

Abstract
TnpX is a site-specific recombinase responsible for the excision and insertion of the transposons Tn4451 and Tn4453 in Clostridium perfringens and Clostridium difficile, respectively. Here, we exploit phenotypic features of TnpX to facilitate genetic mutagenesis and complementation studies. Genetic manipulation of bacteria often relies on the use of antibiotic resistance genes; however, a limited number are available for use in the clostridia. The ability of TnpX to recognize and excise specific DNA fragments was exploited here as the basis of an antibiotic resistance marker recycling system, specifically to remove antibiotic resistance genes from plasmids in Escherichia coli and from marked chromosomal C. perfringens mutants. This methodology enabled the construction of a C. perfringens plc virR double mutant by allowing the removal and subsequent reuse of the same resistance gene to construct a second mutation. Genetic complementation can be challenging when the gene of interest encodes a product toxic to E. coli. We show that TnpX represses expression from its own promoter, PattCI, which can be exploited to facilitate the cloning of recalcitrant genes in E. coli for subsequent expression in the heterologous host C. perfringens. Importantly, this technology expands the repertoire of tools available for the genetic manipulation of the clostridia.
INTRODUCTION
The clostridia are a diverse group of bacteria, incorporating both pathogenic and industrially important species. Genetic manipulation of clostridia can be challenging. The recent introduction of TargeTron technology, using mobile group II introns to disrupt the gene of interest (1–3), has improved researchers' abilities to generate targeted gene disruptions; however, the tools available for use in the clostridia are still limited. Antibiotic resistance cassettes are commonly used to select for relatively rare recombination events in attempts to introduce DNA onto another DNA molecule in vivo. The number of antibiotic resistance markers available for use within a particular species or strain can sometimes be limited, restricting a researcher's ability to manipulate these strains. When using the TargeTron system, there are few antibiotic resistance retrotransposition-activated markers (RAMs) available, and only one is currently widely used in the clostridia (ermB RAM) (1, 4, 5). Without multiple resistance markers at one's disposal, the ability to remove an integrated marker for subsequent reuse becomes essential. The FLP recombinase system to remove antibiotic resistance markers (6) has been used in the nonpathogenic clostridial species Clostridium acetobutylicum (7, 8); however, to date, there have been no reports of this system being used on clinically important clostridial species. Here, we describe the use of an alternative system for marker recycling in the human and animal pathogen Clostridium perfringens based on the clostridial recombinase TnpX.
TnpX is a site-specific serine recombinase encoded by the Tn4451/53 family of clostridial mobilizable transposons (9). This family includes Tn4451 and Tn4453, which confer chloramphenicol resistance to C. perfringens (10) and Clostridium difficile (11, 12), respectively. TnpX is required for the transposition of Tn4451, excising the linear element from surrounding DNA to form a circular intermediate. TnpX then mediates the integration of the circular intermediate back into a replicating DNA molecule in a linear form (12). TnpX recognizes attL and attR sites that flank the linear transposon, leading to the excision of the element (13). The formation of the circular intermediate results in the juxtaposition of these sites to form the attCI site, which also results in the formation of a strong promoter for tnpX expression, PattCI (14). TnpX binds with high affinity to attCI (13), thereby in effect binding to PattCI (13, 14). In this study, we demonstrate the construction of a plc mutant using TargeTron technology, with the subsequent removal of the ermB RAM by TnpX. This process then allowed for the disruption of a second gene, virR, in the same strain by using the ermB RAM.
Complementation is another important aspect of classical mutagenesis studies, particularly when attempting to decipher gene function. Often, complementation may be achieved through cloning the gene of interest, either with or without its native promoter, onto a plasmid and introducing this construct into the mutant. However, attempts to clone clostridial genes in Escherichia coli are occasionally unsuccessful, as the products of these genes may be toxic to E. coli. The use of a controlled inducible expression system can overcome these difficulties. One such system is a tetracycline-inducible system developed for C. difficile which involves promoter repression to control gene expression; the addition of tetracycline relieves repression, allowing the expression of the gene of interest (15). However, if the complemented strain is then to be used in an animal infection model, induction of the gene of interest becomes more difficult. We experienced toxicity problems when attempting to clone the gene encoding a C. perfringens peptidoglycan hydrolase, TcpG, in E. coli and a gene involved in ferrous iron