

Abstract
Clostridium perfringens is a Gram-positive anaerobic pathogen of humans and animals. Although they lack flagella, C. perfringens bacteria can still migrate across surfaces using a type of gliding motility that involves the formation of filaments of bacteria lined up in an end-to-end conformation. In strain SM101, hypermotile variants are often found arising from the edges of colonies on agar plates. Hypermotile cells are longer than wild-type cells, and video microscopy of their gliding motility suggests that they form long, thin filaments that move rapidly away from a colony, analogously to swarmer cells in bacteria with flagella. To identify the cause(s) of the hypermotility phenotype, the genome sequences of normal strains and their direct hypermotile derivatives were determined and compared. Strains SM124 and SM127, hypermotile derivatives of strains SM101 and SM102, respectively, contained 10 and 6 single nucleotide polymorphisms (SNPs) relative to their parent strains. While SNPs were located in different genes in the two sets of strains, one feature in common was mutations in cell division genes, an ftsI homolog in strain SM124 (CPR_1831) and a minE homolog in strain SM127 (CPR_2104). Complementation of these mutations with wild-type copies of each gene restored the normal motility phenotype. A model explaining the principles underlying the hypermotility phenotype is presented.
INTRODUCTION
Clostridium perfringens is a Gram-positive, spore-forming anaerobic bacterium that is capable of causing numerous diseases in humans and animals, including intestinal and invasive tissue infections (1). C. perfringens genome sequencing indicates that the species lacks flagella and any recognizable chemotaxis system (2, 3). However, we reported previously that C. perfringens and other Clostridium species possess type IV pili (TFP) and exhibit a unique type of social gliding motility on agar plates (4). C. perfringens motile cells align themselves in an end-to-end configuration along their long axes to form filaments that move across the surface of an agar plate (4, 5). Most filaments have other filaments attached to them to form thicker elements we refer to as flares. If one end of a flare is attached to a colony, its extension in that direction is blocked, and the filament moves in a direction pointing away from the colony. This motility is dependent on the products of the pilT and pilC1 genes (4), although the precise role that TFP play in this motility is still not understood.
The key factors that seem to support motility are the formation of the end-to-end connections and the growth and division of individual cells within the filament. Evidence in support of these factors comes from our study in which a pool of mariner transposon insertion mutants in strain 13 was screened for mutations that prevented gliding motility on brain heart infusion (BHI) agar plates. Mutations in several different categories of genes were found to be associated with deficient motility, but the most common were in genes encoding proteins likely to be associated with the cell envelope (6). One of these genes encoded an endopeptidase, SagA, predicted to be involved in modifying peptidoglycan (6). This mutant lacked end-to-end connections between the bacteria and was unable to form motile filaments, but the mutant phenotype was reversed by complementation with a wild-type copy of the gene (6). However, the composition of the material that forms the end-to-end connections is still unknown.
Multiple factors appear to play a role in the initiation of filament formation and gliding motility. In liquid cultures composed of the same nutrients as those found in agar plates (e.g., BHI), the cells do not form end-to-end connections and so do not form filaments (4). This implies that the bacteria can sense when they are in contact with a surface and can modify their surface appendages, a feature shared by Pseudomonas aeruginosa, which produces more TFP on agar surfaces than in liquid media (7). Carbohydrates in the form of readily metabolizable hexoses act to suppress motility on agar plates, and this suppression is mediated via the carbohydrate catabolite regulatory protein, CcpA, in C. perfringens strain 13 (8). Interestingly, CcpA was also required for maximum motility in strain 13 in the absence of added sugar (8). We detected similar phenotypes in wild-type strain SM101 and a ccpA mutant (9).
If a concentrated bacterial suspension of C. perfringens strain 13, SM101, or ATCC 13124 is inoculated onto plates containing BHI with 1% agar, the growth of the bacteria at the site of inoculation leads to a slight increase in the diameter of the colony (4, 8). After 12 to 24 h, thick flares can be seen migrating away from the original inoculation site (4, 8), and the cells within the flares are usually aligned in the end-to-end conformation (4). However, of these three strains, only strain SM101 exhibited a third form visible on plates: a rapidly migrating thin film of bacteria that can be seen emerging from the edge of a colony (see Fig. 1). After cells in the hypermotile flares were moved to new plates, they maintained the hypermotile phenotype, suggesting a genetic basis for this phenotype. To identify the mechanism, we sequenced the genomes of two hypermotile derivatives and their parental strains, one from SM101 and the other from a closely related strain, SM102. The hypermotile strains had multiple single nucleotide polymorphisms (SNPs) in different genes. However, two genes that encoded proteins involved in cell division and led to the production of longer cells were solely responsible for the hypermotile phenotype. These results suggest that there is a link between the length of individual cells and the macroscopic appearance of gliding motility in C. perfringens.
FIG 1.

