

Abstract
Pseudomonas aeruginosa is a common nosocomial pathogen that relies on three cell-to-cell signals to regulate multiple virulence factors. The Pseudomonas quinolone signal (PQS; 2-heptyl-3-hydroxy-4-quinolone) is one of these signals, and it is known to be important for P. aeruginosa pathogenesis. PQS is synthesized in a multistep reaction that condenses anthranilate and a fatty acid. In P. aeruginosa, anthranilate is produced via the kynurenine pathway and two separate anthranilate synthases, TrpEG and PhnAB, the latter of which is important for PQS synthesis. Others have previously shown that a P. aeruginosa tryptophan auxotroph could grow on tryptophan-depleted medium with a frequency of 10−5 to 10−6. These revertants produced more pyocyanin and had increased levels of phnA transcript. In this study, we constructed similar tryptophan auxotroph revertants and found that the reversion resulted from a synonymous G-to-A nucleotide mutation within pqsC. This change resulted in increased pyocyanin and decreased PQS, along with an increase in the level of the pqsD, pqsE, and phnAB transcripts. Reporter fusion and reverse transcriptase PCR studies indicated that a novel transcript containing pqsD, pqsE, and phnAB occurs in these revertants, and quantitative real-time PCR experiments suggested that the same transcript appears in the wild-type strain under nutrient-limiting conditions. These results imply that the PQS biosynthetic operon can produce an internal transcript that increases anthranilate production and greatly elevates the expression of the PQS signal response protein PqsE. This suggests a novel mechanism to ensure the production of both anthranilate and PQS-controlled virulence factors.
INTRODUCTION
The Gram-negative bacillus Pseudomonas aeruginosa is a common nosocomial pathogen that causes devastating opportunistic infections in immunocompromised individuals and chronic infections in cystic fibrosis (CF) patients (1–3). P. aeruginosa is notoriously resistant to multiple classes of antibiotics, making many infections difficult to treat. The ability of P. aeruginosa to evade the immune system and cause these serious infections can be attributed to the numerous virulence factors employed by the bacteria. Many of these virulence factors are controlled by three known cell-to-cell signals, which compose a complex communication network that ensures precise expression of many genes in P. aeruginosa. Two of the cell-to-cell signaling systems are mediated by acyl-homoserine lactone signals that are regulated by their cognate transcriptional regulators, LasR and RhlR (4, 5). Together these two signaling systems regulate 6 to 11% of the P. aeruginosa genome (6, 7). The third signal is the Pseudomonas quinolone signal (PQS; 2-heptyl-3-hydroxy-4-quinolone) (8). PQS acts as a coinducer for the transcriptional regulator PqsR, which drives transcription of the PQS biosynthetic gene locus and positively regulates multiple virulence factors (8–13). PQS has also been shown to be important for P. aeruginosa pathogenesis in Caenorhabditis elegans, Drosophila melanogaster, lettuce leaf, and mouse infection models (10, 14–17). PQS has been extracted from airway fluid and sputum of infected CF patients, further implying its importance in P. aeruginosa infections (18).
With the overwhelming evidence that PQS plays a role in virulence and pathogenesis, we set out to understand both the regulation of the genes required for PQS production and the biosynthesis of PQS. The biosynthetic genes required for PQS synthesis, pqsABCD and pqsH, are also responsible for the production of at least 56 other 4-hydroxy-2-alkylquinolines (HAQs) (19, 20). The biosynthesis of all HAQs depends on subsequent condensations of anthranilate with malonyl coenzyme A (malonyl-CoA) and then a fatty acid (Fig. 1), the chain length of which determines the side chain length of the HAQ (21, 22). The precursor anthranilate is an important branch point metabolite in P. aeruginosa that can be synthesized into either PQS or the amino acid tryptophan, as well as catabolized into tricarboxylic acid cycle intermediates (23). The cellular pool of anthranilate in P. aeruginosa is maintained via three separate pathways (24). First, the kynurenine pathway converts tryptophan into anthranilate and is the main source of anthranilate for PQS production when tryptophan is present (24). This pathway is regulated by both LasR and KynR, which induce the pathway genes in the presence of kynurenine (25, 26). Anthranilate is also supplied by the two anthranilate synthases, TrpEG and PhnAB (27), which convert chorismate into anthranilate. TrpEG catalyzes the first step in tryptophan synthesis and is regulated by tryptophan through attenuation and posttranslational competitive inhibition of TrpE (28, 29). Though this enzyme seemingly could provide anthranilate for the cellular pool, TrpEG does not naturally provide anthranilate for PQS production in a phnA mutant grown without tryptophan (24). However, overexpression of TrpEG causes PQS production in a phnAB mutant grown without tryptophan, thus indicating the importance of phnAB regulation for PQS production (30).
FIG 1.
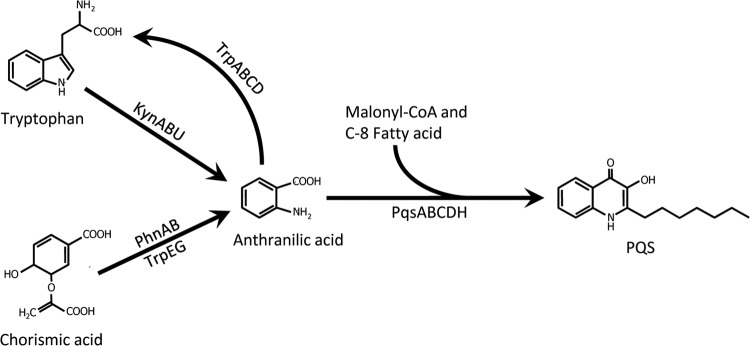
Anthranilate and PQS synthesis in P. aeruginosa. In P. aeruginosa, anthranilate is made via three separate enzymatic pathways. The kynurenine pathway degrades tryptophan into anthranilate via the KynABU enzymatic pathway, while anthranilate is synthesized from chorismate by two separate anthranilate synthases, PhnAB and TrpEG. Anthranilate can be used as a precursor for many compounds, including tryptophan and 4-quinolones.