uptake by C. perfringens, feoB, and developed a new technique to overcome this barrier. Repression of transposon-encoded recombinase promoters by the recombinase itself is a mechanism used by mobile elements to minimize deleterious effects on the host by limiting recombinase expression and, thus, element movement (16). In this study, we demonstrate that TnpX is able to repress expression from its own promoter, PattCI. We then used this ability to repress expression from PattCI to facilitate the cloning of tcpG and feoB in E. coli. By placing these genes under the control of PattCI and repressing their expression with TnpX, we avoided the production of toxic products in E. coli and were able to generate the required recombinant plasmids for complementation. Subsequent transfer of the cloned genes into C. perfringens tcpG or feoB mutants resulted in the expression of the target gene for successful complementation analysis.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Bacterial strains and plasmids are listed in Table 1. E. coli strains were grown at 37°C in 2× YT medium (27) and supplemented with antibiotic selection where appropriate, using ampicillin (100 μg/ml), kanamycin (20 μg/ml), or chloramphenicol (30 μg/ml). C. perfringens strains were cultured at 37°C in TPG (Trypticase-peptone-glucose) broth (28), FTG (fluid thioglycolate) medium (Difco/BD, Sparks, MD, USA), or nutrient agar (NA) (29) supplemented with chloramphenicol (30 μg/ml), erythromycin (50 μg/ml), or tetracycline (10 μg/ml) when required. C. perfringens cultures were grown in an atmosphere of 10% H2, 10% CO2, and 80% N2 at 37°C in anaerobic jars (Oxoid, Basingstoke, United Kingdom).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Characteristic(s)a | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| CB454 | F− ΔlacZ lacY+ galK rpsL thi recA56; Smr | 17 |
| DH5α | F− ϕ80dlacZΔM15 Δ(lacZYA argF)U169 endA1 recA1 hsdR17 (rK− mK−) deoR thi-1 supE44 gyrA96 relA1 | Bethesda Research Laboratories |
| DH12S | ϕ80dlacZΔM15 mcrA Δ(mrr-hsdRMS-mcrBC) araD139 Δ(ara, leu)7697 Δ(lacX74 galU galK rpsL (Strr) nupG recA1/F′ proAB+ lacIqZΔM15 | Life Technologies |
| C. perfringens | ||
| JIR325 | Strain 13; Rifr Nalr | 18 |
| JIR12483 | JIR325 plc::TargetTron; Emr | This study |
| JIR12531 | JIR12483(pDLL3); Emr Cmr | This study |
| JIR12564 | JIR12531 cured of pDLL3; Emr Cms | This study |
| JIR12594 | JIR12483(pDLL3), second independent derivative; Emr Cmr | This study |
| JIR12597 | JIR12594 cured of pDLL3; Emr Cms | This study |
| JIR12600 | JIR12594 virR::TargeTron (plc viR double mutant); Emr | This study |
| JIR12128 | JIR325 (pCW3ΔtcpG::ermQ) | 19 |
| Plasmids | ||
| pCR-Blunt II-TOPO | Cloning vector; Knr | Invitrogen |
| pCW3 | Conjugative plasmid from C. perfringens; Tcr | 20 |
| pSU39 | Cloning vector; Knr | 21 |
| pJIR750 | E. coli-C. perfringens shuttle vector; Cmr | 22 |
| pJIR1456 | E. coli-C. perfringens mobilizable shuttle vector; Cmr | 23 |
| pJIR1457 | E. coli-C. perfringens mobilizable shuttle vector; Emr | 23 |
| pJIR1635 | pJIR1457 carrying tnpX; Emr | 13 |
| pJIR1835 | pCB182ΩattCI from Tn4453a; Apr | 14 |
| pJIR1872 | pSU39ΩtnpX; Knr | 12 |
| pJIR2085 | pSU39ΩtnpXS15L; Knr | 24 |
| pJIR2816 | E. coli-C. perfringens mobilizable shuttle vector; Cmr | 25 |
| pJIR3422 | pJIR750ΩattCI; Cmr | This study |
| pJIR3562 | TargeTron vector targeted to interrupt C. perfringens plc; erm RAM; Cmr | 26 |
| pJIR3589 | pCR-Blunt II-TOPOΩattL-catP-attR fragment generated by SOE-PCR; Cmr Knr | This study |
| pJIR3607 | pJIR3678 retargeted to virR of C. perfringens; Cmr | This study |
| pJIR3629 | pJIR3422ΩtcpG; Cmr | 19 |
| pJIR3678 | pJIR3562 containing erm RAM with flanking attL and attR sites; Cmr | This study |
| pJIR3683 | pJIR3422ΩfeoB; Cmr | This study |
| pDLL3 | TnpX subcloned from pJIR1635 into pJIR2816; Cmr | This study |
Cmr, chloramphenicol resistant; Cms, chloramphenicol sensitive; Apr, ampicillin resistant; Knr, kanamycin resistant; Smr, streptomycin resistant; Rifr, rifampin resistant; Nalr, nalidixic acid resistant.
DNA isolation and molecular techniques.
Plasmid DNA from E. coli was extracted by using QIAprep spin miniprep columns (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Genomic DNA from C. perfringens was prepared from FTG cultures grown overnight, as described previously (25).
Transformation of E. coli (30) and C. perfringens (31) was performed as described previously. Standard methods for the digestion, ligation, and analysis of plasmid DNA and PCR products were used (30).
PCR was performed by using Taq DNA polymerase (Roche Diagnostics, Mannheim, Germany), unless otherwise stated, and 0.5 μM each primer. Denaturation (94°C for 30 s), annealing (50°C for 30 s), and extension (72°C for 3 to 5 min) steps were carried out for 30 to 35 cycles. DNA sequencing was carried out by using a Prism Ready Reaction DyeDeoxy Terminator cycle sequencing kit (Applied Biosystems/Life Technologies, Carlsbad, CA, USA), according to the manufacturer's instructions. The oligonucleotide primers used in this study are listed in Table 2. Sequence analysis was carried out on an Applied Biosystems 3730S genetic analyzer and by using Sequencher 3.1 software (Gene Codes Corporation, Ann Arbor, MI, USA).
TABLE 2.
