

Abstract
Hereditary diffuse leucoencephalopathy with spheroids (HDLS) is a rare autosomal dominantly inherited disease with unknown pathophysiology. Diagnosis of neurodegenerative diseases is increasingly based on biomarkers. Although lumbar puncture is routinely performed during the diagnostic workup of HDLS, reports on alterations of neurodegeneration-specific biochemical markers have not been documented so far. We report a 35-year-old woman with clinical, radiological and neuropathological signs of HDLS. She suffered from a rapidly progressive frontal lobe syndrome. Brain MRI revealed diffuse leucoencephalopathy with predominant involvement of the periventricular white matter and corpus callosum. Although she was severely impaired and leucoencephalopathy was prominent, only cerebrospinal fluid total-τ was moderately elevated. Other markers of neuronal (NSE) and astrocytic (S100B) damage were within normal range. Therefore, biochemical markers of central nervous system damage are not helpful in the diagnosis of HDLS.
Background
Hereditary diffuse leucoencephalopathy with spheroids (HDLS) is a rare neurodegenerative disease, usually affecting adults between 24 and 60 years of age. Although symptoms vary at the beginning of the disease, almost all patients suffer from a rapidly progressive frontal lobe syndrome with depressive symptoms, apathy, aboulia, dementia and apraxia in the later stages of the disease. Further symptoms frequently seen in HDLS include aggressiveness, anxiety, parkinsonism, pyramidal signs and epilepsy.1 2 Owing to its rarity and the wide range of symptoms, the disease is often misdiagnosed. In most cases the correct diagnosis is made after a brain biopsy.
Biochemical biomarkers gain more and more importance for the diagnosis of neurodegenerative diseases. Especially τ and neuron-specific enolase (NSE), as a marker of neuronal damage, as well as S100B as indicator of astrocytic stress are well established.3–5
To the best of our knowledge, alterations of biochemical biomarkers in HDLS have not been reported so far in the literature. In this case report we describe clinical, radiological and histopathological findings of a patient with HDLS, together with a broad investigation of biochemical markers indicative for neurodegeneration.
Case presentation
A 35-year-old, previously healthy woman first presented with gait impairment, ataxia, increased tendon reflexes, a dysmetric finger to nose test and a slight impairment of memory. MRI revealed periventricular white matter lesions. This led to the diagnosis of multiple sclerosis (MS). Yet, cerebrospinal fluid (CSF) cell count was normal and oligoclonal bands were negative. Electrophysiological tests, including peripheral nerve conduction studies, electromyography as well as motor and somatosensory evoked potentials did not show any significant alterations. After an intravenous methyl prednisolone pulse the motor symptoms improved and she was able to walk without help.
Three months later she was readmitted in a dramatically worsened condition. During the examination, she had difficulties in moving her arms and fingers according to the instructions of her physician, suggesting an apraxia. She was no longer able to write. Her gait was insecure with rapid shuffling steps. Only when she was pulled, an accelerated gait with larger steps was possible. The Romberg’s test was normal. She was incontinent for urine and stool. The neurological examination further revealed an atactic and slightly dysmetric finger-to-nose test with a deviation to the left side and bradydiadochokinesis. Tendon reflexes were very brisk and without clonus. The abdominal reflexes were present. Apart from a positive Strümpell sign on the left leg, pyramidal signs were negative. There were no spasticity, no paresis and no pathological reflexes. Also no hypo or dysaesthesia was found. The examination of the cranial nerves was unremarkable. The ophthalmological examination did not reveal any signs of visual disturbances. She was depressed and apathic, suffered indifference and was also noticed to have a labile affect. In the Mini-Mental State Examination, a beginning disorientation, especially in time was obvious. She could only recall one of three items and the 100–7 test had to be abandoned after one item. With a final score of 15/30 her cognitive functioning was deemed as severely impaired. Except from severe obesity and acne the general clinical examination was normal.
X-rays of the left hand and foot were normal. EEG recording revealed generalised slowing (δ waves) without regional slowing, epileptic potentials or triphasic potentials.