The phnAB operon is located between the pqsABCDE operon (same strand) and pqsR (opposite strand), placing it in the center of the PQS synthesis region. Like pqsABCDE, phnAB is positively controlled by PqsR (31–33). Due to both the location and regulation of phnAB, it was assumed that PhnAB provided the majority of anthranilate for PQS synthesis. However, we have shown that PhnAB supplies anthranilate for PQS only under nutrient-limiting conditions (24). With three separate and uniquely regulated pathways to anthranilate, it is clear that the production of anthranilate is a priority for P. aeruginosa. This was obvious when the two initial studies describing TrpEG and PhnAB in P. aeruginosa revealed an interesting aspect of phnAB regulation (27, 34). When a tryptophan auxotroph was plated on tryptophan-depleted media, auxotrophy-negating mutations occurred at a frequency of 10−5 to 10−6 (27). When one of the revertants was investigated, it was discovered that it had constitutive levels of phnAB transcript expression, as opposed to the peak stationary levels exhibited by wild-type P. aeruginosa (27). In addition, the revertant had increased levels of pyocyanin, a virulence factor influenced by the PQS signaling system. In this study, we set out to identify the source of the spontaneous mutation that suppresses the tryptophan auxotrophy and discovered a novel regulatory mechanism for phnAB and PQS-controlled genes.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Escherichia coli and P. aeruginosa strains were freshly plated to begin each experiment and were maintained at −80°C in 30% glycerol and 10% skim milk (Difco), respectively. All strains and plasmids used are listed in Table 1. Bacteria were cultured at 37°C in Luria-Bertani medium (LB) or Vogel-Bonner minimal medium supplemented with 0.5% glycerol (VBG) (35). Growth of bacterial cultures was monitored spectrophotometrically based on the optical density at 600 nm (OD600) for E. coli and OD660 for P. aeruginosa. To maintain plasmids, 100 μg/ml of ampicillin or 200 μg/ml of carbenicillin was added when appropriate. Also, 10 mM l-tryptophan was added to VBG cultures when indicated.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or phenotype | Reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | F′/endA1 hsdR17 supE44 thi-1 recA1 gyrA relA1 (lacZYA-argF) U169 deoR Φ80dlacΔ(lacZ)M15recA1 | 52 |
| P. aeruginosa | ||
| PAO1 | Wild type | |
| PJF-QR1 | pqsR deletion mutant derived from strain PAO1 | 53 |
| PAΔTrpE | trpE deletion mutant derived from strain PAO1 | 48 |
| PJF-QE1 | pqsE deletion mutant derived from strain PAO1 | 48 |
| PJF-PhnA | phnA deletion mutant derived from strain PAO1 | 24 |
| PAQC-G1041A | PAO1 derivative with G-to-A mutation in pqsC at bp 1041 | This study |
| TrpErev1 | Spontaneous mutant of strain PAΔTrpEa | This study |
| TrpErev2 | Spontaneous mutant of strain PAΔTrpEa | This study |
| TrpErev3 | Spontaneous mutant of strain PAΔTrpEa | This study |
| TrpErev4 | Spontaneous mutant of strain PAΔTrpEa | This study |
| TrpErev5 | Spontaneous mutant of strain PAΔTrpEa | This study |
| Plasmids | ||
| pLP170 | lacZ transcriptional fusion vector; Amprb | 54 |
| pPqsDwtTcLacZ | pqsD′-lacZ transcriptional fusion; Ampr | This study |
| pPqsDG1041ATcLacZ | pqsDpqsCG1041A1041′-lacZ transcriptional fusion; Ampr | This study |
| pEX18AP | Suicide vector; Ampr | 39 |
| pPAQC-G1041Adel | G-to-A reversion in pqsC suicide vector; Ampr | This study |
| ptrpEdel | trpE deletion suicide vector; Ampr | This study |
| pΔpqsD-suc | pqsD deletion suicide vector; Ampr | This study |
| pDSW8 | tacp-pqsR on pEX1.8, Ampr | 32 |
| pBluescript II SK+ | Cloning vector; Ampr | Agilent Technologies |
| pSLM100 | phnA expression vector; Ampr | This study |
See Table 3 for the location(s) of mutation(s).
Ampr, ampicillin resistant.
To generate transcriptional fusions for pqsD, DNA fragments of various length that were 3′ nested at 58 bp downstream from the pqsD start codon (see Fig. 7 for diagram) were amplified by PCR using chromosomal DNA as a template from either strain PAO1 or strain TrpErev1. The oligonucleotide primers (Table 2) used for the amplification contained either a XhoI or a HindIII restriction site. The amplified fragment was digested with XhoI and HindIII, purified from an agarose gel, and ligated into pLP170 digested with the same enzymes. All reporter fusions were sequenced to ensure that mutations did not occur during cloning. Plasmids were transformed into P. aeruginosa strains by electroporation (36).
FIG 7.
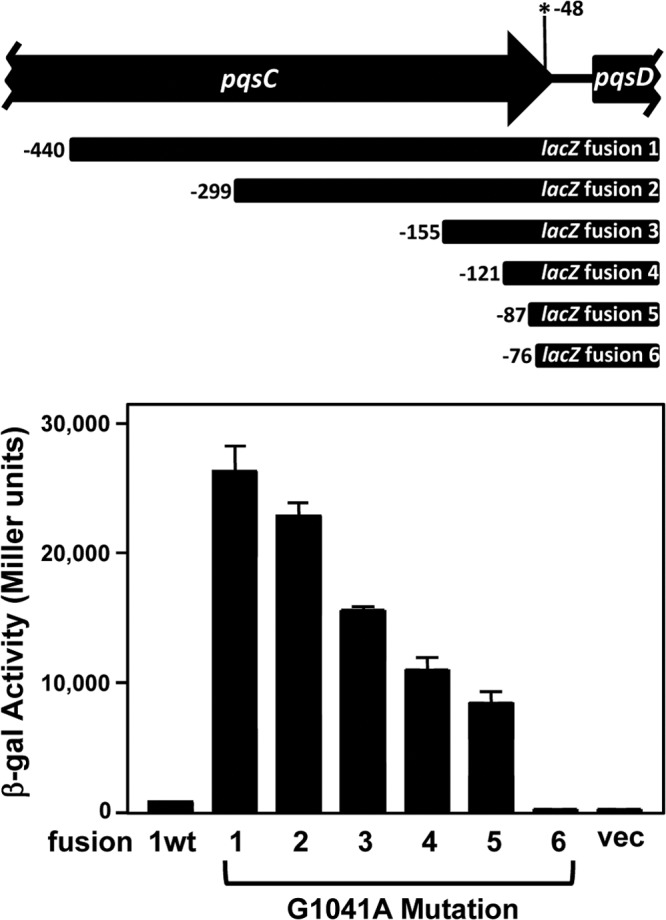
The G1041A mutation results in the expression of a new transcript upstream from pqsD. The P. aeruginosa wild-type strain PAO1 containing a reporter plasmid with either the wild-type sequence (ppqsDwtTcZ, indicated as “1wt”) or nested deletion plasmids with the pqsC point mutation (ppqsDG1041ATcZ 1 through 6) was grown in VBG until an OD660 of 0.2 was reached. The cultures were then assayed for β-Gal activity, which is presented in Miller units as the mean ± σn − 1 of results from at least three separate experiments. The numbers on the fusion diagram indicate the distance of the 5′ end of each fusion from the pqsD start codon. The asterisk indicates the location of the G1041A mutation.
TABLE 2.
Primers used in this study
| Primer use and name | Sequencea |
|---|---|
| Mutagenesis | |
| TrpEF1-HindIII | AAAAAAGCTTCCTGATCGATGCCTCCGGTC |
| TrpEdelF2 | TCACCGTTCTTGATCACCGCAGTCGGCAAGGGTCTCGA |
| TrpEdelR1 | TCGAGACCCTTGCCGACTCGCGGTGATCAAGAACGGTGA |
| TrpER2-HindIII | AAAAAAGCTTACCGGCAACAGCATGGAAC |
| PqsCUp1 | AAAAAAGCTTAGATTCCCTCGCACATGCTG |
| PqsCDown2 | AAAAAAGCTTACCTCCTCAGGTTTGCGGTA |
| Expression plasmids | |
| pqsClacZfwd | AAAAAGAATTCCCGACCGGCGAGGCCAA |
| pqsClacZrev | AAAAAAGCTTTGCTGACCTGGCGTTTCGGC |
| pqsCLacZfwd-2 | AAAAACTCGAGGATTATTTCGTCTTCCACCAG |
| pqsCLacZfwd-3A | AAAAACTCGAGGCCTGCGCGAGGGCAGAGTCCG |
| pqsCLacZfwd-3B | AAAAACTCGAGGTATCGTCATGGCCGGCGCAG |
| pqsCLacZfwd-5 | AAAAACTCGAGTTCGCCGCCCAGGTCTGGC |
| pqsCLacZfwd-6 | AAAAACTCGAGAGGTCTGGCAATTGGGTGA |
| Real-time PCR | |
| RPLU RT1 | GGTGGCAAGCAGCACAAAGTCACCG |
| RPLU RT2 | GCGGACCTTGTCGTGACGGCCGTGG |
| pqsARTF1 | GCCCTTTGCTCGACGATTTCTCG |
| pqsARTR2 | AACCCGAGGTGTATTGCAGGAAACA |
| pqsCRTF1 | CGAGTCCTGGTGGCAATTCT |
| pqsCRTR1 | TCAGCATGTCCACGCTATCC |
| pqsDRTF1 | GTGGATCGTCTGGGCAACAT |
| pqsDRTR1 | CTCCTCAGGTTTGCGGTACA |
| pqsERTF1 | TGATGACCTGTGCCTGTTGG |
| pqsERTR1 | GTCGTAGTGCTTGTGGGTGA |
| phnARTF1 | ACGTGGACAGCAAGGCTGCG |
| phnARTR1 | GGGCCGGACAGTCCTCGCTC |
| RT- PCR | |
| E2 | CCAGACTTTCTTCCAGTCGATAGC |
| F1 | TACCGAGTGTCTGCGCCTGT |
| F2 | GGCCAGGTCGAAGCTGAACA |
Restriction sites are indicated by bold type.