Oligonucleotide primers used in this study
| Oligonucleotide | Sequence (5′–3′) | Purpose | Reference |
|---|---|---|---|
| JRP3803 | GCGCGCAAGCTTTGCGAAGAAGCCTTTGAATAC | Amplification of PattCI for cloning into pJIR750 | This study |
| JRP3804 | GCGCGCGCATGCAGTCCTTGACATTTTTCTTAT | Amplification of PattCI for cloning into pJIR750 | This study |
| JRP3820 | GCGCGGATCCCCTTAAACTATGAGGATGTT | Amplification of tcpG and the upstream region; includes the BamHI site | 19 |
| JRP3821 | GCGCGGTACCTTATTCTACGACTCTTCTTACA | Amplification of tcpG and the upstream region; includes the KpnI site | 19 |
| JRP4321 | GCGGTACCTTCCTTTTGAAGCACATCCACTG | Amplification of feoB and the upstream region; includes the KpnI site | This study |
| JRP4322 | CGGGATCCGAATAATTTAATTAGGGGGAAAACTAATGAC | amplification of feoB and the upstream region; includes the BamHI site | This study |
| JRP4313 | TTGATGCAGGACGATTTGATTG | Forward oligonucleotide for tcpG qRT-PCR analysis | This study |
| JRP4314 | TTTCTCTTGGCCCTAACTGTATTCC | Reverse oligonucleotide for tcpG qRT-PCR analysis | This study |
| JRP2479 | CCATCTGTTTTTATATCTGCTCCAGTA | rpoA, forward oligonucleotide for qRT-PCR analysis | 32 |
| JRP2480 | GGAAGGTGAAGGACCAAAAACTATT | rpoA, reverse oligonucleotide for qRT-PCR analysis | 32 |
| JRP5715 | GTCGGAGAATACAAAATCTC | Amplification of attL from Tn4451 for SOE-PCR | This study |
| JRP5716 | CTTCTTGAACACTTGCCGACCTTTCCGTATGTATTTTTTC | Amplification of attL and SOE-PCR to splice together the attL site with the 5′ end of catP | This study |
| JRP5717 | GAAAAAATACATACGGAAAGGTCGGCAAGTGTTCAAGAAG | SOE-PCR to splice together the attL site with the 5′ end of catP | This study |
| JRP4071 | GTCCGTAAAGAAATCGCCCATTAGTTACAGACAAACCTGAAG | SOE-PCR to splice together the attR site with the 3′ end of catP | This study |
| JRP4072 | CTTCAGGTTTGTCTGTAACTAATGGGCGATTTCTTTACGGAC | SOE-PCR to splice together the attR site with the 3′ end of catP | This study |
| JRP5720 | CACCTATTTTAAAGGTTCC | Amplification of attR from Tn4451 for SOE-PCR | This study |
| JRP2142 | CTCAGTACTGAGAGGGAACTTAGATGGTAT | catP probe for Southern blotting | 33 |
| JRP2143 | CCGGGATCCTTAGGGTAACAAAAAACACC | catP probe for Southern blotting | 33 |
| JRP2980 | AATAAGTAAACAGGTAACGTCT | ermB probe for Southern blotting | 34 |
| JRP2981 | GCTCCTTGGAAGCTGTCAGTAG | ermB probe for Southern blotting | 34 |
| JRP3947 | GGGAACGAAACGAAAGCG | TargeTron probe for Southern blotting | 26 |
| JRP3948 | CGTAATAAATATCTGGAC | TargeTron probe for Southern blotting | 26 |
Construction of the repression shuttle plasmid pJIR3422.
The PattCI promoter fragment was generated by PCR amplification using oligonucleotide primers JRP3803 and JRP3804 to generate a 225-bp product flanked with HindIII and SphI sites. Plasmid pJIR3422 was constructed by cloning the digested PCR product into HindIII/SphI-digested pJIR750 DNA. The plasmid was confirmed by restriction and sequence analysis.
β-Galactosidase assays.
β-Galactosidase assays were conducted as previously described (14, 27). The optical density at 600 nm (OD600) values of cultures grown overnight were adjusted to 0.6. An equal volume of Z buffer (500 μl) (60 mM Na2HPO4 · 7H2O, 40 mM NaH2PO4 · H2O, 10 mM KCl, 1 mM MgSO4 · 7H2O, 50 mM β-mercaptoethanol [pH 7.0]) was added to the diluted cultures, followed by 2 drops from a Pasteur pipette of chloroform and 1 drop of 0.1% (wt/vol) SDS. The sample was mixed, and 100-μl aliquots were added to a microtiter plate and incubated at room temperature. After a 15-min incubation, 200 μl of a 0.7-mg/ml o-nitrophenyl-β-d-galactopyranoside (ONPG) (in Z buffer) solution was added to the microtiter plate. Optical density measurements were made at 410 nm and 570 nm for up to 5 h. Miller units were calculated as {1,000 × [OD410 − (1.75 × OD570)]}/(time × volume × OD600), as previously described (14).
Determination of dependence on the presence of TnpX for stable maintenance of the TcpG expression plasmid.
In separate experiments, plasmid DNA was extracted from strains DH12S(pJIR3422, pJIR2085), DH12S(pJIR3683, pJIR2085), and DH12S(pJIR3629, pJIR2085) in triplicate. Plasmid DNA from each strain was introduced into DH5α, and transformants were selected on chloramphenicol-containing medium. The resultant colonies were patched onto medium containing kanamycin and chloramphenicol, and the number of chloramphenicol- and kanamycin-resistant colonies was calculated.
RNA isolation and qRT-PCR.
Total RNA was obtained from C. perfringens strain JIR325 derivatives grown in 20 ml TPG to an optical density (at 600 nm) of 0.9 to 1.0. Cells were pelleted and stored in RNAwiz (Ambion/Life Technologies, Carlsbad, CA, USA) overnight. RNA extraction was performed by using a Ribopure Bacteria kit (Ambion) according to the manufacturer's instructions. RNA was treated for DNA contamination by using Turbo DNase (Ambion) and was determined to be DNA free by the failure to amplify the tet(A)P gene present on pCW3 (data not shown). cDNA was amplified from 4 μg of RNA by using random hexamers, as described previously (32, 33, 35). Quantitative PCR using cDNA was performed as described previously (32, 33, 35). The oligonucleotides used for reverse transcription-quantitative PCR (qRT-PCR) are listed in Table 2.
Splicing by overlap extension PCR.
Splicing by overlap extension PCR (SOE-PCR) (36) was used to generate att-catP and att-ermB resistance cassettes. The oligonucleotide primer sequences used for SOE-PCR are described in Table 2. Phusion polymerase (Finnzymes/New England BioLabs, Ipswich, MA, USA) was used for all SOE-PCRs, according to the manufacturer's instructions. Primary PCR mixtures were gel purified by using the QIAquick gel extraction kit (Qiagen) before use as the templates in secondary PCRs. Secondary PCRs were performed by using 0.1 pmol of each of the purified primary PCR mixtures as the template.
Oligonucleotide primers were designed to splice together the attL site from Tn4451 and the 5′ end of the catP gene and then the 3′ end of the catP gene and attR from Tn4451. This product was subsequently cloned into pCR-Blunt II-TOPO (Invitrogen/Life Technologies, Carlsbad, CA, USA) to generate pJIR3589.