After a second MRI a brain biopsy was performed and a neuroaxonal leukodystrophy with axonal spheroids was diagnosed (see below). The patient was discharged to a nursing home.
The family of the patient reported on a maternal cousin who was also diagnosed with neuroaxonal leucoencephalopathy after brain biopsy. His first apparent symptoms were that of depression. Later, he also suffered from a rapidly progressive frontal brain syndrome. Motor symptoms were much less pronounced than in our patient. Interestingly no definite signs of dementia were reported from the ancestors of the two patients. The mother of our patient died at the age of 51 years from a tumour-associated bleeding. The family reported that she became very impulsive during her last years. Her half-sister, the mother of the cousin, died at the age of 60 years from a vulva carcinoma. The grandmother of both patients had suffered some kind of personality change between 60 and 70 years of age.
Investigations
Laboratory investigations
All routinely performed laboratory examinations, copper, thyroid-stimulating hormone, thyroidal autoantibodies and a screening for autoimmune diseases by determination of tyroidal autoantibodies, antinuclear antibody, extractable nuclear antigen, antineutrophil cytoplasmic antibody , β2-microglobulin, anti cardiolipin, β2 glycoprotein antibody and immunfixation were unremarkable. Borreliosis, syphilis, toxoplasmosis, mykoplasmosis, cryptococcosis were excluded. Also antibody screening for infectious diseases by cytomegalovirus (CMV), Frühsommer-Meningoenzephalitis (FSME), HSV (herpes simples virus), hepatitis A, B and C, measles, rubella, varicella-zoster virus, HIV, human T-lymphotropic virus type I (HTLV) and John Cunningham virus was negative. Lipid and lysosomal storage diseases like GM2-gangliosidosis, Fabry disease, Schindler disease, fucosidosis, α-mannosidosis/β-mannosidosis, mucopolysacchridoses IIIB, IVB and VII, mucolipidoses II and III, GM1 gangliosidosis, galactosialidosis, metachromatic leucodystrophy, Krabbe disease and aspartylglucosaminuria were excluded by the testing of lysosomal and peroxisomal functioning.
Only a vitamin B12 and folate deficiency as well as an elevation of ferritin were found.
Apart from a slightly increased albumin ratio, CSF examination was normal for cell count, protein, and glucose and negative for oligoclonal bands and the MRZH reaction. The MRZH reaction is an antibody index which considers the neurotropic viruses measles (M), rubella (R), varizella zoster (Z), herpes simplex (H) and which is a sign of a polyspecific intrathecal immune response observed in multiple sclerosis. Antineuronal antibodies (Yo, Hu, Ri, Ma1, Ma2, CV2, amphiphysin) were all negative.
Interestingly, among markers of neurodegeneration, only τ was moderately elevated (343 pg/mL (<300 pg/mL)), whereas phospho-τ (32 pg/mL (<60 pg/mL)), Aβ1–42 (655 pg/mL (>600 pg/mL)), brain protein 14–3–3, Protein S100b (1.2 ng/mL (<2.7 ng/mL)) and NSE (9.4 ng/mL (<20 ng/mL)), were normal.
Neuroradiology
MRI presents global volume loss of brain parenchyma. The deep periventricular white matter of frontal and parietal lobe demonstrates bilateral, symmetrical, confluent abnormal high-signal intensities on fluid-attenuated inversion recovery and T2-weighted sequences sparing the subcortical U-fibers and the cerebral cortex. The parenchymal abnormalities extended into the corpus callosum presenting an extensive lesion load and thinning of all segments.
Within the areas of white matter abnormalities in the frontal lobes scattered foci of decreased diffusion with a decreased apparent diffusion coefficient were observed. No contrast enhancement with gadolinium and no areas of abnormal susceptibility were detected. The brain stem, cerebellum and spinal cord were spared (figure 1). Within a follow-up of 4 months, findings in MRI did not change significantly.
Figure 1.
