

Abstract
We demonstrate a method for the prediction of chemotherapeutic response in patients using only before-treatment baseline tumor gene expression data. First, we fitted models for whole-genome gene expression against drug sensitivity in a large panel of cell lines, using a method that allows every gene to influence the prediction. Following data homogenization and filtering, these models were applied to baseline expression levels from primary tumor biopsies, yielding an in vivo drug sensitivity prediction. We validated this approach in three independent clinical trial datasets, and obtained predictions equally good, or better than, gene signatures derived directly from clinical data.
Background
Identifying and applying molecular biomarkers to predict response to medication is particularly important for drugs with a narrow therapeutic index, for example chemotherapeutic agents, because response is highly variable and side effects are potentially lethal [1,2]. Many studies have been conducted with this objective but only a handful of markers can reproducibly predict chemotherapeutic response in the clinic [3]. It is anticipated that the number of biomarkers discovered will rise as high-throughput sequencing becomes cheaper and more pervasive [3,4]; however, the effect size of these markers is generally small, since drug response is typically a complex trait, usually influenced by many genomic and environmental factors [3,4]. Thus, it has been hypothesized that methods that consider the cumulative effect of many markers, may predict complex phenotypes (like drug response) more accurately. Consequently, some researchers have recently developed sophisticated methods that incorporate all of the data in a genome. For example, there has been some success in using whole-genome SNP or sequence data to predict complex traits [5,6].
In cancer, genomic aberrations and aneuploidy are common, which means that it is difficult to obtain reliable SNP or genome sequences directly from tumors [7]. However, the quantification of whole-genome gene expression levels from primary tumor biopsies is straightforward and has been successfully applied for many years [8,9]. Unfortunately, prediction using gene expression microarray data has traditionally been fraught with reproducibility issues [10]. One of the major concerns is that gene expression estimates, generated on different microarray platforms or even in different batches, are not always consistent [11]. Several analytical approaches have recently been suggested to address this problem and a large-scale comparison has found that some of these methods reliably correct for these biases [12]. Also, multiple studies have compared the performance of various algorithms for predicting survival phenotypes from microarray expression data [13,14]. These have found that ridge regression (a type of regularized linear regression that can include the expression of all genes in the model) performed best, or was consistently amongst the best performing methods. However, gaps remain in the utility of these tools in predicting clinical phenotypes.
Here, we present an approach that integrates several of these recently developed computational and statistical tools to predict in vivo drug response, using models trained on cell line data (see Materials and methods). For model development, the approach was applied to recently released data from the Cancer Genome Project (CGP) [15], consisting of baseline (i.e. before drug treatment) gene expression microarray data and sensitivity to 138 drugs in a panel of almost 700 cell lines. Our results demonstrate that by building a statistical model from these data, it is possible to capture a significant proportion of the variability in drug response in patients. The Cancer Cell Line Encyclopedia [16] has an additional large panel of cell lines, for which it is possible to construct such models, although here, we focus on the CGP, because those cell lines have been screened against more drugs.
To test our approach, we identified clinical trial datasets that had assessed tumor gene expression before drug treatment (using expression microarrays) and had subsequently measured a clear drug response phenotype. Using these data, we can test whether our models derived from cell lines capture a significant proportion of the variability in drug response in patients. The clinical datasets must fulfill the following criteria. Firstly, the clinical trial data (both baseline tumor expression and post-treatment drug response) must be publicly available and easily accessible to allow other researchers to reproduce the results. Patients must have been treated with monotherapy, rather than a combination of drugs, as multi-drug regimes would clearly confound the results. The data must have been published and not retracted. A reasonable number of clinically evaluable samples (>20) are required for statistical power. Finally, sensitivity to the particular drug (as measured by the concentration required for 50% of cellular growth inhibition (IC50)), must have been quantified in the CGP cell lines, because we cannot create suitable models otherwise. To our knowledge, there are four existing datasets that fulfill these criteria [17-20] and the results of our analysis of these data are presented below. Interestingly, the four trials were for three different types of cancers treated with either cytotoxic or targeted agents.
Results
Our goal was to use baseline gene expression and in vitro drug sensitivity derived from cell lines, coupled with in vivo baseline tumor gene expression, to predict patients’ response to drugs. An overview of our approach is shown in Figure 1 (complete details are in Materials and methods; see Data availability for details of how to acquire the R code). Ridge regression models, which allow a small contribution from every gene, have previously been shown to be the best method for predicting survival from gene expression microarray data [13,14]. Our analyses are consistent with these previously published findings. In preliminarily tests, we assessed several of the plethora of available machine learning algorithms, including random forests [21], PAM [22], principal component regression [23], Lasso [24] and ElasticNet regression [25]. Among them, ridge regression was consistently the best performer, with the added advantage of being highly computationally efficient, which is crucial for cross-validation analysis. Furthermore, principal component analysis (PCA) demonstrates that whole-genome gene expression can capture far more information about cancer biology, than may have been previously appreciated. As illustrated in Figure 2 and Additional file 1: Figures S1 and S2, whole-genome gene expression recapitulates tissue of origin, cancer subtype and various genomic aberrations, when the CGP cell lines are plotted on the first two principal components of a whole-genome gene expression matrix. This suggests that whole-genome gene expression acts as a surrogate for unmeasured genetic and non-genetic phenotypes, providing additional support for this approach.
Figure 1.
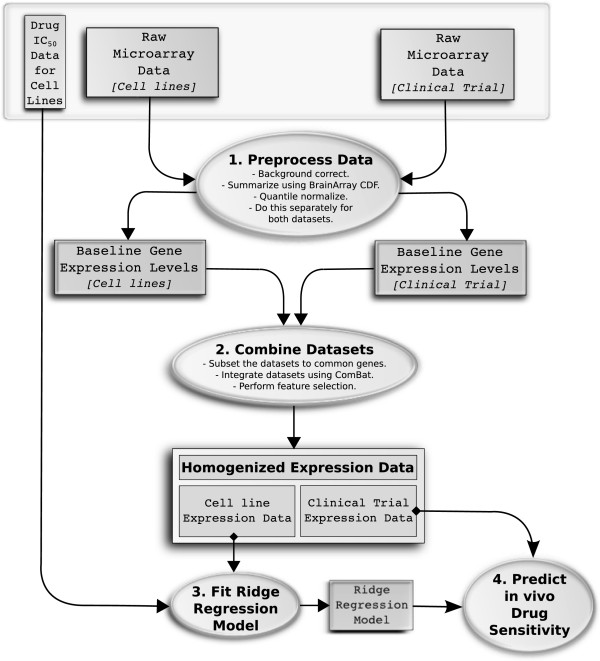
Data flow diagram showing our approach to predicting in vivo drug sensitivity. Data are represented by rectangles and processes by ovals. The input data (baseline expression and drug IC50 in cell lines and in vivo tumor gene expression) are shown in a gray box. The raw microarray data are (1) preprocessed separately using the robust multi-array average [26] method and the CDF files remapped by BrainArray [27] are summarized, (2) then combined and homogenized using ComBat. (3) A ridge regression model is fitted for baseline gene expression levels in the cell lines against the in vitro drug IC50 estimates and (4) this model is then applied to the baseline tumor expression data from the clinical trial, to yield drug sensitivity estimates. Complete details are given in Materials and methods. CDF, chip definition file.