Generation of mutants.
A splicing-by-overlap extension protocol was used to generate the mutant allele for the trpE deletion strain (37). The mutation was constructed to contain an in-frame deletion in the coding DNA sequence corresponding to amino acids 26 to 445 for trpE (85% of protein sequence). Primers were designed to amplify approximately 1 kb of DNA both upstream and downstream from the splice junction, and each primer added a HindIII restriction site to the ends of the PCR product. The PCR fragment and pEX18Ap (suicide vector) were digested with HindIII and ligated together to make the suicide vector ptrpEdel. To generate the PAQC-G1041A mutant, a PCR fragment was created using strain TrpErev1 chromosomal DNA, and it was digested with HindIII and ligated into pEX18AP. The resulting plasmids, ptrpEdel and pPAQC-G1041A, were transformed into strain PAO1 by electroporation (38). Mutants were selected by plating transformants on medium containing carbenicillin and then on medium containing 6% sucrose to select for deletion of the vector sequence (39). PCR was used to screen colonies, and DNA sequencing of PCR products was used to confirm mutations.
Tryptophan auxotroph revertants were made through selective pressure on tryptophan-free media. The tryptophan auxotroph strain PAΔTrpE was grown overnight in VBG supplemented with 10 mM l-tryptophan at 37°C with shaking at approximately 180 rpm. Overnight cultures were washed with fresh, unsupplemented VBG, and 100 μl of washed culture was added to 10 ml of unsupplemented VBG. The culture was grown for 3 h at 37°C with shaking at approximately 180 rpm. After 3 h of growth, cells were harvested by centrifugation at 6,000 × g for 10 min. The cell pellet was resuspended in 1 ml of VBG and then plated onto VBG plates. Individual colonies were then reisolated and stored at −80°C for further analysis.
Whole-genome sequencing.
An Illumina GAIIX at the North Carolina State University Gene Sequencing Laboratory was used to sequence wild-type strain PAO1 and three tryptophan auxotroph revertants (TrpErev1, -2, and -3). The sequencing provided between 5.8 and 7.3 million reads for a high-quality map. To determine the location of the mutation(s), the published PAO1 genome (40) was compared to those of the sequenced PAO1 laboratory strain and the tryptophan revertant strains. The Burrows-Wheeler Aligner (BWA) was used to align the raw reads and provided the high-quality map (41). The trpE deletion in each of the revertants was confirmed by using the Integrative Genomics Viewer (IGV) (42, 43). Both Joint Genotyper for Inbred Lines (JGIL) and SAMtools and BCFtools were used to identify single nucleotide polymorphisms (SNPs), and the SAMtools and BCFtools were used specifically to identify any insertions and/or deletions (44, 45). The JGIL is designed to look for SNPs in inbred lines but is appropriate to use for bacteria due to all strains being derived from the same parent strain.
PQS concentration determination.
Washed cells from overnight cultures were used to inoculate 10-ml cultures of LB or VBG to an OD660 of 0.05. After 24 h of growth, a 300-μl sample of each culture was extracted with 900 μl of acidified ethyl acetate as previously described (18). One-half of the resulting organic phase was evaporated to dryness at 37°C, and 50 μl of 1:1 acidified ethyl acetate-acetonitrile was used to reconstitute the extract (18). Samples were analyzed by thin-layer chromatography (TLC), visualized by long-wave UV light, and photographed (18). The PQS spots were quantified using Bio-Rad Quantity One software compared to a standard curve of synthetic PQS.
RNA isolation.
For all RNA extractions, the cultures were grown at 37°C in either LB medium or VBG until an OD660 of 0.2 or 0.6 was reached. At that point, total RNA for reverse transcriptase PCR (RT-PCR) and quantitative real-time RT-PCR (real-time PCR) was purified using the RNeasy mini-spin columns (Qiagen). All RNA samples were treated with RQ1 DNase (Promega) for 2 h at 37°C to remove any contaminating DNA. To remove RQ1 DNase, a phenol-chloroform extraction was performed, followed by an ethanol precipitation. RNA was stored in diethyl pyrocarbonate (DEPC)-treated water at −80°C.
Quantitative real-time PCR.
cDNA synthesis was performed on a Mastercycler (Eppendorf) using 2.5 μg of DNA-free RNA and Superscript III (Invitrogen) by following the manufacturer's instructions. Primers to generate cDNA were random primers (Invitrogen) as well as random hexamers, 72% GC (Gene Link). Real-time PCR was performed on a CFX96 (Bio-Rad) using the PerfeCTa SYBR green FastMix for iQ (Quanta Biosystems). The reaction mixtures contained 5 μl of cDNA diluted (1:500) in a 15-μl volume. Relative transcript levels were determined using the Pfaffl method by comparing experimental gene expression (phnA, pqsA, pqsC, pqsD, or pqsE) to that of the control gene, rplU (46). Standard curve and efficiency values were determined by using gene-specific primers with 10 ng to 0.1 pg of RNA-free genomic DNA from strain PAO1. A melting-curve step was included at the end of each PCR to ensure that primer dimers and nonspecific variation between samples did not occur. Each real-time PCR was performed on at least three biological replicates of each RNA sample, with technical replicates performed in duplicate.
RT-PCR.
For reverse transcriptase PCR, the AccessQuick kit from Promega was used. PAO1 chromosomal DNA (100 ng) was used as a positive control, and 300 ng of RNA was used in all experimental samples. Two biological RNA samples per strain were used to verify results.
β-Gal assays in P. aeruginosa.
Cells from overnight cultures of P. aeruginosa grown in VBG were washed and resuspended in fresh medium to an OD660 of 0.05 and incubated at 37°C with shaking at approximately 180 rpm until the OD660 had reached 0.2. At that time, cultures were assayed for β-galactosidase (β-Gal) activity in duplicate. Data are presented in Miller units as the mean ± the standard deviation (σn − 1) of at least three separate experiments.
RESULTS
Tryptophan starvation reverts tryptophan auxotrophs by permanently increasing phnAB expression.