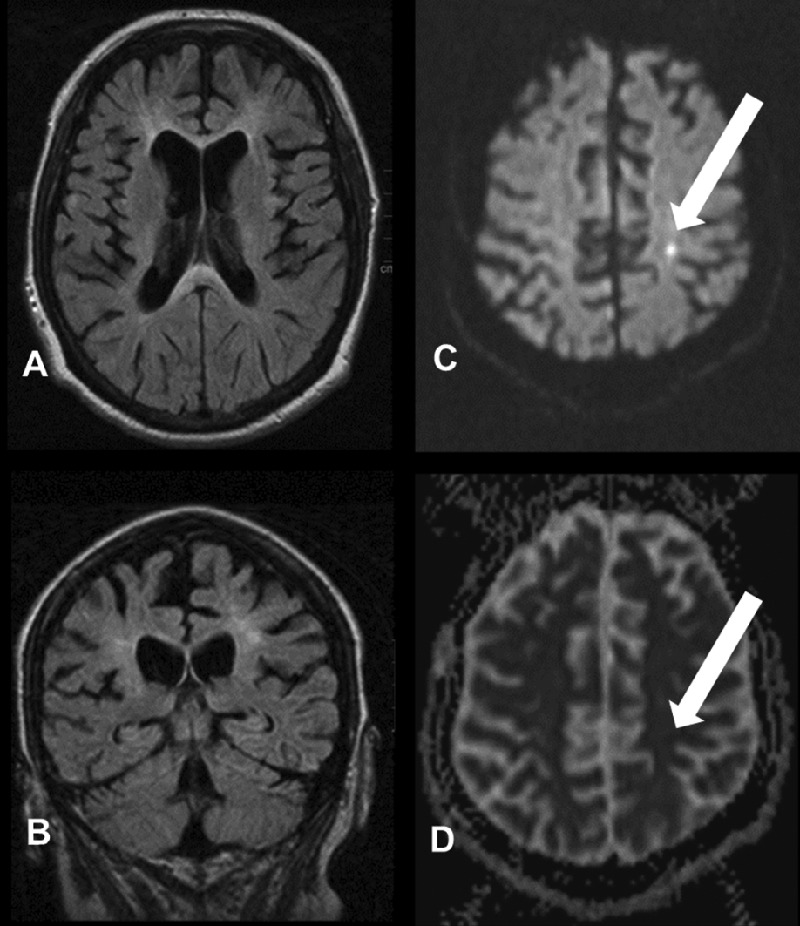
MRI findings: axial (A) and coronal (B) fluid-attenuated inversion recovery sequence showing marked atrophy and periventricular leukodystrophy. Diffusion-weighted imaging (C) reveal areas of decreased diffusion correlating with a decreased apparent diffusion coefficient (D).
Neuropathology
Microscopic examination of the tissue specimens obtained from frontal lobe biopsy showed cortical grey and subcortical white matter with pronounced pathological changes within the white matter. Here, numerous axonal swellings/spheroids could be observed. There was a faint reduction of myelinated fibres with loosening of the tissue as well as reactive gliosis. The histomorphological aspects, as well as the histochemical and immunohistochemical stainings were consistent with a neuroaxonal dystrophy. We could exclude the presence of other neurodegenerative or demyelinating diseases. Besides, there were no signs of acute inflammation or tumour (figure 2).
Figure 2.