Figure 2.
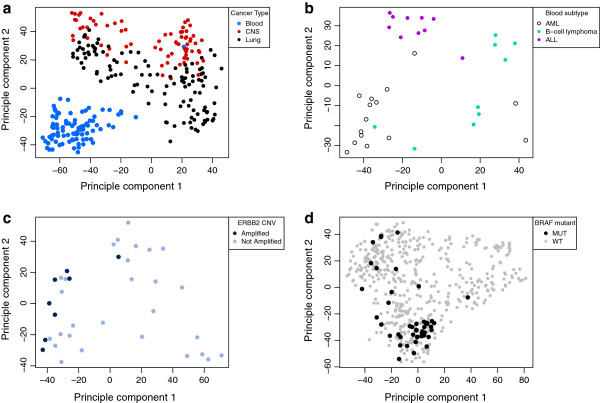
Whole-genome gene expression data capture molecular phenotypes in cancer cell lines. (a) Clustering of cancer types on principal component (PC) 1 and PC2 of a gene expression matrix from the CGP cell lines. There is clear clustering of blood, central nervous system and lung cancers. For clarity, only these three cancer types are shown here. Additional file 1: Figure S1 shows all cancer types. (b) Clustering of subtypes of hematological cancers on PC1 and PC2 of a gene expression matrix of CGP hematological cancer cell lines. For clarity only acute myeloid leukemia, acute lymphoblastic leukemia and B-cell lymphoma are shown here. All data is shown in Additional file 1: Figure S2. (c) Clustering of ERBB2 amplified breast cancers on PC1 and PC2 of a gene expression matrix of CGP breast cancer cell lines. (d) Clustering of BRAF mutated cancers on PC1 and PC2 of a gene expression matrix from all CGP cell lines. ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CGP, Cancer Genome Project; CNS, central nervous system; CNV, copy number variation; MT, mutated; PC, principal component; WT, wild type.
Docetaxel and cisplatin treatment of breast cancer
We first applied our method to gene expression microarray data obtained from 24 breast cancer tumor biopsies through a clinical trial, which measured the response of patients to docetaxel neoadjuvant treatment [18]. Tumor size, measured before and after four cycles of docetaxel, was used to calculate the percentage of residual disease. The authors designated individuals as ‘sensitive’ or ‘resistant’ to docetaxel, depending on whether there was ≤25% or >25% of the tumor remaining. Tumor gene expression levels were measured from biopsies using Affymetrix microarrays (GEO accession number [GEO:GSE6434]). In the original study, receiver operating characteristic (ROC) curve [28,29] analysis reported an area under the curve (AUC) of 0.96 (from leave-one-out cross-validation (LOOCV)) using a 92-gene signature derived from the 24 samples. However, given that this signature was generated on the same data on which it was evaluated, this is likely to represent an inflated estimate of the classification accuracy.
To compare our method to these results, we used the CGP cell lines to build a ridge regression model, which related whole-genome gene expression to docetaxel sensitivity. We applied the model to the in vivo pretreatment breast cancer tumor expression data. The predicted drug sensitivity value was lower in the patients who were defined (by the trial) to be sensitive to docetaxel, compared to the patients defined as resistant (Figure 3a; P = 4.0 × 10−3 from t-test). Of the seven individuals who were predicted to be most sensitive, six are in the trial-defined sensitive group. ROC curve analysis revealed an AUC of 0.81 (Figure 3b; P = 5.0 × 10−3). Notably, training (cell lines) and test (clinical trial) data were assessed using different microarray platforms and the training set contained only 24 breast cancer cell lines. Interestingly, when the models were trained on these 24 breast cancer cell lines alone, there was no difference in predicted drug sensitivity between the trial-defined sensitive/resistant groups (P = 0.65 from a t-test). This suggests that the non-breast cancer cell lines included in the full training panel are informative for predicting the in vivo drug response for breast cancer. For comparison, ElasticNet and Lasso regression models were also applied to this data, but both underperformed when compared to ridge regression (P = 0.01 from t-tests for both models; Additional file 1: Table S1; see Materials and methods for details).
Figure 3.
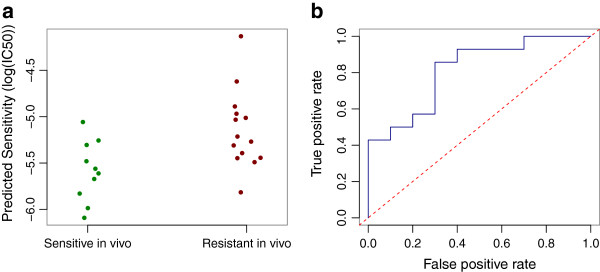
Prediction of docetaxel sensitivity in breast cancer patients. (a) Strip chart showing the difference in predicted drug sensitivity for individuals sensitive or resistant to docetaxel treatment in vivo. (b) ROC curve showing the proportion of true positives against the proportion of false positives as the classification threshold is varied. ROC, receiver operating characteristic.
Next, we applied our method to a second breast cancer dataset, which assessed the response of 24 triple-negative patients to neoadjuvant cisplatin therapy [20]. We downloaded the raw data from ArrayExpress (accession number E-GEOD-18864) and processed it as described in Materials and methods. The authors assigned patients to one of four drug response categories based on RECIST [30] criteria. This time, our models did not capture variability in clinical response (Additional file 1: Figure S3; P = 0.26 from a linear regression model). LOOCV (see Materials and methods) indicated that, for the cell line panel, our models captured approximately the same proportion of variability in cellular response to cisplatin as they had for docetaxel (r = 0.35, P = 2.6 × 10−15 for docetaxel and r = 0.32, P = 1.4 × 10−13 for cisplatin from Pearson’s correlation test between LOOCV estimated log IC50 and measured log IC50 values). Thus, it is surprising that we could not also predict the in vivo response to cisplatin. Notably, the authors of the original trial could not generate a gene signature from their data, or show that any signature in the literature captured cisplatin response in vivo. Furthermore, they found that no genes were significantly correlated with response, following correction for multiple testing [20]. Therefore, it is possible that we (and the original authors) could not achieve statistical significance, because of the lack of variability in drug response among a small group of patients, as cisplatin is not routinely used to treat breast cancer [20]. Encouragingly, patients showing a ‘complete response’ or ‘progressive disease’ had the lowest and highest median predicted drug sensitivity values, respectively (Additional file 1: Figure S3); but given that there were only three individuals in each of these groups, it is not surprising that we did not establish significance. Consequently, a larger clinical cohort may be required to assess rigorously whether our models capture variability in cisplatin response for triple-negative breast cancer.
