (el Mouelhi et al., 1987). In this study, kinetic parameters of S-ibuprofen, R-ibuprofen, and rac-ibuprofen were similar in microsomes of humans, hUGT1 mice, and regular mice (Table 2). Therefore, we believe that glucuronidation of ibuprofen is not enantioselective. Although the metabolic profile of the microsomal glucuronidation activity toward etodolac and ibuprofen in hUGT1 mice was similar to that in humans, considering the intrinsic clearance parameters (Table 2), diclofenac glucuronidations in hUGT1 mice were not very similar to those in humans (Table 2). Although etodolac, ibuprofen, and diclofenac were glucuronidated by multiple UGT isoforms (Table 1), the major isozyme involved in catalyzing the glucuronidation of diclofenac in humans was UGT2B7 because the Km value for diclofenac glucuronidation in human liver microsomes was similar to that in stably expressed human UGT2B7 (King et al., 2001). In humans, multiple UGT2B genes encode UGT2B4, UGT2B7, UGT2B10, UGT2B11, UGT2B15, UGT2B17, and UGT2B28 proteins. In mice, seven Ugt2b genes include Ugt2b1, Ugt2b5, Ugt2b34, Ugt2b35, Ugt2b36, Ugt2b37, and Ugt2b38. The species difference in the function of human UGT2B and mouse Ugt2b family enzymes has not been fully understood. However, it was previously reported that there are species differences in drug glucuronidations that are catalyzed by UGT2B7 (Cai et al., 2010). Therefore, the differences of diclofenac glucuronidations between hUGT1 mice and humans might be explained by the functional difference between human UGT2 families and mouse Ugt2 families.
Although rodents such as rats and mice are commonly used as experimental models for studies in toxicology, there are also species differences between rodents and humans in the toxicity based on metabolism of xenobiotics (O'Brien et al., 1983; Eberhart et al., 1991). The humanized mouse models are useful because they offer the knowledge to evaluate and predict the toxicological risk, which may aid in the development of safer drugs. For instance, humanized CYP2E1 mice were created to compare the functional differences in response to xenobiotics between human and mouse CYP2E1 (Cheung et al., 2005). Hepatotoxicity resulting from the CYP2E1-mediated activation of acetaminophen (APAP) was investigated in the humanized CYP2E1 mice. After 200 mg/kg APAP dosing, the levels of ALT were unchanged in humanized CYP2E1 mice, but were significantly elevated by approximately 12-fold in regular mice. These results showed that there was a difference in response to APAP between humanized CYP2E1 mice and regular mice (Cheung and Gonzalez, 2008). This study investigated ibuprofen-induced liver injury and pharmacokinetics of ibuprofen in hUGT1 and regular mice. Our results showed that there were differences in ibuprofen-induced liver injury between hUGT1 mice and regular mice (Fig. 2). In hUGT1 mice, rac-ibuprofen increased ALT levels 6 hours after dosing. Rac-Ibuprofen clearance in hUGT1 mice was much lower than that in regular mice (Table 2) and AUC0–8 h values of ibuprofen in hUGT1 mice were statistically higher than those in regular mice. These data suggest that hUGT1 mice, which had lower intrinsic clearance of ibuprofen, had higher exposure to ibuprofen than regular mice. By contrast, ALT levels were increased 24 hours after dosing in regular mice. Our data indicate that slower intrinsic clearance of ibuprofen in hUGT1 mice still resulted in the higher AUC0–8 h values of ibuprofen acyl-glucuronides. It is possible that hUGT1 mice have transporters that may be coordinately modulated and thus change exposure of the acyl-glucuronides. The underlying mechanism of different patterns of liver injury expression induced by ibuprofen would be very complicated due to the complexity of acyl-glucuronide disposition with enterohepatic recycling, systemic reversible metabolism, and renal clearance of the glucuronide. The differences in the pharmacokinetics of ibuprofen might be associated with the onset of the liver injury in hUGT1 mice and regular mice.
This study demonstrated that the glucuronidations of etodolac and ibuprofen in hUGT1 mice were almost comparable to those in humans. These findings indicate that in vitro and in vivo studies utilizing hUGT1 mice are useful for predicting in vivo human glucuronidation of drugs that are mainly catalyzed UGT1As. Furthermore, although only one compound, rac-ibuprofen, was investigated for the toxicity study, the onset pattern of ibuprofen-induced liver toxicity in hUGT1 mice was different from that in regular mice. Although further investigations need to be conducted, hUGT1 mice can also be valuable to understand drug-induced toxicity in humans.