To introduce attL and attR sites flanking the ermB RAM into pJIR3678, SOE-PCR was used to introduce an MluI site between the fused attL and attR sites, which were also flanked by AscI sites. The AscI-attL-MluI-attR-AscI fragment was introduced into the vector pJIR3562 by digestion with MluI to remove the existing ermB RAM in this vector. The ermB RAM was subsequently reinserted between the att sites by digestion with MluI and ligation of the MluI ermB RAM fragment from pJIR3562 to construct pJIR3678.
Construction of plc TargeTron mutants and removal of the att-ermB cassette.
Strains containing the group II intron derived from pJIR3678 were constructed as described previously (26). To remove the att-ermB cassette from the integrated group II intron, the tnpX gene, carried by plasmid pDLL3, was introduced into a plc-negative, erythromycin-resistant strain, designated JIR12483, via electroporation (37). The resultant strains, JIR12531 and JIR12594, were plated onto NA containing chloramphenicol to select for pDLL3 and subsequently onto NA containing erythromycin or chloramphenicol. Erythromycin-susceptible derivatives were obtained and passaged in the absence of chloramphenicol to cure the strains of pDLL3. Loss of the plasmid was determined by patching onto medium containing chloramphenicol to identify strains that were no longer resistant. Southern hybridization analysis was subsequently conducted to illustrate the genomic content of the derived strains at each stage. A plc virR double mutant was constructed by introducing plasmid pJIR3607 into the strains and selecting for erythromycin-resistant colonies. Further characterization involved antibiotic resistance screening to demonstrate chloramphenicol sensitivity and PCR to confirm the interruption of the virR gene and the loss of pJIR3607.
Southern hybridization.
Randomly digoxigenin (DIG)-labeled probes were generated by PCR according to the manufacturer's instructions (Roche), using the oligonucleotide primers listed in Table 2. Genomic DNA was extracted from C. perfringens cultures, digested with HaeII, and separated by electrophoresis at a low voltage of 30 V overnight on a 0.8% agarose gel, alongside DIG-labeled HindIII-digested Lambda standards. DNA transfer and high-stringency washes were conducted as described previously (30). Detection of DIG-labeled probes was performed according to the manufacturer's instructions (Roche), using CDP-Star (Roche) as the chemiluminescent substrate.
RESULTS AND DISCUSSION
TnpX-mediated excision of antibiotic resistance cassettes allows for the reuse of resistance markers.
TnpX-mediated excision of Tn4451 relies on the recognition of attL and attR sites flanking the transposon (13, 14). Thus, we introduced the attL and attR sites on either side of the catP gene, which confers chloramphenicol resistance, by using splicing by overlap extension PCR (SOE-PCR). The resultant att-catP resistance cassette was cloned into pCR-Blunt II-TOPO (Invitrogen) (encoding kanamycin resistance) to produce pJIR3589. In the presence of TnpX, we would expect to see a loss of chloramphenicol resistance, as TnpX excises the att-catP cassette from pJIR3589. To demonstrate this property, we initially performed an excision assay with the E. coli host before attempting this strategy in C. perfringens. The TnpX-carrying vector pJIR1635 (encoding erythromycin resistance) was introduced into DH5α(pJIR3589). The resultant strain was grown overnight in the presence of kanamycin and erythromycin, to allow for the loss of the att-catP fragment, and plasmid DNA was then extracted. This plasmid DNA was then reintroduced into DH5α, selecting transformants on kanamycin only. Colonies were patched onto chloramphenicol and kanamycin to determine the percent loss of the att-catP cassette. This experiment was also performed by using pJIR1457 instead of pJIR1635; pJIR1457 encodes erythromycin resistance but does not carry the tnpX gene. In the absence of TnpX, there was no detectable loss of chloramphenicol resistance. However, in the presence of TnpX, the frequency of loss of chloramphenicol resistance was 10.7% ± 3.7%, demonstrating TnpX-mediated excision of the att-catP cassette.
We next decided to test this system further in C. perfringens, coupled with TargeTron technology. The attL and attR sites were introduced on either side of the ermB RAM to produce an att-ermB cassette. The ermB RAM is used to select for the integration of a TargeTron derivative into a gene of interest. Following the construction of the mutant via TargeTron insertion, the introduction of the TnpX-encoding gene onto a plasmid allows the att-ermB cassette to be removed. The plasmid encoding TnpX can then be lost via passage, while the group II intron (disrupting the gene of interest) is still present and stable but no longer marked with an antibiotic resistance gene. We used this method to introduce mutations into both the plc and virR genes of C. perfringens strain JIR325. The plc gene was chosen as it encodes alpha-toxin, a phospholipase C; colonies that no longer express functional alpha-toxin can be screened for a loss of phospholipase activity on egg yolk agar (EYA) (37). The virR gene encodes a global regulator that controls the expression of the cholesterol-dependent cytolysin perfringolysin O (PFO), and mutants of this gene no longer demonstrate hemolytic activity on horse blood agar (HBA) (26, 38).
Initially, the plc gene in strain JIR325 was disrupted with the TargeTron insertion containing the att-ermB RAM. Potential mutants were screened for erythromycin resistance and for a loss of phospholipase C activity. The integration of the TargeTron insertion into plc and the loss of the TargeTron vector were subsequently confirmed by Southern hybridization (strain designated JIR12483) (Fig. 1, lanes 1 and 2 in each panel). Plasmid pDLL3, carrying the tnpX gene, was introduced into this plc mutant via electroporation. Transformants were selected on chloramphenicol (resistance encoded by catP on pDLL3) and erythromycin (resistance encoded on the TargeTron vector). Colonies were then selected and grown overnight before plating out for single colonies on nutrient agar (NA) with chloramphenicol. To check for the loss of the att-ermB cassette, single colonies were patched onto NA with and without erythromycin. The frequency of excision of att-ermB was 1%. One strain found to be erythromycin sensitive was confirmed by Southern hybridization (JIR12531) (Fig. 1, third panel, lane 3). This strain was further passaged to select for chloramphenicol-sensitive colonies cured of pDLL3 and confirmed by Southern hybridization (JIR12564) (Fig. 1, third panel, lane 4). An identical independent unmarked plc mutant was also constructed and confirmed by using the same methodology, designated JIR12597 (data not shown), and used for subsequent experiments.