P. aeruginosa possesses two anthranilate synthases, TrpEG and PhnAB. The existence of two anthranilate synthases and the kynurenine pathway ensures that the cellular pool of anthranilate can be maintained for tryptophan synthesis and PQS production. In two studies describing both TrpEG and PhnAB, Essar et al. discovered that a trpE mutant is a tryptophan auxotroph that will revert to tryptophan prototrophy at a high frequency (10−5 to 10−6) when grown on tryptophan-depleted medium (27, 34). The tryptophan auxotroph revertant exhibited increased pyocyanin levels and a constitutive high induction of the phnAB transcript, which apparently complemented the loss of anthranilate production caused by the mutation of trpE (27). These strains are no longer available (D. Essar, personal communication), so our laboratory selected revertants in the same manner and set out to identify the mutation(s) responsible for the loss of tryptophan auxotrophy. In order to do this, we constructed a trpE mutant (PAΔTrpE) and grew it on minimal media depleted of tryptophan to select for a revertant. This was repeated three times and we chose to analyze five colonies that grew well, indicating that they were tryptophan auxotroph revertants. All five revertants showed increased pyocyanin levels, which was also reported by Essar et al. (27) (Fig. 2A). In addition, the auxotrophy revertants all produced a greatly decreased amount of PQS (Fig. 2B), which was unexpected because PQS positively controls pyocyanin production (10). (This phenomenon is addressed below.) Quantitative real-time PCR analysis of phnA showed that all five revertants had at least a 3.5-fold increase in phnA expression compared to strain PAO1 when grown in either rich medium (Fig. 3A) or minimal medium without tryptophan (Fig. 3B). This is similar to data reported previously (27), and taken together, our data lead us to believe that we were studying the same phenomenon as first reported by Essar et al. (27) and that an unknown regulatory event occurs when cells are starved for tryptophan.
FIG 2.
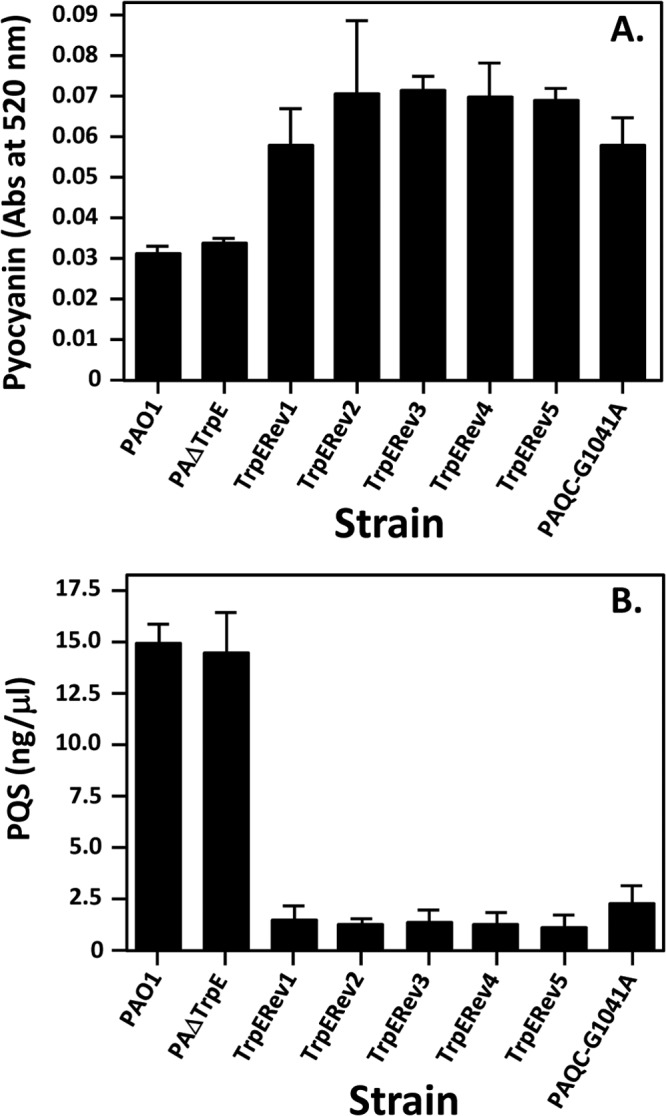
Pyocyanin and PQS production by tryptophan auxotroph revertants. (A) Pyocyanin assays were performed after 18 h of growth in LB broth. Results are shown as light absorbance at 520 nm and are the mean ± σn − 1 of results from duplicate assays from at least three separate experiments. (B) Ethyl acetate extracts from cultures grown in LB broth for 24 h were analyzed by TLC. Equal volumes of extracts were resolved in each lane. The TLC plates were photographed under UV light, and densitometric readings of PQS spots were compared to those from a standard curve of synthetic PQS in order to determine the PQS concentration in extracts. Results shown are the average ± σn − 1 of results from at least three separate experiments.
FIG 3.
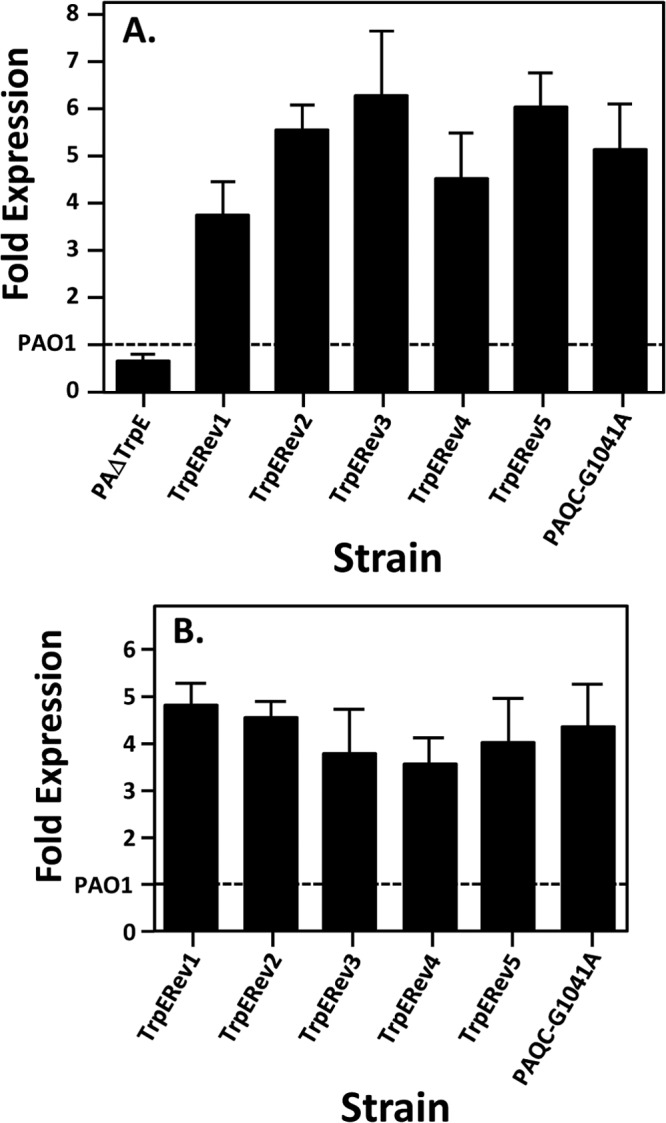
Tryptophan auxotroph revertants have increased phnA transcript levels. Quantitative real-time PCR was used to measure the expression of phnA mRNA from the indicated strains grown at 37°C in LB medium (A) and VBG (B). Wild-type strain PAO1 transcript levels are set at 1, which is indicated by the dashed line. Strain PAΔTrpE was not included in panel B because it will not grow in VBG medium without tryptophan supplementation. Error bars indicate the standard deviations of three independent experiments performed in duplicate.
Discovery of a conserved point mutation in tryptophan auxotroph revertants.
To understand the mechanism of change in the revertant strains, the full genome sequences of three tryptophan auxotroph revertants (TrpErev1, -2, and -3) were determined. This experiment identified two locations that had a single nucleotide change. One change was found in all three strains, and the other was found in just two strains, which were derived from the same selection experiment and likely were clones (Table 3). No other substitution, insertion, or deletion mutations were found. One mutation was a G-to-A change at nucleotide 1041 of pqsC that did not cause a change in the encoded amino acid. The other mutation was in the ntrC gene (nucleotide 1227), and it also did not cause an amino acid change. Once these mutations were identified, the same regions of two other revertant strains (TrpErev4 and TrpErev5) were amplified by PCR and sequenced. These other two strains were derived from independent experiments; both had the same pqsC mutation as described above (referred to henceforth as G1041A), and neither had a mutation in ntrC (Table 3). These data led us to further examine the G1041A mutation.