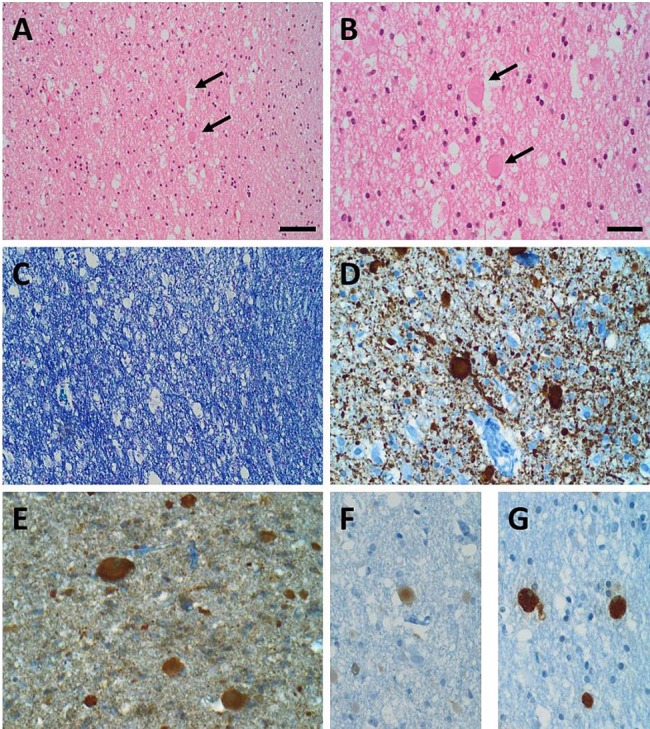
Neuropathological findings: (A and B)H&E staining showed subcortical white matter with matrix loosening containing several eosinophilic axonal swellings/spheroids (black arrows in A and B; B: higher magnification of A). (C) A myelin stain demonstrated a faint reduction of fibres within the subcortical white matter (Nissl-Luxol Fast Blue (LFB) staining). Immunohistochemical stainings (D–G): the axonal spheroids were highlighted in a neurofilament staining (D) and were immunoreactive for α-Synuclein (E) and amyloid precursor protein (F). Macrophages with CD68-immunoreactivity are shown in (G). Scale bar in A=40 μm, applies also for (C). Scale bar in (B)=20 μm, applies also for (D–G).
Outcome and follow-up
Three months after the diagnosis of HDLS, cognitive functioning of the patient was further deteriorated (MMSE: 7/30) and a dystonia of the right arm was seen. She was unable to stand or walk. Only easy commands could be followed. One month later the patient died due to acute sepsis.
Discussion
HDLS is a very rare but important differential diagnosis of leukoencephalopathies. Especially in the beginning of the disease MRI findings resemble very much that of MS. Also motor symptoms of HDLS, like increased tendon reflexes, ataxia, dystonia and dyskinesia are frequently found in patients with MS. Therefore many cases of HDLS in the literature were originally diagnosed as MS.1 6
We present the case of a patient suffering from a presenile neurodegenerative disease that was also primarily misdiagnosed as MS. The diagnosis of an adult onset neuroaxonal leukodystrophy with spheroids could only be made after brain biopsy. This entity encompasses diseases such as HDLS, pigmentary orthochromatic leukodystrophy (POLD), pantothenate kinase-associated neurodegeneration, polycystic lipomembranous osteodysplasia with sclerosing leucoencephalopathy (PLOSL/Nasu-Hakola disease) or vanishing white matter disease.7
Indicative for HDLS was the frontal brain syndrome together with MRI where scattered foci of decreased diffusion with a decreased apparent diffusion coefficient were found. This finding is unusual in other leukoencephalopathies but has repeatedly been found in HDLS.8–10
POLD, another hereditary leukodystrophy is very similar in clinical features to HDLS but can be neuropathologically distinguished by the presence of pigmented, autofluorescent glia.7 Nasu-Hakola disease, vanishing white matter disease and pantothenate kinase-associated neurodegeneration present with very similar neuropathological abnormalities but can be distinguished by radiological examinations. Characteristic for pantothenate kinase-associated neurodegeneration would be the ‘eye of the tiger’ sign in MRI representing iron deposits in the basal ganglia. Cystic degenerations of the bone and repeated fractures of extremities in the past are crucial for the diagnosis of Nasu-Hakola disease.11 On the other hand, vanishing white matter disease is characterised by degeneration of the white matter.
Interestingly, despite the rapid cognitive and functional deterioration and the advanced cortical brain atrophy, among markers of neurodegeneration and astroglial degeneration, only total-τ was moderately elevated. NSE and S100b, were not altered in the CSF. This finding fits the theory that HDLS is rather a microgliopathy or an oligodendrogliopathy than a neuropathy. This has been recently hypothesised when mutations in the CSF1R gene were found by a genome-wide association study in 14 HDLS families.12 CSF1R is a receptor expressed mainly by mononuclear phagocytes such as microglia and regulates their survival, proliferation and differentiation.