Bortezomib in myeloma
Next, we applied our approach to a larger publicly available clinical phase II/III trial dataset, which assessed response to bortezomib in relapsed multiple myeloma patients [19]. In the original study, a pretreatment bone marrow aspirate was collected and enriched for tumor cells, which underwent microarray expression profiling. It was found that 168 patients had a clinically evaluable bortezomib response, which was classified as complete response (CR), partial response (PR), minimal response (MR), no change (NC) or progressive disease (PD) [19] using European Group for Bone Marrow Transplantation criteria [31]. CR, PR and MR patients were defined as responders and NC and PD patients as non-responders. Expression in tumor cells was measured using either Affymetrix Human Genome U133A or U133B arrays in triplicate. The same samples were interrogated using both A and B arrays. Data were processed using the Affymetrix MAS5.0 algorithm and the median expression value of the replicates that passed quality control was reported.
This clinical dataset presents some obstacles. Firstly, only the preprocessed data is publicly available (GEO accession number [GEO:GSE9782]). The fact that the raw microarray data are not available is problematic because the lack of standardized raw data processing likely lowers the performance of our model. Also, clinical samples were collected as part of three different clinical trials from various sites, and they were also hybridized in different batches, reflecting the types of issues that may be encountered were one to apply such a method in the clinic. Furthermore, there is only a single myeloma cell line in the training panel.
LOOCV in the cell line training set revealed similar correlations to other drugs (r = 0.45; P = 2.6 × 10−15). Despite the suboptimal clinical data, our method captured substantial variability in bortezomib response. There was a statistically significant difference between the predicted drug sensitivity in patients between the trial-defined responder and non-responder groups (Figure 4a; P = 8.9 × 10−4 for samples quantified using U133A and Additional file 1: Figure S4; P = 1.5 × 10−6 for samples quantified using U133B from t-tests). In the U133A dataset, the nine patients who were predicted to be most sensitive were all drug responders (Figure 4a). The AUCs from ROC curve analysis are 0.63 and 0.71 for U133A and U133B measurements, respectively (Figure 4b; P = 1.3 × 10−3 and Additional file 1: Figure S5; P = 1.0 × 10−5). Strikingly, when the response was further subdivided (as CR, PR, MR, NC and PD), the median predicted drug sensitivity in each of these five groups was in exactly the correct order (Figure 4c and Additional file 1: Figure S6) in the U133B samples.
Figure 4.
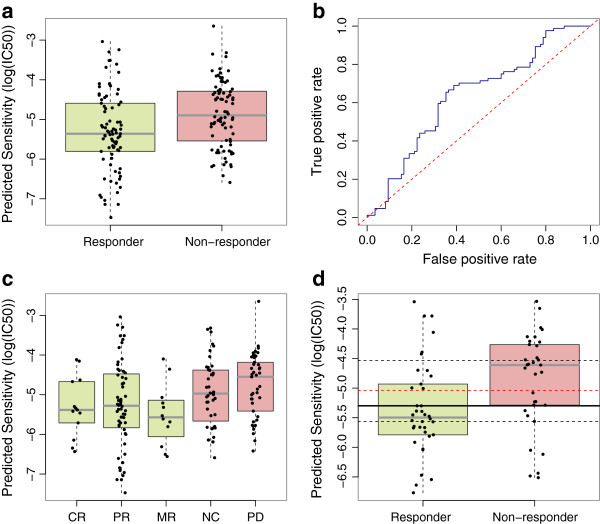
Prediction of bortezomib sensitivity in multiple myeloma patients. (a) Strip chart and boxplot of predicted drug sensitivity for in vivo responders and non-responders to bortezomib. (b) ROC curve illustrating estimated prediction accuracy. (c) Strip chart and boxplot with responders and non-responders further broken down as showing CR, PR, MR, NC or PD. (d) Strip chart and boxplot illustrating our predictions for the (039) arm of the bortezomib trial. The horizontal black line indicates the optimal cut-point, where classification accuracy is 72%. The horizontal dashed black lines indicate the range of cut-points for which classification accuracy is >63%, which was the accuracy reported for the trial-derived 100-gene signature on this dataset. The horizontal dashed red line is the mean IC50 value in the cell line training set, which could be used as an unbiased cut-point. CR, complete response; MR, minimal response; NC, no change; PD, progressive disease; PR, partial response.
The authors of the original clinical study reported that a 100-gene signature model [32], built on two arms of the trial (025 and 040), could predict bortezomib response in the third (039) arm of the trial with 63% accuracy. To compare our predictions with those originally reported, we assessed the performance of our model on only this third arm of the trial. Our models generate a continuous variable and to compare the results previously reported directly, we must dichotomize this variable (i.e. split the data into ‘sensitive’ and ‘resistant’ at an arbitrary cut-point). At the optimal cut-point (−5.29), 51 of 71 patients were correctly classified, meaning that our method achieved a classification accuracy of 72%. For a large range (−5.57 to −4.53) of possible cut-points, our accuracy was greater than the 63% achieved by the trial-derived gene signature (Figure 4d). While dichotomizing clinical response data is not ideal [33], the original clinical data is again not available, thus this is the only means of directly comparing the predictions. Nevertheless, the results indicate that our models offer a substantial performance improvement.
This study also contained a group of 70 patients who were treated with dexamethasone (and had a clinically evaluable drug response). It was not possible to construct a dexamethasone specific model as this drug was not screened against the CGP cell lines. This group is still suitable as a negative control and thus we applied the bortezomib model in this cohort. Encouragingly, there was no difference in predicted bortezomib sensitivity between responders and non-responders to dexamethasone (P = 0.81 from a t-test), suggesting that the models applied to bortezomib-treated patients are drug specific.
Erlotinib in non-small cell lung cancer
Finally, we applied our approach to a dataset from the Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) study (trial registration ID: NCT00409968) [17,34]. A subset of patients with recurrent or metastatic non-small cell lung cancer (NSCLC) were treated with either erlotinib (n = 25), an EGFR inhibitor, or sorafenib (n = 37), a VEGFR inhibitor, in a second-line setting. Raw microarray and drug sensitivity data were downloaded from GEO ([GEO:GSE33072]).