TABLE 3.
Location(s) of the mutation(s) in tryptophan auxotroph revertants
| Revertant strain | Mutation |
|
|---|---|---|
| pqsC | ntrC | |
| TrpErev1 | Base 1041, G to A | No change |
| TrpErev2 | Base 1041, G to A | Base 1227, G to A |
| TrpErev3 | Base 1041, G to A | Base 1227, G to A |
| TrpErev4 | Base 1041, G to A | No change |
| TrpErev5 | Base 1041, G to A | No change |
The G1041A mutation is two codons from the pqsC stop codon and is in the third base of a valine codon, resulting in a codon that still encodes valine and therefore does not alter PqsC. There is a 43-bp intergenic region between the end of the pqsC coding sequence and the start codon of pqsD, so this change does not affect the PqsD protein (see the asterisk in Fig. 5 for a visual representation of the location of this mutated nucleotide). To ensure that the G1041A mutation in pqsC resulted in the effects observed in the tryptophan auxotroph revertants, we constructed a mutant using wild-type strain PAO1, which contained only the G1041A mutation. Alteration of this single base in strain PAO1 resulted in the same phenotype as in the revertant strains: pyocyanin increased (Fig. 2A), PQS decreased (Fig. 2B), and the phnA transcript increased (Fig. 3). This confirmed that a single base change at nucleotide 1041 of pqsC caused the observed results and is a mechanism by which P. aeruginosa can survive when starved for tryptophan (and probably anthranilate). Our next goal was to try to understand the mechanism through which this single base change brought about the observed effects.
FIG 5.

Analysis of the pqsE and phnA transcripts by RT-PCR. (A) Diagram of the pqsD through phnB genetic region with location of the primers used for RT-PCR experiments. The asterisk indicates the location of the G1041A mutation in pqsC, of which only the 3′ region is shown. (B and C) Agarose gel with products from RT-PCR experiments performed with the indicated oligonucleotide primer pairs and RNA isolated from cultures grown in LB medium (B) or VBG (C). Lane 1 contains molecular size standards (1 Kb Plus DNA ladder; Invitrogen); size, in base pairs, is marked on the left side of each panel. Lane 2 contains reactions performed with chromosomal DNA as a template for positive-control reactions. Lanes 3 and 4 contain total RNA from wild-type strain PAO1, and lanes 5 and 6 contain total RNA from strain PAQC-G1041A. RNA was isolated from VBG at an OD660 of 0.2 and was used as a template for experimental reactions (lanes 3 and 5). For negative controls, reverse transcriptase was omitted from reactions shown in lanes 4 and 6.
A synonymous mutation in pqsC results in differential expression of a novel transcript within the PQS biosynthetic operon.
The G1041A mutation in pqsC resulted in increased pyocyanin and decreased PQS, which seems contradictory since PQS is required for the production of pyocyanin (10). However, PQS has not been directly linked to pyocyanin control, and it has been shown that the last gene of the pqsABCDE operon, pqsE, can independently activate pyocyanin production (47, 48). In addition, it is known that the overexpression of pqsE causes PQS production to stop (49). This led us to speculate that the G1041A mutation was causing pqsE expression to increase. We therefore utilized quantitative real-time PCR to compare transcript levels of genes within the pqsABCDE and phnAB operons. Analysis of RNA from a trpE auxotroph revertant (Fig. 4A) and our constructed G1041A mutant (Fig. 4B) showed that both strains have a decreased level of pqsA and pqsC transcript compared to the wild-type strain, PAO1. These decreases are most likely due to the lower PQS levels described above, which would result in less coinducer for the transcriptional regulator PqsR to drive transcription of the pqsABCDE operon (32). Most interestingly, levels of the pqsD, pqsE, and phnA mRNA were increased at least 3-fold in both strain TrpErev1 and strain PAQC-G1041A compared to the wild-type strain (Fig. 4). This result was unexpected because previous data suggested that the PQS biosynthetic operon transcript is derived from the pqsA promoter and that the transcript included only pqsABCDE (20). However, the data of Fig. 4 led us hypothesize that the G1041A mutation caused the induction of a transcript within pqsC and that this transcript contains at least pqsD, pqsE, and phnA. To investigate this possibility, we analyzed transcripts by reverse transcriptase PCR (RT-PCR). The oligonucleotides used for RT-PCR are illustrated in Fig. 5A and included a set used by McGrath et al. (primer set F1 and F2) when they suggested that the PQS biosynthetic operon included only pqsABCDE and not phnAB (20). We obtained results similar to those we reported previously (20) when mRNA derived from a wild-type culture grown in LB medium was used in the RT-PCR experiments. The wild-type strain PAO1 mRNA (Fig. 5B, lane 3) did not produce a product that would indicate mRNA continuity between pqsE and phnA. However, an RT-PCR product indicative of a continuous transcript was evident with both primer sets from the analysis of G1041A mutant mRNA (Fig. 5B, lane 5). (The E1-F2 primer set starts and ends, respectively, 556 nucleotides upstream from and 468 nucleotides downstream from the phnA ATG start codon.) This suggested that the G1041A mutant strain does express a novel transcript that includes at least part of pqsE and phnA. Most interestingly, the same products appeared in the analysis of either the wild-type strain PAO1 or mutant G1041A mRNA from cultures grown in minimal medium (Fig. 5C, lanes 3 and 5, respectively). Considering these data along with those in Fig. 4, it appears that under nutrient-limiting conditions, a novel transcript that includes pqsD, pqsE, and phnA is induced in the wild-type strain and that this transcript is constitutively induced in the G1041A mutant.
FIG 4.
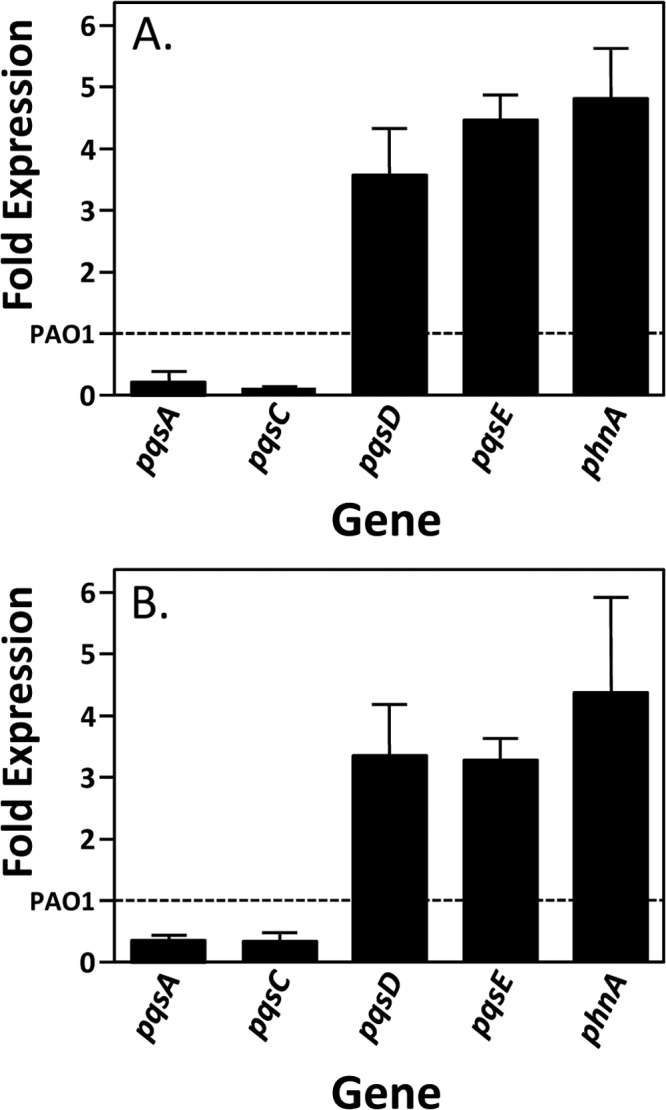
A single nucleotide mutation in pqsC induces pqsD, pqsE, and phnA. Quantitative real-time PCR was used to measure the expression of pqsA, pqsC, pqsD, pqsE, and phnA mRNA from strains TrpErev1 (A) and PAQC-G1041A (B) grown at 37°C in VBG to an OD660 of 0.2 (mid-log phase). Wild-type strain PAO1 transcript levels are set at 1, which is indicated by the dashed line. Error bars indicate the standard deviations of three independent experiments performed in duplicate.