Supplementary Material
Abbreviations
- ADR
adverse drug reaction
- AIC
Akaike information criterion
- ALT
alanine amino transferase
- APAP
acetaminophen
- AUC
area under the plasma concentration-time curve
- CLint
intrinsic clearance
- HPLC
high-performance liquid chromatography
- hUGT1
humanized UGT1
- NSAID
nonsteroidal anti-inflammatory drug
- UDPGA
UDP-glucuronic acid
- UGT
UDP-glucuronosyltransferase
Authorship Contributions
Participated in research design: Itoh, Tukey, Fujiwara.
Conducted experiments: Kutsuno, Fujiwara.
Performed data analysis: Kutsuno, Fujiwara.
Wrote or contributed to the writing of the manuscript: Kutsuno, Itoh, Tukey, Fujiwara.
Footnotes
This research was supported in part by the National Institutes of Health National Institute of Environmental Health Sciences [Grant P42-ES010337] and National Institute of General Medical Sciences [Grant R01-GM100481]; and the Mochida Memorial Foundation for Medical and Pharmaceutical Research.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Bogaards JJP, Bertrand M, Jackson P, Oudshoorn MJ, Weaver RJ, van Bladeren PJ, Walther B. (2000) Determining the best animal model for human cytochrome P450 activities: a comparison of mouse, rat, rabbit, dog, micropig, monkey and man. Xenobiotica 30:1131–1152 [DOI] [PubMed] [Google Scholar]
- Bradford MM. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254 [DOI] [PubMed] [Google Scholar]
- Brocks DR, Jamali F. (1991) Enantioselective pharmacokinetics of etodolac in the rat: tissue distribution, tissue binding, and in vitro metabolism. J Pharm Sci 80:1058–1061 [DOI] [PubMed] [Google Scholar]
- Cai H, Nguyen N, Peterkin V, Yang YS, Hotz K, La Placa DB, Chen S, Tukey RH, Stevens JC. (2010) A humanized UGT1 mouse model expressing the UGT1A1*28 allele for assessing drug clearance by UGT1A1-dependent glucuronidation. Drug Metab Dispos 38:879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C, Gonzalez FJ. (2008) Humanized mouse lines and their application for prediction of human drug metabolism and toxicological risk assessment. J Pharmacol Exp Ther 327:288–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C, Yu AM, Ward JM, Krausz KW, Akiyama TE, Feigenbaum L, Gonzalez FJ. (2005) The cyp2e1-humanized transgenic mouse: role of cyp2e1 in acetaminophen hepatotoxicity. Drug Metab Dispos 33:449–457 [DOI] [PubMed] [Google Scholar]
- Deguchi T, Watanabe N, Kurihara A, Igeta K, Ikenaga H, Fusegawa K, Suzuki N, Murata S, Hirouchi M, Furuta Y, et al. (2011) Human pharmacokinetic prediction of UDP-glucuronosyltransferase substrates with an animal scale-up approach. Drug Metab Dispos 39:820–829 [DOI] [PubMed] [Google Scholar]
- Dutton GJ. (1980) Acceptor substrates of UDP glucuronosyltransferase and their assay, in Glucuronidation of Drugs and Other Compounds (Dutton GJ. ed) pp 69–78, CRC Press, Boca Raton, FL [Google Scholar]
- Eberhart DC, Gemzik B, Halvorson MR, Parkinson A. (1991) Species differences in the toxicity and cytochrome P450 IIIA-dependent metabolism of digitoxin. Mol Pharmacol 40:859–867 [PubMed] [Google Scholar]
- el Mouelhi M, Ruelius HW, Fenselau C, Dulik DM. (1987) Species-dependent enantioselective glucuronidation of three 2-arylpropionic acids. Naproxen, ibuprofen, and benoxaprofen. Drug Metab Dispos 15:767–772 [PubMed] [Google Scholar]