Inspection of the training data revealed that only a very small proportion of the cell lines assessed for sensitivity to these drugs were within the drug screening concentration used by the CGP. This is the case for many targeted agents. In contrast, most cell lines treated with cytotoxic agents, for example docetaxel, have more accurately quantified IC50 values, because a much larger proportion of cell lines tended to respond within the sensitivity screening window. The drastically different response of cell lines to cytotoxic or targeted agents is illustrated in Additional file 1: Figure S7. This can be rigorously demonstrated by segmenting all drugs into two groups (cytotoxic or targeted) and comparing the median size of the confidence intervals associated with the IC50 values of each drug. Unsurprisingly, the confidence intervals are larger for targeted agents (average of 1.9 for cytotoxic compared to 4.5 for targeted agents; P = 1.4 × 10−5 from a Wilcoxon rank sum test). Consistent with this, the signal-to-noise ratio is also significantly different for cytotoxic and targeted drugs (P = 7.1 × 10−7 from a Wilcoxon rank sum test). In light of this, it is not reasonable to fit a linear ridge regression for most targeted agents, because IC50 values for most cell lines were derived using extrapolated data, and thus have very large associated confidence intervals. An approach that reduces the level of noise fitted in the model will be more suited for targeted agent sensitivity prediction. Consequently, we fitted logistic ridge regression models for the 15 most sensitive (which had reliably measured IC50 values) versus the 55 most resistant CGP cell lines (see Materials and methods). In LOOCV, this method provided 89% classification accuracy on the training set and separated sensitive and resistant groups with P = 9.3 × 10−5, providing additional support for applying this approach to clinical samples.
When applied to the clinical trial data, this modified approach captured a large proportion of variability in the in vivo erlotinib response (Figure 5a; rho = 0.64 and P = 5.3 × 10−4 from a Spearman’s correlation test). All patients were EGFR wild-type. Since KRAS mutation is a known biomarker of resistance to EGFR inhibitors [35-37], we evaluated performance in the subset of 20 individuals who were both EGFR and KRAS wild-type. The results remained highly significant (rho = 0.59 and P = 6.4 × 10−3 from a Spearman’s correlation test). This means that we enriched for drug responders, even in the absence of any known biomarker. A further interesting observation is that one patient, who was among the most sensitive to erlotinib, had a KRAS mutation, normally a biomarker of EGFR inhibitor resistance. Our approach predicted this would be the fifth most sensitive individual in the cohort, and they were in fact, the fifth most sensitive individual.
Figure 5.
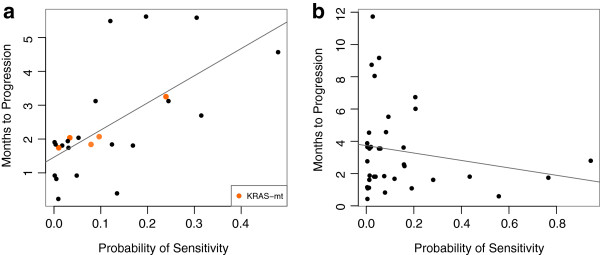
Prediction of erlotinib sensitivity in NSCLC patients. (a) Months-to-progression plotted against predicted probability of erlotinib sensitivity. All patients are EGFR wild-type. KRAS mutations are highlighted and a linear regression line is shown. (b) The predicted probability of sensitivity to erlotinib plotted against months-to-progression for individuals treated with sorafenib. NSCLC, non-small cell lung cancer.
The authors of the original study developed a 76-gene epithelial-mesenchymal transition (EMT) gene expression signature using both NSCLC cell lines and patient data. They reasoned, as EMT had been previously shown to be associated with EGFR inhibitor resistance, that this signature may capture variability in erlotinib response in vivo. The gene signature was applied to the 20 EGFR and KRAS wild-type NSCLC patients treated with erlotinib. They found that individuals with disease control at eight weeks showed a more epithelial-like signature and the result was of borderline significance (P = 0.052 from a t-test). For comparison with our approach, we assessed the difference in predicted probability of erlotinib sensitivity (from the logistic ridge regression model) between individuals with disease progression and those without disease progression at two months. In our case, the difference was highly statistically significant (P = 4.9 × 10−4 from a t-test). This suggests that whole-genome gene expression models, derived from a large panel of cell lines, have superior power to predict erlotinib sensitivity, compared to the 76-gene EMT signature.
Since the drug sensitivity phenotype evaluated in this trial is ‘months-to-progression’, it is possible that our models are capturing a prognostic phenotype, rather than drug sensitivity. To test this, we applied our cell-line-derived erlotinib sensitivity prediction model to predict months-to-progression for an independent arm of the same trial in which NSCLC patients were treated with sorafenib. The erlotinib-specific model was not predictive of months-to-progression after sorafenib treatment (Figure 5b; P = 0.83 from a t-test), suggesting that the model is drug specific, rather than a general predictor of disease progression or prognosis.
Discussion
We have shown that models constructed using baseline gene expression and drug IC50 values, from a large panel of cell lines, can predict the chemotherapeutic response in patients. Our method uses whole-genome ridge regression, and the expression of every gene contributes a small amount to the prediction. The ridge regression penalty parameter is automatically selected, meaning that no user input is required to tune the algorithm. Our approach captures a statistically significant proportion of variability in drug response in three of four clinical trials, regardless of the drugs’ mechanism of action or the patients’ cancer type and in all cases, performance was comparable to that of the gene signatures derived directly from clinical data. To our knowledge, this is the first time that a method capable of this has been described.
It may seem surprising that whole-genome gene expression alone has such remarkable power in enriching for drug responders, as the approach ignores what are thought to be important factors in drug sensitivity, such as cancer type, genomic aberrations or any other specific markers. We showed that the impressive performance may (at least in part) stem from the fact that whole-genome gene expression may act as a surrogate for unmeasured phenotypes that are directly relevant to chemotherapeutic sensitivity (Figure 2), and capture aspects of both germ line and tumor-specific genome variation. This observation is further supported by multiple studies that have shown that gene expression can be used to characterize novel cancer subtypes and has been shown to have predictive and prognostic value [38-43].