Evidence for a novel transcript.
In order to confirm that the putative transcript described above was present in wild-type strain PAO1, we used quantitative real-time PCR to compare pqsA and pqsD expression in minimal and rich media. The data showed that the ratio of expression for pqsA and our control gene (rplU) was approximately 1:1 in minimal versus rich medium, indicating no change in relative expression (Fig. 6). However, we found that pqsD expression was over 2-fold higher in minimal medium than in rich medium (Fig. 6). These data confirmed the data of Fig. 5 and indicated that in wild-type strain PAO1, the pqsA and pqsD transcripts are differentially regulated and therefore most likely on two differentially expressed mRNAs.
FIG 6.
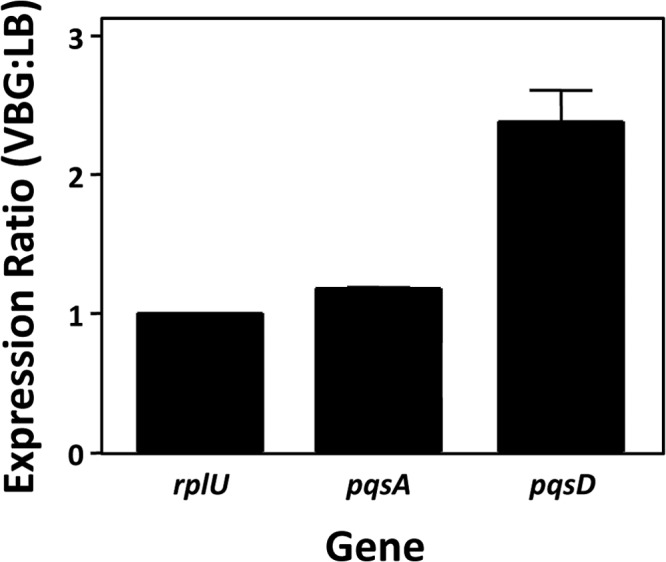
The pqsA and pqsD genes are differentially regulated in wild-type strain PAO1 when grown in rich (LB) or nutrient-limiting (VBG) medium. Cultures of strain PAO1 were grown to mid-log phase, at which time RNA was purified. Quantitative real-time PCR was used to measure expression of the indicated genes, and the ratio of expression in VBG versus LB was determined. The expression of the housekeeping gene rplU was measured as a control and set to a value of 1 for the purpose of comparison. Error bars indicate the standard deviations of three independent experiments performed in duplicate.
Despite using multiple different protocols and both natural and enriched mRNA, repeated attempts to map the 5′ end of this novel transcript did not succeed in reproducibly identifying a transcriptional start site (data not shown). Therefore, to understand the sequence elements required to control this transcript, we constructed a series of lacZ reporter fusions that were nested within the 5′ region of pqsD and contained the pqsC G1041A mutation and progressive deletions of portions of pqsC (Fig. 7). This experiment showed that the region of pqsC that is upstream from nucleotide −87 relative to the pqsD start codon was required for transcription. Deleting an additional 11 bp from this sequence produced the fusion that ended at nucleotide −76 relative to the pqsD start codon and caused a complete loss of transcriptional activity. This suggests that a promoter element is located between 76 and 87 bp upstream from the pqsD start codon. The G1041A mutation is at position −48 relative to the pqsD start codon, which places the mutation close to the location where one would expect to find a transcriptional start site if a promoter begins just inside position −87. This allows us to speculate that the G1041A mutation is either at or near the transcriptional start site or is within a −10 region of a promoter. The latter option seems more likely, as the large positive effect on transcription would be more simply explained by the mutation leading to the creation of an optimal promoter sequence. It is also interesting that as the 5′ region of the fusions was removed, a progressive decrease in transcriptional activity occurred from a maximum of 26,230 ± 2,015 down to 8,366 ± 925 U of β-Gal activity, indicating that the promoter of the novel transcript is modulated by elements well upstream from the −35 region. Nonetheless, the promoter was still highly induced when only 87 nucleotides upstream from the pqsD start codon were included. Taken together, the data suggest that a conserved synonymous mutation near the end of pqsC is selected for when a tryptophan auxotroph is grown under tryptophan-limiting conditions. This mutation leads to the overexpression of a transcript that rescues the tryptophan auxotroph due to the induction of phnAB, and to the induction of pqsE, which induces PQS-controlled genes in the absence of PQS.
DISCUSSION
The data we present here point to a mechanism that explains how tryptophan auxotroph revertants alter their ability to produce PQS-controlled virulence factors. The intriguing results reported in 1990 by Essar et al. (27) on the link between phnA dysregulation and phenazine production led us to investigate this natural phenomenon. Our isolation of five tryptophan auxotroph revertants, which mirrored the prior results (27), suggested that we had recreated the mutation that allowed for survival of a tryptophan auxotroph on tryptophan-depleted medium (Fig. 2 and 3). Full-genome sequencing of the revertants revealed a point mutation in pqsC at nucleotide 1041 that occurred in all five strains. The single nucleotide pqsC G1041A mutation was interesting because it was within the PQS biosynthetic operon upstream from phnAB, causing us to focus on it as a likely cause of the observed phenotype. The construction of a G1041A mutant strain (PAQC-G1041A) then confirmed that the single nucleotide change in pqsC was responsible for the increase in pyocyanin production and phnA transcription exhibited by our revertants (Fig. 2 and 3) and likely those originally reported by Essar et al. (27).
Further investigation of the G1041A mutation in pqsC led us to discover that not only was the single nucleotide change responsible for elevated levels of pyocyanin production and the phnA transcript (Fig. 2 and 3) but also it resulted in dysregulation of the PQS biosynthetic operon (Fig. 4). The G1014A mutation resulted in decreased levels of pqsA and pqsC transcripts, which is not surprising, as they are likely due to decreased coinducer (PQS) (Fig. 2) for PqsR, the transcriptional regulator of the PQS biosynthetic operon (32). What was perplexing was that the G1041A mutant had elevated transcript levels of the last two genes in the PQS operon, pqsD and pqsE (Fig. 4). Though these results were unexpected, they provided an explanation for the phenotypic results we had obtained (Fig. 2). The significant increase of the pqsE transcript would explain the observed increase in pyocyanin (Fig. 2), as this effect has been shown previously (10, 48). Furthermore, the elevated transcription of pqsE, and likely overproduction of the PqsE protein, may also account for the decrease in PQS production in the revertants (Fig. 2), as overproduction of PqsE results in decreased levels of PQS (49, 50). Though the overall phenotypic results were explained by the increase in pqsE transcript overexpression, we were still uncertain of what was causing the elevation of the transcript and how pqsD, pqsE, and phnA are linked.