Learning points.
The diagnosis of hereditary diffuse leucoencephalopathy with spheroids (HDLS) is difficult because of the heterogeneity of symptoms and its similarity to other neurodegenerative and neuroinflammatory diseases.
The fast progressive neurodegenerative process in our case did not result in an elevation of established markers of neuronal or astrocytic damage.
Scattered foci of decreased diffusion with a decreased apparent diffusion coefficient in MRI can point to HDLS in patients with an expected neurodegenerative process.
The correct diagnosis in our case could only be made after brain biopsy. Therefore, a brain biopsy remains an essential intervention in cases of non-distinctive neurodegeneration.
Acknowledgments
The authors thank Professor Stefan Schwab, Department of Neurology, Erlangen and Professor Jürgen Winkler, Department of Molecular Neurology, Erlangen for their counselling and their critical review of the manuscript. The authors are also grateful to Professor Piotr Lewczuk Department of Psychiatry, Erlangen for providing the neurochemical diagnostics.
Footnotes
Contributors: PS was involved in the treatment of the patient and prepared the manuscript. ZK, PG, RC, IB, WB, AD and CM were also involved in the treatment of the patient. PG and AD contributed by providing expertise in neuroradiology. RC, IB and WB contributed by providing expertise in neuropathology. ZK and CM contributed by providing expertise in neurology. All authors reviewed the article carefully and gave their final approval.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Baba Y, Ghetti B, Baker MC, et al. Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol 2006;111:300–11 [DOI] [PubMed] [Google Scholar]
- 2.Wider C, Van Gerpen JA, DeArmond S, et al. Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology 2009;72:1953–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Harten AC, Kester MI, Visser PJ, et al. Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin Chem Lab Med 2011;49:353–66 [DOI] [PubMed] [Google Scholar]
- 4.Lamers KJ, Vos P, Verbeek MM, et al. Protein S-100B, neuron-specific enolase (NSE), myelin basic protein (MBP) and glial fibrillary acidic protein (GFAP) in cerebrospinal fluid (CSF) and blood of neurological patients. Brain Res Bull 2003;61:261–4 [DOI] [PubMed] [Google Scholar]
- 5.Steiner J, Bogerts B, Schroeter ML, et al. S100B protein in neurodegenerative disorders. Clin Chem Lab Med 2011;49:409–24 [DOI] [PubMed] [Google Scholar]
- 6.Keegan BM, Giannini C, Parisi JE, et al. Sporadic adult-onset leukoencephalopathy with neuroaxonal spheroids mimicking cerebral MS. Neurology 2008;70(13 Pt 2): 1128–33 [DOI] [PubMed] [Google Scholar]
- 7.Ali ZS, Van Der Voorn JP, et al. A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia—a role for oxidative damage. J Neuropathol Exp Neurol 2007;66:660–72 [DOI] [PubMed] [Google Scholar]
- 8.Mateen FJ, Keegan BM, Krecke K, et al. Sporadic leucodystrophy with neuroaxonal spheroids: persistence of DWI changes and neurocognitive profiles: a case study. J Neurol Neurosurg Psychiatry 2010;81:619–22 [DOI] [PubMed] [Google Scholar]
- 9.Maillart E, Rousseau A, Galanaud D, et al. Rapid onset frontal leukodystrophy with decreased diffusion coefficient and neuroaxonal spheroids. J Neurol 2009;256:1649–54 [DOI] [PubMed] [Google Scholar]
- 10.Freeman SH, Hyman BT, Sims KB, et al. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol 2009;19:39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paloneva J, Autti T, Raininko R, et al. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology 2001;56:1552–8 [DOI] [PubMed] [Google Scholar]
- 12.Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 2012;44:200–5 [DOI] [PMC free article] [PubMed] [Google Scholar]