- Fujiwara R, Chen S, Karin M, Tukey RH. (2012) Reduced expression of UGT1A1 in intestines of humanized UGT1 mice via inactivation of NF-κB leads to hyperbilirubinemia. Gastroenterology 142:109–118, e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara R, Nguyen N, Chen S, Tukey RH. (2010) Developmental hyperbilirubinemia and CNS toxicity in mice humanized with the UDP glucuronosyltransferase 1 (UGT1) locus. Proc Natl Acad Sci USA 107:5024–5029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara R, Nakajima M, Yamanaka H, Nakamura A, Katoh M, Ikushiro SI, Sakaki T, Yokoi T. (2007) Effects of coexpression of UGT1A9 on enzymatic activities of human UGT1A isoforms. Drug Metab Dispos 35:747–757 [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ. (2003) Role of gene knockout and transgenic mice in the study of xenobiotic metabolism. Drug Metab Rev 35:319–335 [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Wolf CR. (2003) Transgenic analysis of human drug-metabolizing enzymes: preclinical drug development and toxicology. Mol Interv 3:331–343 [DOI] [PubMed] [Google Scholar]
- Hutt AJ, Caldwell J. (1983) The metabolic chiral inversion of 2-arylpropionic acids—a novel route with pharmacological consequences. J Pharm Pharmacol 35:693–704 [DOI] [PubMed] [Google Scholar]
- Jefferys DB, Leakey D, Lewis JA, Payne S, Rawlins MD. (1998) New active substances authorized in the United Kingdom between 1972 and 1994. Br J Clin Pharmacol 45:151–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M, Matsui T, Yokoi T. (2007) Glucuronidation of antiallergic drug, Tranilast: identification of human UDP-glucuronosyltransferase isoforms and effect of its phase I metabolite. Drug Metab Dispos 35:583–589 [DOI] [PubMed] [Google Scholar]
- King C, Tang W, Ngui J, Tephly T, Braun M. (2001) Characterization of rat and human UDP-glucuronosyltransferases responsible for the in vitro glucuronidation of diclofenac. Toxicol Sci 61:49–53 [DOI] [PubMed] [Google Scholar]
- Kretz-Rommel A, Boelsterli UA. (1994) Mechanism of covalent adduct formation of diclofenac to rat hepatic microsomal proteins. Retention of the glucuronic acid moiety in the adduct. Drug Metab Dispos 22:956–961 [PubMed] [Google Scholar]
- Kuehl GE, Lampe JW, Potter JD, Bigler J. (2005) Glucuronidation of nonsteroidal anti-inflammatory drugs: identifying the enzymes responsible in human liver microsomes. Drug Metab Dispos 33:1027–1035 [DOI] [PubMed] [Google Scholar]
- Kutsuno Y, Sumida K, Itoh T, Tukey RH, Fujiwara R. (2013). Glucuronidation of drugs in humanized UDP‐glucuronosyltransferase 1 mice: Similarity with glucuronidation in human liver microsomes. Pharmacol Res Perspect 1:1–11, e00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapeyre-Mestre M, de Castro AMR, Bareille MP, Del Pozo JG, Requejo AA, Arias LM, Montastruc JL, Carvajal A. (2006) Non-steroidal anti-inflammatory drug-related hepatic damage in France and Spain: analysis from national spontaneous reporting systems. Fundam Clin Pharmacol 20:391–395 [DOI] [PubMed] [Google Scholar]
- Lazarou J, Pomeranz BH, Corey PN. (1998) Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 279:1200–1205 [DOI] [PubMed] [Google Scholar]
- Mackenzie PI, Bock KW, Burchell B, Guillemette C, Ikushiro S, Iyanagi T, Miners JO, Owens IS, Nebert DW. (2005) Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics 15:677–685 [DOI] [PubMed] [Google Scholar]
- Magdalou J, Chajes V, Lafaurie C, Siest G. (1990) Glucuronidation of 2-arylpropionic acids pirprofen, flurbiprofen, and ibuprofen by liver microsomes. Drug Metab Dispos 18:692–697 [PubMed] [Google Scholar]
- Miyashita T, Kimura K, Fukami T, Nakajima M, Yokoi T. (2014) Evaluation and mechanistic analysis of the cytotoxicity of the acyl glucuronide of nonsteroidal anti-inflammatory drugs. Drug Metab Dispos 42:1–8 [DOI] [PubMed] [Google Scholar]