In order to identify the possible causes for the elevation of the transcript, we first focused on the link between pqsDE and phnA. Northern blot analysis of these transcripts were inconclusive, so we utilized RT-PCR to determine whether there was a transcriptional link between pqsE and phnAB, which had been previously reported to be separate (20). We confirmed that in rich medium, there is no transcriptional link between pqsE and phnA in the wild-type strain, PAO1, and that there is a link in the G1041A mutant (Fig. 5). Most interestingly, this link was present in both the wild-type and mutant strains grown under nutrient-limiting conditions (Fig. 5). These data suggested that an alternate transcript is activated under nutrient-limiting conditions and that tryptophan starvation causes a mutation that greatly induces this transcript. This was demonstrated by showing that in the wild-type strain, a transcript containing pqsD was 2.5-fold higher in cultures grown under nutrient-limiting conditions than in one grown in rich medium (Fig. 6). These results suggest that the transcript between pqsD, pqsE, and phnA is likely expressed only under nutritionally stressed conditions and further explained our previous results which indicated that PhnA could restore PQS to a kynurenine pathway mutant only under nutrient-limiting conditions (24). These data also nicely corroborate those of Palmer et al. (30), who showed that overexpression of phnAB complements tryptophan auxotrophy and PQS production in a trpE mutant. Considering all of this, we believe that we have identified a unique natural transcript that allows for differential expression of phnAB when grown under different conditions. An alternative explanation for our data is that the G1041A mutation could change mRNA stability or result in antitermination of the pqsABCDE transcript. This seems unlikely, as the novel transcript we describe which connects pqsE and phnA was observed in the wild-type strain under minimal-medium growth conditions, but a change in message stability and/or termination in the mutant is possible. Nevertheless, it is clear that the mutation does provide for a significant increase in the transcript, and the exact mechanism through which this happens is unknown.
We attempted to directly locate the start site of the novel transcript but did not succeed and had to resort to a reporter deletion analysis to roughly map the minimal transcriptional control element. These data showed that the positive regulatory element was at least 87 bp upstream from the pqsD start codon, which causes us to speculate that the G1041A mutation is probably in a −10 region of a promoter (Fig. 7). The promoter is apparently naturally responsive to low-nutrient conditions (Fig. 5) and selective pressure, such as tryptophan auxotrophy, will cause it to be mutated so that it is highly induced. It should also be noted here that a previous transcriptome study on P. aeruginosa strain PA14 suggested that there is a potential transcriptional start near the stop codon of pqsC, which would be very close to the G1041A mutation, but this start site was not confirmed (51).
Our data clearly show that the G1041A mutation resulted in the upregulation of an alternate transcript that includes pqsD, pqsE, and phnA. A large increase in PqsE would cause PQS production to stop while activating PQS-controlled genes in the absence of PQS (47–49). At the same time, an increase in PhnAB will complement the loss of TrpE. The lack of 4-quinolone production would also ensure that anthranilate, for which the cell is starved, can be utilized in pathways that are important for survival. In general, these interesting findings indicate that anthranilate production is highly important to P. aeruginosa and that multiple layers of regulatory mechanisms exist to ensure the production of this metabolite.
ACKNOWLEDGMENTS
This work was supported by research grants from the National Institute of Allergy and Infectious Disease (grants R01-AI076272 and R56-AI076272).
We thank J. Farrow and K. Tipton for help with manuscript preparation and thoughtful insight.
Footnotes
Published ahead of print 18 April 2014
REFERENCES
- 1.Lyczak JB, Cannon CL, Pier GB. 2000. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2:1051–1060. 10.1016/S1286-4579(00)01259-4 [DOI] [PubMed] [Google Scholar]
- 2.Stuart B, Lin JH, Mogayzel PJ., Jr 2010. Early eradication of Pseudomonas aeruginosa in patients with cystic fibrosis. Paediatr. Respir. Rev. 11:177–184. 10.1016/j.prrv.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, Rosenfeld M, Hiatt P, McCoy K, Castile R, Smith AL, Ramsey BW. 2001. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J. Infect. Dis. 183:444–452. 10.1086/318075 [DOI] [PubMed] [Google Scholar]
- 4.Gambello MJ, Iglewski BH. 1991. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J. Bacteriol. 173:3000–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearson JP, Gray KM, Passador L, Tucker KD, Eberhard A, Iglewski BH, Greenberg EP. 1994. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc. Natl. Acad. Sci. U. S. A. 91:197–201. 10.1073/pnas.91.1.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuster M, Lostroh CP, Ogi T, Greenberg EP. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J. Bacteriol. 185:2066–2079. 10.1128/JB.185.7.2066-2079.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH. 2003. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J. Bacteriol. 185:2080–2095. 10.1128/JB.185.7.2080-2095.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pesci EC, Milbank JB, Pearson JP, McKnight S, Kende AS, Greenberg EP, Iglewski BH. 1999. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 96:11229–11234. 10.1073/pnas.96.20.11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diggle SP, Winzer K, Chhabra SR, Worrall KE, Camara M, Williams P. 2003. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol. Microbiol. 50:29–43. 10.1046/j.1365-2958.2003.03672.x [DOI] [PubMed] [Google Scholar]
- 10.Gallagher LA, McKnight SL, Kuznetsova MS, Pesci EC, Manoil C. 2002. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J. Bacteriol. 184:6472–6480. 10.1128/JB.184.23.6472-6480.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau GW, Hassett DJ, Ran H, Kong F. 2004. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol. Med. 10:599–606. 10.1016/j.molmed.2004.10.002 [DOI] [PubMed] [Google Scholar]
- 12.McKnight SL, Iglewski BH, Pesci EC. 2000. The Pseudomonas quinolone signal regulates rhl quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 182:2702–2708. 10.1128/JB.182.10.2702-2708.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palmer KL, Mashburn LM, Singh PK, Whiteley M. 2005. Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J. Bacteriol. 187:5267–5277. 10.1128/JB.187.15.5267-5277.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Déziel E, Gopalan S, Tampakaki AP, Lepine F, Padfield KE, Saucier M, Xiao G, Rahme LG. 2005. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-L-homoserine lactones. Mol. Microbiol. 55:998–1014. 10.1111/j.1365-2958.2004.04448.x [DOI] [PubMed] [Google Scholar]
- 15.Lau GW, Goumnerov BC, Walendziewicz CL, Hewitson J, Xiao W, Mahajan-Miklos S, Tompkins RG, Perkins LA, Rahme LG. 2003. The Drosophila melanogaster toll pathway participates in resistance to infection by the gram-negative human pathogen Pseudomonas aeruginosa. Infect. Immun. 71:4059–4066. 10.1128/IAI.71.7.4059-4066.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lau GW, Ran H, Kong F, Hassett DJ, Mavrodi D. 2004. Pseudomonas aeruginosa pyocyanin is critical for lung infection in mice. Infect. Immun. 72:4275–4278. 10.1128/IAI.72.7.4275-4278.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahme LG, Tan MW, Le L, Wong SM, Tompkins RG, Calderwood SB, Ausubel FM. 1997. Use of model plant hosts to identify Pseudomonas aeruginosa virulence factors. Proc. Natl. Acad. Sci. U. S. A. 94:13245–13250. 10.1073/pnas.94.24.13245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collier DN, Anderson L, McKnight SL, Noah TL, Knowles M, Boucher R, Schwab U, Gilligan P, Pesci EC. 2002. A bacterial cell to cell signal in the lungs of cystic fibrosis patients. FEMS Microbiol. Lett. 215:41–46. 10.1111/j.1574-6968.2002.tb11367.x [DOI] [PubMed] [Google Scholar]