Our method attained classification accuracy approaching, or even surpassing that of the gene signatures derived directly from clinical trials. In these trials, gene signatures were derived using in vivo samples. In the case of docetaxel, the signature was generated on the same set of samples on which it was tested, which inevitably inflates their estimate of prediction accuracy. It would only be possible to compare performance fairly using an independent dataset. Nevertheless, our method significantly enriched for docetaxel responders in the trial dataset. Our approach predicted in vivo drug sensitivity more accurately than a 100-gene signature derived from the bortezomib clinical trial. We also outperformed the 76-gene EMT signature (generated using both cell lines and patients) in predicting sensitivity to erlotinib. There was no evidence that KRAS mutation, previously identified as a biomarker of EGFR inhibitor resistance, had predictive power in this data, although the number of samples was small and this result does not discount KRAS mutation as a biomarker for this drug. The fact remains that our method also outperformed this (already established) biomarker in this dataset. We modified the original algorithm (to use logistic instead of linear ridge regression) in the analysis of erlotinib data. This is justifiable given the severe noise associated with the IC50 values for these types of targeted agents in the CGP cell lines. Overall, the results suggest that models created on very large panels of cell lines, can rival, or even surpass the performance of similar in vivo approaches.
In all studies discussed here, the data are suboptimal, because all of the clinical trials used a different microarray platform than was used on the cell line panel training set. Also, the cell line panel often only contained a very small number of samples from the actual cancer type that the clinical trial evaluated. For example, only one myeloma cell line was treated with bortezomib in our panel of training cell lines. We anticipate that if training and test microarray platforms were the same and if the cell line panel contained more relevant cancer types, accuracy would be further improved. These results are congruent with the emerging view that -omics characterization of tumors may rival traditional tissue-of-origin and pathological descriptors for a variety of clinically important classifications. This was supported by our finding that, unlike the full cell line panel, the 24 available breast cancer cell lines could not predict docetaxel response for breast cancer patients (although it would be difficult to achieve accurate prediction using such a small training set).
Performance would also likely be enhanced by a more detailed assessment of the transcriptome, for example, by quantifying transcript expression levels with RNA sequencing (RNA-seq), which has been shown to provide better estimates of expression than microarrays [44]. A recent study has also found that results of transcriptome sequencing using RNA-seq were highly reproducible between different laboratories, if procedures are standardized (which is not generally the case for expression microarrays) [45]. This provides further evidence that incorporating RNA-seq would increase power and the widespread utility of these types of expression-based prediction assays. However, a different machine learning algorithm may be better suited to the distribution of RNA-seq data.
We have recently completed a separate study that provides additional support for emphasizing transcriptomics in pharmacogenomic prediction. In that analysis, we used whole-genome models, similar to the ridge regression models used here, to compare the relative contribution of whole-genome SNPs, gene expression or microRNA (miRNA) expression, to inter-individual variability in cellular growth rate [46]. We found that, in lymphoblastoid cell lines, far more of the variability in growth rate (between cell lines isolated from different individuals) can be explained by the transcriptome than by genome-wide SNPs. Using gene and miRNA expression data, we constructed statistical models that explained 48% of variability in growth rate, compared to just 2% when using models based on only whole-genome SNP data. Given that a substantial proportion of chemotherapeutic agents target fast-growing cells (for example, docetaxel), these results provide a strong rationale for prioritizing the transcriptome when predicting clinical response to this class of drugs. The result also showed that combining miRNA and gene expression data significantly improved prediction (from 38% to 48% of variability explained) over mRNA expression alone, suggesting that including miRNA and other non-coding RNAs may improve the prediction of clinical drug sensitivity, although no data is currently available that would allow us to test this hypothesis.
Here, we have demonstrated that models derived from a very large panel of cell lines achieve equal or better performance for clinical drug sensitivity prediction than those derived directly from patients and these findings can have a profound impact on patient care. For example, screening for drug sensitivity in vitro is far less costly and time-consuming than conducting large clinical trials. A much larger number of samples can be screened against any given drug using cell lines than would be feasible (either practically or ethically) in a clinical setting. Furthermore, in vitro drug sensitivity screening is usually conducted under controlled experimental conditions to achieve a greater degree of accuracy. The fact that many more samples can be screened, with more accuracy, may lead to improved statistical power, compared to in vivo methods, which inevitably rely on smaller sample sizes and noisy clinical response phenotypes. This, and the fact that recently developed statistical methods robustly correct for intrinsic differences in gene expression between cell lines and in vivo tumors, suggest that this type of approach is a very promising option for personalizing drug treatment. There is no limit to the number of drugs that could be screened against a panel of cell lines. In theory, every existing chemotherapeutic compound could be tested, meaning that given a tumor biopsy, it would be trivial to use this approach to estimate sensitivity to every drug prior to any course of treatment. The falling price of gene expression microarrays makes it feasible to incorporate this technology into patient care. The cell line training set could also be expanded by including data on cellular sensitivity within different microenvironments and cell lines under different simulated stromal conditions, as it is known that the tumor microenvironment plays a key role in tumor development [47].
The results also have important implications in drug development, where our approach could be used to enrich for likely drug responders, prior to carrying out clinical trials. There has been much recent interest in developing drugs in conjunction with companion diagnostic tests [48]. The benefits of enriching for likely drug responders are obvious, but it is normally difficult to develop accurate biomarkers without exposing the drug to patients. However, our approach, because of its ability to enrich for drug responders in a clinical cohort, has enormous potential as a companion diagnostic. There is also a clear ethical benefit, in that such a diagnostic could be developed without ever exposing potentially unresponsive patients to toxic chemotherapeutic agents.
Finally, we highlight the work of Menden et al., who recently constructed models using the CGP cell line data, with the aim of predicting in vitro drug sensitivity [49]. The authors achieved impressive prediction (R2 = 0.72 from eightfold cross-validation) by using models that consider the effectiveness of drugs with similar mechanisms of action. These results are better than those achieved using our models (based on expression data alone), but currently cannot be extended to in vivo data because clinical response to similar drugs is almost never known a priori. However, the results suggest that in future, in vivo prediction could be improved by considering multiple drugs, using either prior information on what is known about mechanism of drug action, or simply the empirical correlation of drug sensitivities.
Conclusions
In summary, we have shown for the first time that it is possible to enrich for drug responders in a clinical cohort using only baseline tumor gene expression levels, by applying models generated from a large panel of cell lines. We have also demonstrated that this approach outperforms several existing biomarkers in the available clinical datasets. These findings have profound implications for personalized medicine and drug development. Future work will focus on improving predictions using more rigorous transcriptome quantification and further testing in prospective clinical trials. The R code [50] needed to reproduce all figures and results in this paper, is provided (for academic use) on our website in Sweave [51] format (see Data availability).