- O’Brien K, Moss E, Judah D, Neal G. (1983) Metabolic basis of the species difference to aflatoxin B1 induced hepatotoxicity. Biochem Biophys Res Commun 114:813–821 [DOI] [PubMed] [Google Scholar]
- Ozer J, Ratner M, Shaw M, Bailey W, Schomaker S. (2008) The current state of serum biomarkers of hepatotoxicity. Toxicology 245:194–205 [DOI] [PubMed] [Google Scholar]
- Park BK, Naisbitt DJ, Gordon SF, Kitteringham NR, Pirmohamed M. (2001) Metabolic activation in drug allergies. Toxicology 158:11–23 [DOI] [PubMed] [Google Scholar]
- Pirmohamed M, Breckenridge AM, Kitteringham NR, Park BK. (1998) Fortnightly review: adverse drug reactions. BMJ 316:1295–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirmohamed M, Madden S, Park BK. (1996) Idiosyncratic drug reactions. Metabolic bioactivation as a pathogenic mechanism. Clin Pharmacokinet 31:215–230 [DOI] [PubMed] [Google Scholar]
- Pumford NR, Myers TG, Davila JC, Highet RJ, Pohl LR. (1993) Immunochemical detection of liver protein adducts of the nonsteroidal antiinflammatory drug diclofenac. Chem Res Toxicol 6:147–150 [DOI] [PubMed] [Google Scholar]
- Ritter JK, Chen F, Sheen YY, Tran HM, Kimura S, Yeatman MT, Owens IS. (1992) A novel complex locus UGT1 encodes human bilirubin, phenol, and other UDP-glucuronosyltransferase isozymes with identical carboxyl termini. J Biol Chem 267:3257–3261 [PubMed] [Google Scholar]
- Ross-Degnan D, Soumerai SB, Fortess EE, Gurwitz JH. (1993) Examining product risk in context. Market withdrawal of zomepirac as a case study. JAMA 270:1937–1942 [PubMed] [Google Scholar]
- Rubenstein JH, Laine L. (2004) Systematic review: the hepatotoxicity of non-steroidal anti-inflammatory drugs. Aliment Pharmacol Ther 20:373–380 [DOI] [PubMed] [Google Scholar]
- Shiratani H, Katoh M, Nakajima M, Yokoi T. (2008) Species differences in UDP-glucuronosyltransferase activities in mice and rats. Drug Metab Dispos 36:1745–1752 [DOI] [PubMed] [Google Scholar]
- Silber B, Holford NH, Riegelman S. (1982) Stereoselective disposition and glucuronidation of propranolol in humans. J Pharm Sci 71:699–704 [DOI] [PubMed] [Google Scholar]
- Spahn-Langguth H, Benet LZ. (1992) Acyl glucuronides revisited: is the glucuronidation process a toxification as well as a detoxification mechanism? Drug Metab Rev 24:5–47 [DOI] [PubMed] [Google Scholar]
- Tougou K, Gotou H, Ohno Y, Nakamura A. (2004) Stereoselective glucuronidation and hydroxylation of etodolac by UGT1A9 and CYP2C9 in man. Xenobiotica 34:449–461 [DOI] [PubMed] [Google Scholar]
- Tukey RH, Strassburg CP. (2000) Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol 40:581–616 [DOI] [PubMed] [Google Scholar]
- Uchaipichat V, Mackenzie PI, Elliot DJ, Miners JO. (2006) Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) “probes” for human udp-glucuronosyltransferases. Drug Metab Dispos 34:449–456 [DOI] [PubMed] [Google Scholar]
- Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, Ball SE. (2004) Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos 32:1201–1208 [DOI] [PubMed] [Google Scholar]
- Xu L, Krenitsky DM, Seacat AM, Butenhoff JL, Tephly TR, Anders MW. (2006) N-glucuronidation of perfluorooctanesulfonamide by human, rat, dog, and monkey liver microsomes and by expressed rat and human UDP-glucuronosyltransferases. Drug Metab Dispos 34:1406–1410 [DOI] [PubMed] [Google Scholar]
- Zhang M, Fawcett JP, Kennedy JM, Shaw JP. (2000) Stereoselective glucuronidation of formoterol by human liver microsomes. Br J Clin Pharmacol 49:152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.