- 19.Lépine F, Milot S, Deziel E, He J, Rahme LG. 2004. Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa. J. Am. Soc. Mass Spectrom. 15:862–869. 10.1016/j.jasms.2004.02.012 [DOI] [PubMed] [Google Scholar]
- 20.McGrath S, Wade DS, Pesci EC. 2004. Dueling quorum sensing systems in Pseudomonas aeruginosa control the production of the Pseudomonas quinolone signal (PQS). FEMS Microbiol. Lett. 230:27–34. 10.1016/S0378-1097(03)00849-8 [DOI] [PubMed] [Google Scholar]
- 21.Coleman JP, Hudson LL, McKnight SL, Farrow JM, III, Calfee MW, Lindsey CA, Pesci EC. 2008. Pseudomonas aeruginosa PqsA is an anthranilate-coenzyme A ligase. J. Bacteriol. 190:1247–1255. 10.1128/JB.01140-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dulcey CE, Dekimpe V, Fauvelle DA, Milot S, Groleau MC, Doucet N, Rahme LG, Lepine F, Deziel E. 2013. The end of an old hypothesis: the Pseudomonas signaling molecules 4-hydroxy-2-alkylquinolines derive from fatty acids, not 3-ketofatty acids. Chem. Biol. 20:1481–1491. 10.1016/j.chembiol.2013.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oglesby AG, Farrow JM, III, Lee JH, Tomaras AP, Greenberg EP, Pesci EC, Vasil ML. 2008. The influence of iron on Pseudomonas aeruginosa physiology: a regulatory link between iron and quorum sensing. J. Biol. Chem. 283:15558–15567. 10.1074/jbc.M707840200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farrow JM, III, Pesci EC. 2007. Two distinct pathways supply anthranilate as a precursor of the Pseudomonas quinolone signal. J. Bacteriol. 189:3425–3433. 10.1128/JB.00209-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knoten CA, Hudson LL, Coleman JP, Farrow JM, III, Pesci EC. 2011. KynR, a Lrp/AsnC-type transcriptional regulator, directly controls the kynurenine pathway in Pseudomonas aeruginosa. J. Bacteriol. 193:6567–6575. 10.1128/JB.05803-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilbert KB, Kim TH, Gupta R, Greenberg EP, Schuster M. 2009. Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR. Mol. Microbiol. 73:1072–1085. 10.1111/j.1365-2958.2009.06832.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Essar DW, Eberly L, Hadero A, Crawford IP. 1990. Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications. J. Bacteriol. 172:884–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamir H, Srinivasan PR. 1971. Studies of the mechanism of anthranilate synthase. Effect of hydroxylamine and N-methylhydroxylamine. J. Biol. Chem. 246:3024–3029 [PubMed] [Google Scholar]
- 29.Queener SF, Gunsalus IC. 1970. Anthranilate synthase enzyme system and complementation in Pseudomonas species. Proc. Natl. Acad. Sci. U. S. A. 67:1225–1232. 10.1073/pnas.67.3.1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer GC, Jorth PA, Whiteley M. 2013. The role of two Pseudomonas aeruginosa anthranilate synthases in tryptophan and quorum signal production. Microbiology 159:959–969. 10.1099/mic.0.063065-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao H, Krishnan G, Goumnerov B, Tsongalis J, Tompkins R, Rahme LG. 2001. A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proc. Natl. Acad. Sci. U. S. A. 98:14613–14618. 10.1073/pnas.251465298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wade DS, Calfee MW, Rocha ER, Ling EA, Engstrom E, Coleman JP, Pesci EC. 2005. Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. J. Bacteriol. 187:4372–4380. 10.1128/JB.187.13.4372-4380.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Déziel E, Lepine F, Milot S, He J, Mindrinos MN, Tompkins RG, Rahme LG. 2004. Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc. Natl. Acad. Sci. U. S. A. 101:1339–1344. 10.1073/pnas.0307694100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Essar DW, Eberly L, Han CY, Crawford IP. 1990. DNA sequences and characterization of four early genes of the tryptophan pathway in Pseudomonas aeruginosa. J. Bacteriol. 172:853–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogel HJ, Bonner DM. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97–106 [PubMed] [Google Scholar]
- 36.Enderle PJ, Farwell MA. 1998. Electroporation of freshly plated Escherichia coli and Pseudomonas aeruginosa cells. Biotechniques 25:954–956, 958 [DOI] [PubMed] [Google Scholar]
- 37.Warrens AN, Jones MD, Lechler RI. 1997. Splicing by overlap extension by PCR using asymmetric amplification: an improved technique for the generation of hybrid proteins of immunological interest. Gene 186:29–35. 10.1016/S0378-1119(96)00674-9 [DOI] [PubMed] [Google Scholar]
- 38.Choi KH, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64:391–397. 10.1016/j.mimet.2005.06.001 [DOI] [PubMed] [Google Scholar]
- 39.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. 10.1016/S0378-1119(98)00130-9 [DOI] [PubMed] [Google Scholar]
- 40.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39:D596–D600. 10.1093/nar/gkq869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol. 29:24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14:178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stone EA. 2012. Joint genotyping on the fly: identifying variation among a sequenced panel of inbred lines. Genome Res. 22:966–974. 10.1101/gr.129122.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diggle SP, Cornelis P, Williams P, Camara M. 2006. 4-Quinolone signalling in Pseudomonas aeruginosa: old molecules, new perspectives. Int. J. Med. Microbiol. 296:83–91. 10.1016/j.ijmm.2006.01.038 [DOI] [PubMed] [Google Scholar]
- 48.Farrow JM, III, Sund ZM, Ellison ML, Wade DS, Coleman JP, Pesci EC. 2008. PqsE functions independently of PqsR-Pseudomonas quinolone signal and enhances the rhl quorum-sensing system. J. Bacteriol. 190:7043–7051. 10.1128/JB.00753-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hazan R, He J, Xiao G, Dekimpe V, Apidianakis Y, Lesic B, Astrakas C, Deziel E, Lepine F, Rahme LG. 2010. Homeostatic interplay between bacterial cell-cell signaling and iron in virulence. PLoS Pathog. 6:e1000810. 10.1371/journal.ppat.1000810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rampioni G, Pustelny C, Fletcher MP, Wright VJ, Bruce M, Rumbaugh KP, Heeb S, Camara M, Williams P. 2010. Transcriptomic analysis reveals a global alkyl-quinolone-independent regulatory role for PqsE in facilitating the environmental adaptation of Pseudomonas aeruginosa to plant and animal hosts. Environ. Microbiol. 12:1659–1673. 10.1111/j.1462-2920.2010.02214.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dötsch A, Eckweiler D, Schniederjans M, Zimmermann A, Jensen V, Scharfe M, Geffers R, Haussler S. 2012. The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS One 7:e31092. 10.1371/journal.pone.0031092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woodcock DM, Crowther PJ, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, Smith SS, Michael MZ, Graham MW. 1989. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 17:3469–3478. 10.1093/nar/17.9.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holloway BW, Krishnapillai V, Morgan AF. 1979. Chromosomal genetics of Pseudomonas. Microbiol. Rev. 43:73–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Preston MJ, Seed PC, Toder DS, Iglewski BH, Ohman DE, Gustin JK, Goldberg JB, Pier GB. 1997. Contribution of proteases and LasR to the virulence of Pseudomonas aeruginosa during corneal infections. Infect. Immun. 65:3086–3090 [DOI] [PMC free article] [PubMed] [Google Scholar]