Materials and methods
Bioinformatics analysis overview
Bioinformatics analyses were performed in R. Our implementation is extremely fast (typically running in <30 s on a standard desktop computer) and easy to use. Once the data are correctly loaded, the user need only provide the baseline gene expression and drug sensitivity data (i.e. IC50) from the cell line panel and baseline gene expression data from the clinical trial. The predicted drug sensitivity is then calculated, requiring no further user input. All R code is provided in annotated Sweave format on our website (see Data availability), enabling other investigators to reproduce easily all the results and figures presented in this paper.
Obtaining gene expression and drug sensitivity data
Drug IC50 values for docetaxel, bortezomib and erlotinib were downloaded from the CGP website ([52]; accessed August 2013). The raw CGP gene expression microarray data (CEL files) were downloaded from ArrayExpress under accession number E-MTAB-783. These data were preprocessed using the robust multi-array average algorithm (implemented by the rma() function in the affy [53] library in R). This algorithm does background correction, quantile normalization and median-polish summarization in one step. For summarization, we used the updated probeset annotation chip definition file (CDF) provided by BrainArray (version 17.0.0 for Affymetrix HT Human Genome U133A arrays, probesets mapped to Entrez gene IDs). We followed the same set of steps to preprocess the docetaxel, cisplatin and erlotinib/sorafenib clinical trial gene expression data (using the appropriate BrainArray CDF file in each case). The bortezomib expression data were obtained directly from GEO using the getGEO() function implemented in the R library GEOquery [54]. All in vivo drug response data were obtained from GEO or directly from the relevant publication.
Combining and homogenizing cell line and clinical trial gene expression datasets
Training (cell lines) and test (clinical trial) datasets were mapped to official gene symbols. Probesets that mapped to more than one gene symbol were summarized by their mean expression value. In all cases, both datasets were generated on different microarray platforms, thus we used a subset of genes represented on both platforms. This typically left approximately 10,000 gene symbols remaining. These two datasets were then homogenized using the ComBat() function from the sva library in R. Finally, we filtered out genes whose expression did not vary substantially in the homogenized dataset, because if technical variability is greater than biological variability, these can never add predictive value; we removed the 20% of genes with lowest variability in expression across all samples.
Predicting in vivo drug sensitivity using linear ridge regression for docetaxel, cisplatin and bortezomib clinical trials
Once the data were prepared as outlined above, a linear ridge regression model was fitted for in vitro drug sensitivity dependent on the homogenized whole-genome expression levels in the CGP cell lines (for which both drug sensitivity and expression data were available). To do this, we used the linearRidge() function from the ridge [55] package in R. This function implements a method to choose the ridge regression tuning parameter automatically. Before fitting the model, the drug sensitivity phenotype data (IC50 values) were power transformed using the powerTransform() function in the R package car. After the model was fitted, it was then applied to the homogenized gene expression data from the clinical trial, using the predict.linearRidge() function from the ridge package in R, thus yielding a drug sensitivity estimate for each patient.
Leave-one-out cross-validation
For LOOCV, all data were first preprocessed and homogenized as described above. Then, ridge regression models were fitted (as above) on all of the available cell line data, but with one sample omitted. Next, these models were used to calculate a predicted IC50 value, using the gene expression data of the single omitted cell line. This process was repeated iteratively for every sample thus yielding a predicted IC50 for every cell line. These predicted IC50 values were then compared to the measured IC50 values, using a Pearson’s correlation test, giving an estimate of prediction accuracy.
ElasticNet and Lasso models
ElasticNet and Lasso regression models were fitted using the glmnet package in R. The Lasso penalty parameter was selected using the automatic cross-validation feature (i.e. the cv.glmnet() function). ElasticNet penalty parameters (alpha and lambda) were selected using the caret package in R. Optimal parameters were selected using a grid search on the cell line training set, which takes approximately one day to run on a standard PC. Parameters were selected based on an optimal R2 value or Cohen’s kappa (in cross-validation) for linear and logistic models, respectively.
Predicting in vivo drug sensitivity using logistic ridge regression for an erlotinib clinical trial
The data were first prepared as described above. Next, we divided the cell line training data into sensitive (15 samples) or resistant (55 samples) groups and fitted a logistic ridge regression model using the logisticRidge() function from the R package ridge. Again, the ridge regression tuning parameter was automatically selected. As this implementation of logistic ridge regression is extremely computationally intensive, we implemented a feature selection step, where only the 1,000 genes that were most differentially expressed between the 15 sensitive and 55 resistant samples were fitted in the model. These genes were selected using t-tests, specifically using the rowttests() function in the R library genefilter [56]. This step enables a standard desktop computer to fit a model in approximately ten minutes (as opposed to days). Once the model is fitted, it is applied to the homogenized gene expression data from the clinical trial, using the predict.logisticRidge() function, which calculates the predicted log-odds of drug sensitivity.
Statistical analysis of results
ROC curve analysis was performed using the ROCR [28] package in R. Empirical P-values were generated using 100,000 sample label permutations and computing the proportion of permutations for which the AUC was more extreme than that observed in the original data. Linear regression, t-tests and Spearman’s correlation tests were performed using the base functions in R. Figure 1 was generated using the Inkscape software and the remaining figures were generated in R.
Data availability
Annotated R code (in Sweave format) to reproduce all of the analysis in this paper is available from our website [57]. The CGP gene expression data are available from ArrayExpress under accession number E-MTAB-783. The IC50 data for the drugs is available from the CGP website [52]. The docetaxel data are available from GEO under accession numbers [GEO:GSE349] and [GEO:GSE350]. The cisplatin data are available from ArrayExpress under accession number E-GEOD-18864. The bortezomib data are available from GEO under accession number [GEO:GSE9782]. The erlotinib data are available from GEO under accession number [GEO:GSE33072]. Complete details and R code showing how to acquire and preprocess all of these data, as well as the associated clinical data are available in Sweave format on our website.
Abbreviations
AUC: area under the curve; CDF: chip definition file; CGP: Cancer Genome Project; CNS: central nervous system; CR: complete response; EMT: epithelial-mesenchymal transition; LOOCV: leave-one-out cross-validation; miRNA: microRNA; MR: minimal response; NC: no change; NSCLC: non-small cell lung cancer; PCA: principal component analysis; PD: progressive disease; PR: partial response; RNA-seq: RNA sequencing; ROC: receiver operating characteristic; SNP: single nucleotide polymorphism.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PG developed the method, performed the analysis and drafted the paper. RSH initiated and supervised the project. NJC assisted in supervising the project. All authors edited and approved the final manuscript.
Supplementary Material
PDF file containing all supplementary figures, tables and their associated legends.
Contributor Information
Paul Geeleher, Email: paulgeeleher@gmail.com.
Nancy J Cox, Email: ncox@bsd.uchicago.edu.