FIG 1.

Southern hybridization analysis of C. perfringens plc mutants. Genomic DNA was prepared from C. perfringens strains and digested with HaeII. Digested DNA was separated by electrophoresis in a 0.8% agarose gel, transferred, and probed with the respective probes indicated below each panel. M indicates the DIG-labeled HindIII-digested lambda marker lanes, and sizes are indicated on the left. Lanes contained DNA from the following strains: JIR325 (wild type) (lane 1), JIR12483 (plc mutant) (lane 2), JIR12531 (plc mutant with the att-erm cassette excised by TnpX, encoded by pDLL3) (lane 3), and JIR12531 cured of pDLL3, designated JIR12564 (lane 4). The lettered arrows on the right indicate the plc gene containing the intron with the ermB gene (A), the plc gene containing the inserted intron without the ermB gene (B), the wild-type plc gene (C), and the pDLL3 fragment containing the catP gene (D).
The unmarked plc mutant strain JIR12597 was used for experiments in which a second TargeTron mutation was introduced, into the regulator gene virR. Putative mutants were isolated and confirmed by PCR and Southern analyses (data not shown). Plasmid pDLL3 was then introduced into the double mutant strain for TnpX-mediated excision of the att-ermB RAM from the inactivated virR gene. The frequency of excision of att-ermB from the double mutant was 5%, providing proof of principle that the TnpX-mediated excision strategy employed here resulted in the construction of an unmarked double mutant, designated JIR12600. Patching onto HBA confirmed the loss of hemolytic activity in the double mutant compared to single mutant strain JIR12597, as expected (data not shown).
In this study, we have used the excision properties of TnpX to facilitate the removal of antibiotic resistance cassettes, flanked by attL and attR sites, from a plasmid in E. coli and from chromosomal mutants constructed in C. perfringens. While providing evidence that TnpX-mediated excision can be used to remove antibiotic resistance markers, the initial proof of principle using E. coli also demonstrated that the system works outside a clostridial genetic background. This result suggests that the TnpX-mediated excision system could be applied to species other than clostridia, provided that TnpX can be introduced and expressed in that strain background. Although the frequency of excision was not high in C. perfringens (1 to 5%), it was sufficient to ensure that arduous amounts of screening were not required to obtain colonies in which excision of the resistance element had occurred.
TnpX represses expression from its own promoter, PattCI.
Attempts to clone clostridial genes in E. coli are occasionally unsuccessful, as the products of these genes may be toxic to E. coli. Recombinases may repress transcription from their own promoter, in order to limit recombinase expression and, thus, element movement, which may be detrimental to the host cell (16). Whether TnpX represses its own expression was not known. However, we hypothesized that if TnpX did repress expression from PattCI, we could exploit this repression to enable the cloning of genes encoding toxic products in E. coli.
To determine the effect of TnpX on PattCI, we used a β-galactosidase reporter assay. In a previous study, we cloned PattCI into the promoter probe vector pCB182, constructing pJIR1835 (14). To introduce TnpX into this system, we used pJIR2085, which contains a tnpXS15L allele, the product of which is catalytically inert (24), as the use of a plasmid expressing wild-type TnpX in E. coli resulted in the integration of the plasmid into the E. coli chromosome, disrupting PattCI (data not shown). The two plasmids pJIR1835 and pJIR2085 were introduced into E. coli CB454 (17), and β-galactosidase assays were performed as described previously (14, 27). In the absence of TnpXS15L, activity from PattCI resulted in 4,600 ± 290 Miller units of β-galactosidase activity; however, when TnpXS15L was introduced into this system, the levels of β-galactosidase activity decreased to 34 ± 21 Miller units (Fig. 2). These data suggest that TnpX very efficiently represses expression from its own promoter.
FIG 2.
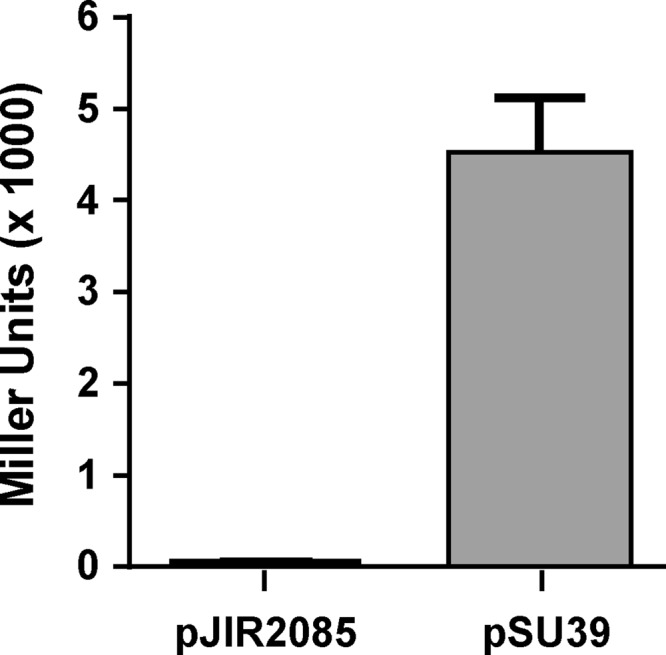
TnpX-mediated repression of PattCI. β-Galactosidase assays were performed by using strains CB454(pJIR2085, pJIR1835), labeled pJIR2085, and CB454(pSU39, pJIR1835), labeled pSU39. Assays were performed as described previously (14), with data represented in Miller units. Data are the averages of three biological replicates. Error bars represent standard deviations.
The reporter experiments described here show that the catalytically inactive derivative TnpXS15L repressed expression from PattCI and support our previous findings; strong binding of TnpX to the PattCI region presumably blocks RNA polymerase binding to the promoter and results in reduced transcription initiated from this promoter.
TnpXS15L-mediated repression can facilitate cloning of recalcitrant genes in E. coli.