R Stephanie Huang, Email: rhuang@medicine.bsd.uchicago.edu.
Acknowledgements
The authors thank Keston Acquino-Michaels and Zaya Amgaabaatar for verifying the R code and reproducing all results and figures. The authors also thank Jana Heitmann, Dr Brian Stewart and Prof Cathal Seoighe for their critical review of the manuscript. The Cancer Genome Project funded by the Wellcome Trust Sanger Institute generated and made all CGP cell line drug sensitivity and baseline expression data publicly available. This study is supported by the National Institutes of Health/National Institute of General Medical Science (Pharmacogenomics of Anticancer Agents grant U01GM61393). RSH also received support from the National Institute of General Medical Science K08 (GM089941), a Circle of Service Foundation Early Career Investigator award, the National Cancer Institute R21 (CA139278), a University of Chicago Cancer Center Support Grant (#P30 CA14599), a University of Chicago Breast Cancer SPORE Career Development Award (CA125183), a Conquer Cancer Foundation of ASCO Translational Research Professorship award in memory of Merrill J Egorin, MD and the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1RR024999).
References
- Mishra A, Verma M. Cancer biomarkers: are we ready for the prime time? Cancers (Basel) 2010;2:190–208. doi: 10.3390/cancers2010190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Wang M. Personalized medicine in oncology: tailoring the right drug to the right patient. Biomark Med. 2010;4:523–533. doi: 10.2217/bmm.10.66. [DOI] [PubMed] [Google Scholar]
- Simon R, Roychowdhury S. Implementing personalized cancer genomics in clinical trials. Nat Rev Drug Discov. 2013;12:358–369. doi: 10.1038/nrd3979. [DOI] [PubMed] [Google Scholar]
- Sawyers CL. The cancer biomarker problem. Nature. 2008;452:548–552. doi: 10.1038/nature06913. [DOI] [PubMed] [Google Scholar]
- Lee SH, Van Der Werf JHJ, Hayes BJ, Goddard ME, Visscher PM. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLoS Genet. 2008;4:e1000231. doi: 10.1371/journal.pgen.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober U, Ayroles JF, Stone EA, Richards S, Zhu D, Gibbs RA, Stricker C, Gianola D, Schlather M, Mackay TFC, Simianer H. Using whole-genome sequence data to predict quantitative trait phenotypes in Drosophila melanogaster. PLoS Genet. 2012;8:e1002685. doi: 10.1371/journal.pgen.1002685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature. 2013;500:207–211. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AAM, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, Desmedt C, Larsimont D, Cardoso F, Peterse H, Nuyten D, Buyse M, Van de Vijver MJ, Bergh J, Piccart M, Delorenzi M. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006;98:262–272. doi: 10.1093/jnci/djj052. [DOI] [PubMed] [Google Scholar]
- Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, Su Z, Chu T-M, Goodsaid FM, Pusztai L, Shaughnessy JD, Oberthuer A, Thomas RS, Paules RS, Fielden M, Barlogie B, Chen W, Du P, Fischer M, Furlanello C, Gallas BD, Ge X, Megherbi DB, Symmans WF, Wang MD, Zhang J, Bitter H, Brors B, Bushel PR, Bylesjo M. et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol. 2010;28:827–838. doi: 10.1038/nbt.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, Luo Y, Sun YA, Willey JC, Setterquist RA, Fischer GM, Tong W, Dragan YP, Dix DJ, Frueh FW, Goodsaid FM, Herman D, Jensen RV, Johnson CD, Lobenhofer EK, Puri RK, Schrf U, Thierry-Mieg J, Wang C, Wilson M, Wolber PK. et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy J, Valafar F. Empirical comparison of cross-platform normalization methods for gene expression data. BMC Bioinformatics. 2011;12:467. doi: 10.1186/1471-2105-12-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wieringen WN, Kun D, Hampel R, Boulesteix A-L. Survival prediction using gene expression data: a review and comparison. Comput Stat Data Anal. 2009;53:1590–1603. doi: 10.1016/j.csda.2008.05.021. [DOI] [Google Scholar]
- Bøvelstad HM, Nygård S, Størvold HL, Aldrin M, Borgan Ø, Frigessi A, Lingjaerde OC. Predicting survival from microarray data – a comparative study. Bioinformatics. 2007;23:2080–2087. doi: 10.1093/bioinformatics/btm305. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F. et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, de Silva M. et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, Nilsson MB, Gudikote J, Tran H, Cardnell RJG, Bearss DJ, Warner SL, Foulks JM, Kanner SB, Gandhi V, Krett N, Rosen ST, Kim ES, Herbst RS, Blumenschein GR, Lee JJ, Lippman SM, Ang KK, Mills GB, Hong WK, Weinstein JN. et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Wooten EC, Tsimelzon A, Hilsenbeck SG, Gutierrez MC, Elledge R, Mohsin S, Osborne CK, Chamness GC, Allred DC, O’Connell P. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 2003;362:362–369. doi: 10.1016/S0140-6736(03)14023-8. [DOI] [PubMed] [Google Scholar]
- Mulligan G, Mitsiades C, Bryant B, Zhan F, Chng WJ, Roels S, Koenig E, Fergus A, Huang Y, Richardson P, Trepicchio WL, Broyl A, Sonneveld P, Shaughnessy JD, Bergsagel PL, Schenkein D, Esseltine D-L, Boral A, Anderson KC. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood. 2007;109:3177–3188. doi: 10.1182/blood-2006-09-044974. [DOI] [PubMed] [Google Scholar]
- Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong C-O, Calogrias D, Buraimoh A, Fatima A, Gelman RS, Ryan PD, Tung NM, De Nicolo A, Ganesan S, Miron A, Colin C, Sgroi DC, Ellisen LW, Winer EP, Garber JE. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J Clin Oncol. 2010;28:1145–1153. doi: 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach Learn. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolliffe IT. A note on the use of principal components in regression. Appl Stat. 1982;31:300. doi: 10.2307/2348005. [DOI] [Google Scholar]
- Tibshirani R. Regression shrinkage and selection via the Lasso. J R Stat Soc. 1994;58:267–288. [Google Scholar]
- Hui Zou TH. Regularization and variable selection via the elastic net. J R Stat Soc. 2005;67:301–320. doi: 10.1111/j.1467-9868.2005.00503.x. [DOI] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- Fawcett T. ROC Graphs: Notes and Practical Considerations for Researchers. 2004.