Having established that TnpXS15L represses expression from PattCI, we moved on to test whether this system could be used for the cloning of recalcitrant genes in E. coli. We tested this approach for the cloning of tcpG, which encodes a peptidoglycan hydrolase from C. perfringens (19). For classical mutagenesis studies, a tcpG mutant was made, and complementation was required to confirm the mutant phenotype. Many attempts at cloning tcpG had been made, but all were unsuccessful, resulting in the isolation of truncated or mutated DNA fragments (data not shown), suggesting that the production of TcpG was not tolerated by E. coli.
The TnpX-repressible PattCI fragment was cloned into the clostridial vector pJIR750 to construct shuttle plasmid pJIR3422. The tcpG gene, together with 25 bp of upstream DNA that included the ribosome binding site, was amplified by PCR. A ligation mixture containing the tcpG PCR product and pJIR3422 was introduced into E. coli strain DH12S, which already contained TnpXS15L plasmid pJIR2085. The resultant TcpG expression plasmid was designated pJIR3629 and was confirmed by restriction digestion and sequence analysis (data not shown).
To investigate the maintenance of pJIR3629 in the absence of TnpX in E. coli, plasmid DNA was extracted from DH12S(pJIR3629, pJIR2085) and used to transform DH5α. Transformants were selected on chloramphenicol to select for transformants carrying TcpG plasmid pJIR3629. Subsequent colonies were then patched onto medium containing either chloramphenicol or kanamycin (resistance encoded by tnpX-carrying plasmid pJIR2085) to determine the proportion of transformants containing pJIR3629 alone or both pJIR3629 and pJIR2085. All of these colonies were resistant to both kanamycin and chloramphenicol, demonstrating that they all contained both pJIR3629 and TnpXS15L plasmid pJIR2085. No colonies that contained the TcpG expression plasmid alone were isolated. As a control, plasmid DNA from DH12S(pJIR3422, pJIR2085) was also used to transform DH5α, with selection on chloramphenicol for the vector pJIR3422 and subsequent patching onto chloramphenicol and kanamycin. All of the colonies were chloramphenicol resistant, while 67% ± 5% were resistant to both chloramphenicol and kanamycin, suggesting cotransformation of both plasmids in this proportion of transformants. These data suggest that TcpG expression plasmid pJIR3629 can be stably maintained only in the presence of TnpXS15L-mediated repression, while the base vector pJIR3422 can be maintained in the absence of tnpX-carrying plasmid pJIR2085. This finding further demonstrates the toxicity of TcpG to E. coli, which is consistent with its function as a peptidoglycan hydrolase (19).
Plasmid pJIR3629 was then introduced into a C. perfringens tcpG mutant, which showed a decrease in the conjugative transfer of C. perfringens plasmid pCW3 compared to the wild type. The introduction of pJIR3629 into this mutant, without TnpXS15L-mediated repression, restored the conjugative transfer levels back to that of the wild-type strain (19). To confirm the expression of tcpG from pJIR3629 in the complemented derivative, the production of tcpG mRNA was examined by using qRT-PCR. RNA was extracted from the wild-type strain [JIR325(pCW3)] and the tcpG-complemented strain [JIR325(pCW3ΔtcpG, pJIR3629)]. Levels of tcpG transcript were determined relative to rpoA transcript levels (Fig. 3). The results indicated that tcpG transcript levels were 6.2 ± 2.5 times those of rpoA in the complemented mutant strain, demonstrating high-level transcription of tcpG from pJIR3629 in C. perfringens. In the wild-type strain, where tcpG is carried on plasmid pCW3, tcpG transcript levels were 0.005 ± 0.0008 relative to those of rpoA (Fig. 3).
FIG 3.
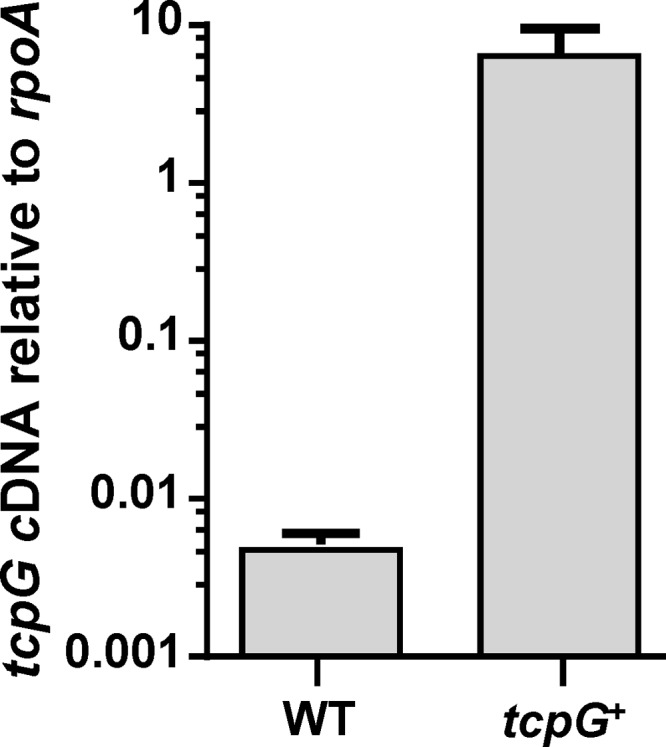
Transcriptional analysis of tcpG expression. Transcription of mRNA from the tcpG gene was quantified by using qRT-PCR, in comparison to rpoA expression levels from each corresponding strain. WT is wild-type strain JIR325(pCW3), and tcpG+ is complemented tcpG mutant strain JIR325(pCW3tcpG, pJIR3629). Results are the averages of three biological replicates, and the error bars represent standard deviations.
Having demonstrated the effectiveness of the TnpXS15L repression system in the cloning of tcpG, we subsequently used the system to clone another clostridial gene, feoB, the product of which is involved in the uptake of ferrous ions (39, 40). Again needed for complementation studies, many unsuccessful attempts were made to clone the gene, with or without its native promoter. However, the gene was successfully cloned here by using pJIR3422 in the presence of TnpXS15L. The requirement of TnpXS15L for the maintenance of FeoB expression plasmid pJIR3683 in E. coli was assessed as described above for pJIR3629. All colonies were resistant to both kanamycin and chloramphenicol, demonstrating that all colonies contained both pJIR3683 and pJIR2085. Thus, as we observed for the TcpG expression plasmid, FeoB plasmid pJIR3683 was successfully introduced into DH5α only in the presence of TnpXS15L encoded on pJIR2085. In contrast, DH5α transformed with the vector control pJIR3422 demonstrated resistance to kanamycin only 76% ± 9% of the time, indicating that pJIR2085 was not cotransformed every time in the absence of selection and that pJIR3422 could be stably maintained in the absence of TnpXS15L. The FeoB plasmid was subsequently shown to complement a C. perfringens strain JIR325 feoB mutant and to restore ferrous iron uptake (data not shown) (M. M. Awad, J. K. Cheung, D. Lyras, and J. I. Rood, unpublished results).
The exploitation of TnpX-mediated repression for successful cloning of otherwise recalcitrant genes represents an important addition to the C. perfringens genetic arsenal. Coupled with the ability of TnpX to specifically excise fragments such as antibiotic resistance markers, we have developed tools that will further enable the genetic modification of the clostridia. The construction of a plc virR double mutant in C. perfringens and the cloning of the tcpG and feoB genes into E. coli provide direct practical examples of how this technology can be used for the construction and subsequent analysis of clostridial mutants. One clear advantage of this system is that it does not require the expression of the cloned gene to be induced in the clostridial background. While other systems which facilitate the control of gene expression via the controlled alleviation of promoter repression are available (15, 41, 42), they often require the addition of specific compounds to cultures to achieve this outcome. This requirement may not be ideal and may interfere with experimental protocols, such as when conducting infection studies with animal models. The technology described here provides new options that may be suitable for these purposes. Furthermore, while we have demonstrated proof of principle that this TnpX-centered strategy is effective for clostridial genes, this system should provide a feasible method for the cloning of recalcitrant genes from other bacterial species.
ACKNOWLEDGMENTS
Funding was provided by grants from the Australian National Health and Medical Research Council, Cancer Council Victoria, and the Australian Research Council.
Footnotes
Published ahead of print 28 March 2014
REFERENCES
- 1.Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron: a universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods 70:452–464. 10.1016/j.mimet.2007.05.021 [DOI] [PubMed] [Google Scholar]
- 2.Zhong J, Karberg M, Lambowitz AM. 2003. Targeted and random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Res. 31:1656–1664. 10.1093/nar/gkg248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, McClane BA, Fisher DJ, Rood JI, Gupta P. 2005. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron. Appl. Environ. Microbiol. 71:7542–7547. 10.1128/AEM.71.11.7542-7547.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuehne SA, Minton NP. 2012. ClosTron-mediated engineering of Clostridium. Bioengineered 3:247–254. 10.4161/bioe.21004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackin KE, Carter GP, Howarth P, Rood JI, Lyras D. 2013. Spo0A differentially regulates toxin production in evolutionarily diverse strains of Clostridium difficile. PLoS One 8:e79666. 10.1371/journal.pone.0079666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 7.Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. 2010. The ClosTron: mutagenesis in Clostridium refined and streamlined. J. Microbiol. Methods 80:49–55. 10.1016/j.mimet.2009.10.018 [DOI] [PubMed] [Google Scholar]
- 8.Al-Hinai MA, Fast AG, Papoutsakis ET. 2012. Novel system for efficient isolation of Clostridium double-crossover allelic exchange mutants enabling markerless chromosomal gene deletions and DNA integration. Appl. Environ. Microbiol. 78:8112–8121. 10.1128/AEM.02214-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams V, Lyras D, Farrow K, Rood J. 2002. The clostridial mobilisable transposons. Cell. Mol. Life Sci. 59:2033–2043. 10.1007/s000180200003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abraham LJ, Rood JI. 1987. Identification of Tn4451 and Tn4452, chloramphenicol resistance transposons from Clostridium perfringens. J. Bacteriol. 169:1579–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyras D, Storie C, Huggins AS, Crellin PK, Bannam TL, Rood JI. 1998. Chloramphenicol resistance in Clostridium difficile is encoded on Tn4453 transposons that are closely related to Tn4451 from Clostridium perfringens. Antimicrob. Agents Chemother. 42:1563–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyras D, Adams V, Lucet I, Rood JI. 2004. The large resolvase TnpX is the only transposon-encoded protein required for transposition of the Tn4451/3 family of integrative mobilizable elements. Mol. Microbiol. 51:1787–1800. 10.1111/j.1365-2958.2003.03950.x [DOI] [PubMed] [Google Scholar]
- 13.Adams V, Lucet IS, Lyras D, Rood JI. 2004. DNA binding properties of TnpX indicate that different synapses are formed in the excision and integration of the Tn4451 family. Mol. Microbiol. 53:1195–1207. 10.1111/j.1365-2958.2004.04198.x [DOI] [PubMed] [Google Scholar]
- 14.Lyras D, Rood J. 2000. Transposition of Tn4451 and Tn4453 involves a circular intermediate that forms a promoter for the large resolvase, TnpX. Mol. Microbiol. 38:588–601. 10.1046/j.1365-2958.2000.02154.x [DOI] [PubMed] [Google Scholar]