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Bladé J, Samson D, Reece D, Apperley J, Björkstrand B, Gahrton G, Gertz M, Giralt S, Jagannath S, Vesole D. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102:1115–1123. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100:9991–9996. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royston P, Altman DG, Sauerbrei W. Dichotomizing continuous predictors in multiple regression: a bad idea. Stat Med. 2006;25:127–141. doi: 10.1002/sim.2331. [DOI] [PubMed] [Google Scholar]
- Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR, Tsao A, Stewart DJ, Hicks ME, Erasmus J, Gupta S, Alden CM, Liu S, Tang X, Khuri FR, Tran HT, Johnson BE, Heymach JV, Mao L, Fossella F, Kies MS, Papadimitrakopoulou V, Davis SE, Lippman SM, Hong WK. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Jänne PA, Januario T, Johnson DH, Klein P, Miller VA, Ostland MA, Ramies DA, Sebisanovic D, Stinson JA, Zhang YR, Seshagiri S, Hillan KJ. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–5909. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, Anaissie E, Hollmig K, Pineda-Roman M, Tricot G, van Rhee F, Walker R, Zangari M, Crowley J, Barlogie B, Shaughnessy JD. The molecular classification of multiple myeloma. Blood. 2006;108:2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnelli L, Bicciato S, Mattioli M, Fabris S, Intini D, Verdelli D, Baldini L, Morabito F, Callea V, Lombardi L, Neri A. Molecular classification of multiple myeloma: a distinct transcriptional profile characterizes patients expressing CCND1 and negative for 14q32 translocations. J Clin Oncol. 2005;23:7296–7306. doi: 10.1200/JCO.2005.01.3870. [DOI] [PubMed] [Google Scholar]
- Tan IB, Ivanova T, Lim KH, Ong CW, Deng N, Lee J, Tan SH, Wu J, Lee MH, Ooi CH, Rha SY, Wong WK, Boussioutas A, Yeoh KG, So J, Yong WP, Tsuburaya A, Grabsch H, Toh HC, Rozen S, Cheong JH, Noh SH, Wan WK, Ajani JA, Lee J-S, Tellez MS, Tan P. Intrinsic subtypes of gastric cancer, based on gene expression pattern, predict survival and respond differently to chemotherapy. Gastroenterology. 2011;141:476–85–485. doi: 10.1053/j.gastro.2011.04.042. e1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci F, Finetti P, Rougemont J, Charafe-Jauffret E, Cervera N, Tarpin C, Nguyen C, Xerri L, Houlgatte R, Jacquemier J, Viens P, Birnbaum D. Gene expression profiling identifies molecular subtypes of inflammatory breast cancer. Cancer Res. 2005;65:2170–2178. doi: 10.1158/0008-5472.CAN-04-4115. [DOI] [PubMed] [Google Scholar]
- Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi M-C, Schiappa R, Guenot D, Ayadi M, Kirzin S, Chazal M, Fléjou J-F, Benchimol D, Berger A, Lagarde A, Pencreach E, Piard F, Elias D, Parc Y, Olschwang S, Milano G, Laurent-Puig P, Boige V. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, Ekman P, DeMarzo AM, Tibshirani R, Botstein D, Brown PO, Brooks JD, Pollack JR. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci USA. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zhang Y, Williams J, Antoniou E, McCombie WR, Wu S, Zhu W, Davidson NO, Denoya P, Li E. Parallel comparison of Illumina RNA-Seq and Affymetrix microarray platforms on transcriptomic profiles generated from 5-aza-deoxy-cytidine treated HT-29 colon cancer cells and simulated datasets. BMC Bioinformatics. 2013;14:S1. doi: 10.1186/1471-2105-14-S9-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ’t Hoen PAC, Friedländer MR, Almlöf J, Sammeth M, Pulyakhina I, Anvar SY, Laros JFJ, Buermans HPJ, Karlberg O, Brännvall M, van Ommen G-JB, Estivill X, Guigó R, Syvänen A-C, Gut IG, Dermitzakis ET, Antonorakis SE, Brazma A, Flicek P, Schreiber S, Rosenstiel P, Meitinger T, Strom TM, Lehrach H, Sudbrak R, Carracedo A, van Iterson M, Monlong J, Lizano E, Bertier G. et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat Biotechnol. 2013;31:1015–1022. doi: 10.1038/nbt.2702. [DOI] [PubMed] [Google Scholar]
- Wheeler HE, Aquino-Michaels K, Gamazon ER, Trubetskoy VV, Dolan ME, Huang RS, Cox NJ, Im HK. Poly-omic prediction of complex traits: OmicKriging. 2013. [DOI] [PMC free article] [PubMed]
- Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, Camps C, Marinez I, Busund L-T. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol. 2011;6:209–217. doi: 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- Zhang CH, Zhang YP. Maximizing the commercial value of personalized therapeutics and companion diagnostics. Nat Biotechnol. 2013;31:803–805. doi: 10.1038/nbt.2679. [DOI] [PubMed] [Google Scholar]
- Menden MP, Iorio F, Garnett M, McDermott U, Benes CH, Ballester PJ, Saez-Rodriguez J. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS One. 2013;8:e61318. doi: 10.1371/journal.pone.0061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RDC. R: A Language and Environment for Statistical Computing. Austria, Vienna; 2008. [Google Scholar]
- Leisch F. Dynamic generation of statistical reports using literate data analysis. Proc Comput Stat. 2002. pp. 575–580. http://link.springer.com/chapter/10.1007%2F978-3-642-57489-4_89#.
- The cancer genome project. http://www.cancerrxgene.org.
- Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23:1846–1847. doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- Cule E, De Iorio M. Ridge regression in prediction problems: automatic choice of the ridge parameter. Genet Epidemiol. 2013;37:704–714. doi: 10.1002/gepi.21750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R, Carey V, Huber W, Hahne F. genefilter: methods for filtering genes from microarray experiments. http://cobra20.fhcrc.org/packages/release/bioc/html/genefilter.html.
- University of Chicago GeneMed Server. http://genemed.uchicago.edu/~pgeeleher/cgpPrediction/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDF file containing all supplementary figures, tables and their associated legends.
Data Availability Statement
Annotated R code (in Sweave format) to reproduce all of the analysis in this paper is available from our website [57]. The CGP gene expression data are available from ArrayExpress under accession number E-MTAB-783. The IC50 data for the drugs is available from the CGP website [52]. The docetaxel data are available from GEO under accession numbers [GEO:GSE349] and [GEO:GSE350]. The cisplatin data are available from ArrayExpress under accession number E-GEOD-18864. The bortezomib data are available from GEO under accession number [GEO:GSE9782]. The erlotinib data are available from GEO under accession number [GEO:GSE33072]. Complete details and R code showing how to acquire and preprocess all of these data, as well as the associated clinical data are available in Sweave format on our website.