- 15.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J. Biol. Chem. 286:27483–27493. 10.1074/jbc.M111.263889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahillon J, Chandler M. 1998. Insertion sequences. Microbiol. Mol. Biol. Rev. 62:725–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider K, Beck CF. 1986. Promoter-probe vectors for the analysis of divergently arranged promoters. Gene 42:37–48. 10.1016/0378-1119(86)90148-4 [DOI] [PubMed] [Google Scholar]
- 18.Lyristis M, Bryant AE, Sloan J, Awad MM, Nisbet IT, Stevens DL, Rood JI. 1994. Identification and molecular analysis of a locus that regulates extracellular toxin production in Clostridium perfringens. Mol. Microbiol. 12:761–777. 10.1111/j.1365-2958.1994.tb01063.x [DOI] [PubMed] [Google Scholar]
- 19.Bantwal R, Bannam TL, Porter CJ, Quinsey NS, Lyras D, Adams V, Rood JI. 2012. The peptidoglycan hydrolase TcpG is required for efficient conjugative transfer of pCW3 in Clostridium perfringens. Plasmid 67:139–147. 10.1016/j.plasmid.2011.12.016 [DOI] [PubMed] [Google Scholar]
- 20.Abraham LJ, Rood JI. 1985. Cloning and analysis of the Clostridium perfringens tetracycline resistance plasmid, pCW3. Plasmid 13:155–162. 10.1016/0147-619X(85)90038-1 [DOI] [PubMed] [Google Scholar]
- 21.Bartolomé B, Jubete Y, Martinez E, de la Cruz F. 1991. Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322 and its derivatives. Gene 102:75–78. 10.1016/0378-1119(91)90541-I [DOI] [PubMed] [Google Scholar]
- 22.Bannam TL, Rood JI. 1993. Clostridium perfringens-Escherichia coli shuttle vectors that carry single antibiotic resistance determinants. Plasmid 29:233–235 [DOI] [PubMed] [Google Scholar]
- 23.Lyras D, Rood JI. 1998. Conjugative transfer of RP4-oriT shuttle vectors from Escherichia coli to Clostridium perfringens. Plasmid 39:160–164. 10.1006/plas.1997.1325 [DOI] [PubMed] [Google Scholar]
- 24.Adams V, Lucet IS, Tynan FE, Chiarezza M, Howarth PM, Kim J, Rossjohn J, Lyras D, Rood JI. 2006. Two distinct regions of the large serine recombinase TnpX are required for DNA binding and biological function. Mol. Microbiol. 60:591–601. 10.1111/j.1365-2958.2006.05120.x [DOI] [PubMed] [Google Scholar]
- 25.O'Connor JR, Lyras D, Farrow KA, Adams V, Powell DR, Hinds J, Cheung JK, Rood JI. 2006. Construction and analysis of chromosomal Clostridium difficile mutants. Mol. Microbiol. 61:1335–1351. 10.1111/j.1365-2958.2006.05315.x [DOI] [PubMed] [Google Scholar]
- 26.Cheung JK, Keyburn AL, Carter GP, Lanckriet AL, Van Immerseel F, Moore RJ, Rood JI. 2010. The VirSR two-component signal transduction system regulates NetB toxin production in Clostridium perfringens. Infect. Immun. 78:3064–3072. 10.1128/IAI.00123-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 28.Rood JI, Maher EA, Somers EB, Campos E, Duncan CL. 1978. Isolation and characterization of multiply antibiotic-resistant Clostridium perfringens strains from porcine feces. Antimicrob. Agents Chemother. 13:871–880. 10.1128/AAC.13.5.871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rood JI. 1983. Transferable tetracycline resistance in Clostridium perfringens strains of porcine origin. Can. J. Microbiol. 29:1241–1246. 10.1139/m83-193 [DOI] [PubMed] [Google Scholar]
- 30.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 31.Scott PT, Rood JI. 1989. Electroporation-mediated transformation of lysostaphin-treated Clostridium perfringens. Gene 82:327–333. 10.1016/0378-1119(89)90059-0 [DOI] [PubMed] [Google Scholar]
- 32.Hiscox TJ, Chakravorty A, Choo JM, Ohtani K, Shimizu T, Cheung JK, Rood JI. 2011. Regulation of virulence by the RevR response regulator in Clostridium perfringens. Infect. Immun. 79:2145–2153. 10.1128/IAI.00060-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyras D, O'Connor JR, Howarth PK, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179. 10.1038/nature07822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farrow KA, Lyras D, Rood JI. 2001. Genomic analysis of the erythromycin resistance element Tn5398 from Clostridium difficile. Microbiology 147:2717–2728 http://mic.sgmjournals.org/content/147/10/2717.long [DOI] [PubMed] [Google Scholar]
- 35.Carter GP, Douce GR, Govind R, Howarth PM, Mackin KE, Spencer J, Buckley AM, Antunes A, Kotsanas D, Jenkin GA, Dupuy B, Rood JI, Lyras D. 2011. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog. 7:e1002317. 10.1371/journal.ppat.1002317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. 10.1016/0378-1119(89)90359-4 [DOI] [PubMed] [Google Scholar]
- 37.Awad MM, Bryant AE, Stevens DL, Rood JI. 1995. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of α-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 15:191–202. 10.1111/j.1365-2958.1995.tb02234.x [DOI] [PubMed] [Google Scholar]
- 38.Shimizu T, Ba-Thein W, Tamaki M, Hayashi H. 1994. The virR gene, a member of a class of two-component response regulators, regulates the production of perfringolysin O, collagenase, and hemagglutinin in Clostridium perfringens. J. Bacteriol. 176:1616–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naikare H, Palyada K, Panciera R, Marlow D, Stintzi A. 2006. Major role for FeoB in Campylobacter jejuni ferrous iron acquisition, gut colonization, and intracellular survival. Infect. Immun. 74:5433–5444. 10.1128/IAI.00052-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weaver EA, Wyckoff EE, Mey AR, Morrison R, Payne SM. 2013. FeoA and FeoC are essential components of Vibrio cholerae ferrous iron uptake system, and FeoC interacts with FeoB. J. Bacteriol. 195:4826–4835. 10.1128/JB.00738-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartman AH, Liu H, Melville SB. 2011. Construction and characterization of a lactose-inducible promoter system for controlled gene expression in Clostridium perfringens. Appl. Environ. Microbiol. 77:471–478. 10.1128/AEM.01536-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nariya H, Miyata S, Kuwahara T, Okabe A. 2011. Development and characterization of a xylose-inducible gene expression system for Clostridium perfringens. Appl. Environ. Microbiol. 77:8439–8441. 10.1128/AEM.05668-11 [DOI] [PMC free article] [PubMed] [Google Scholar]