Representative images showing the migration of different strains of C. perfringens on BHI plates (1% agar). A suspension of cells was spotted onto plates at the beginning of the experiment; the extents of the original inocula are indicated by dotted lines in each image. (A) NCTC 8798; (B) SM101; (C) SM102; (D) SM124; (E) SM124(pHLL59); (F) SM124(pKRAH1); (G) SM127; (H) SM127(pHLL61); (I) SM127(pKRAH1). For panels E, F, H, and I, 0.5 mM lactose and 20 μg/ml chloramphenicol were added to the medium. Arrows point to migration flares moving away from the site of inoculation. Bars, 1 cm.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains, plasmids, and primers used in this study are listed in Table 1. Escherichia coli was grown in Luria-Bertani (LB) medium supplemented with antibiotics as needed (100 μg/ml ampicillin, 20 μg/ml chloramphenicol) and 1% agar for plates. Three different media were used for C. perfringens: BHI (Difco), fluid thioglycolate (FTG) (Difco), and Duncan-Strong sporulation medium with 0.4% raffinose (DSSM) (10). BHI was supplemented with 20 μg/ml chloramphenicol and 0.5 mM lactose for the following four C. perfringens strains: SM124(pKRAH1), SM124(pHLL59), SM127(pKRAH1), and SM127(pHLL61). All C. perfringens cultures were incubated in a Coy anaerobic chamber at 37°C under an atmosphere of 85% N2, 10% CO2, and 5% H2.
TABLE 1.
Strains and plasmids used in this study
| Strain, plasmid, or primer | Relevant characteristic(s) or 5′-to-3′ sequence | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH10B | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 lacX74 deoR recA1 araD139 Δ(ara-leu)7697 galU galK ΔrpsL endA1 nupG | Gibco/BRL |
| C. perfringens | ||
| NCTC 8798 | Acute food poisoning strain | R. Labbe |
| SM101 | Electroporation-efficient derivative of strain NCTC 8798 | 18 |
| SM102 | Electroporation-efficient derivative of strain NCTC 8798 | 18 |
| SM124 | SM101 with mutation in gene CPR_1831 | This study |
| SM127 | SM102 with mutation in gene CPR_2104 | This study |
| Plasmids | ||
| pGEM-T Easy | Cloning vector | Promega |
| pKRAH1 | Contains bgaR-PbgaL and polylinker; chloramphenicol resistance | 13 |
| pHLL58 | Contains the CPR_1831 coding sequence in pGEM-T Easy | This study |
| pHLL59 | Contains the CPR_1831 coding sequence in pKRAH1 | This study |
| pHLL60 | Contains the CPR_2104 coding sequence in pGEM-T Easy | This study |
| pHLL61 | Contains the CPR_2104 coding sequence in pKRAH1 | This study |
| Primers | ||
| OHL141 | GTCGACGAACTCATTTTATTTAACTTACTACGGAGGG | This study |
| OHL142 | GGATCCCACAAAATACTAGCATGTGAAATTACTC | This study |
| OKM13 | GTCGACTCTATATTAAAGTTGTTTAGAAGAGGG | This study |
| OKM14 | GGATCCTTATCTAGCTTTACCCTTTATATTTTTTATTG | This study |
To measure growth rates and yields of C. perfringens strains SM101, SM124, SM124(pKRAH1), and SM124(pHLL59), 5 ml of an overnight liquid culture of each strain was adjusted to an optical density at 600 nm (OD600) of ≈1 and was then inoculated into 45 ml of BHI liquid medium. Because strain SM127 cells tended to lyse after several hours in stationary phase (data not shown), for strains SM102, SM127, SM127(pKRAH1), and SM127(pHLL61), 5 ml of a mid-log-phase liquid culture of each strain was inoculated into BHI liquid medium at the start of the experiment. One milliliter of culture was removed every hour, and the OD600 value was measured in 1-cm cuvettes in a Genesys 10S spectrophotometer (Thermo Scientific) until the culture reached stationary phase. Since the complementing plasmids have the gene of interest behind a lactose-inducible promoter (Table 1), lactose was added every 2 h to bring the final concentration to 0.5 mM. One of two duplicate curves for each strain is shown in Fig. S1 in the supplemental material.
Genome sequencing.
Chromosomal DNA was isolated from nonmotile and hypermotile strains by using a modified version of the method of Pospiech and Neumann (11). For C. perfringens strains, instead of a single chloroform extraction, the cell lysates were extracted once with phenol, twice with phenol-chloroform-isoamyl alcohol (25:24:1), and once with chloroform. The sequence library was prepared as follows. Genomic DNA was sheared to 500 bp using a Covaris M220 focused ultrasonicator (Covaris, Woburn, MA). Sonication was carried out at a peak incident power of 50 W, a duty factor of 20%, and 100 cycles per burst for 50 s. DNA-Seq libraries were constructed using Illumina's TruSeq DNA PCR-free sample preparation kit, version 2, set A/B (part no. FC-121-3001). A 650-bp library was selected by a double SPRI AMPure XP protocol. Each library was individually bar coded to enable multiplexing on a single MiSeq sequencing run. Each library was quantitated using a Quant-iT dsDNA HS kit (Invitrogen), and the library size was validated on a BioAnalyzer instrument, model 2200. The libraries were further quantitated by using quantitative PCR (qPCR) to generate optimal sequencing densities. For MiSeq sequencing, individual libraries were pooled in equimolar amounts, denatured, and loaded onto a MiSeq sequencer at 10 pM. The pooled library was sequenced to 2 × 250 paired ends (PE) on the MiSeq sequencer by using a 500-cycle MiSeq reagent kit, version 2 (part no. MS-102-2003).
Sequence analysis.
The FASTQ files containing 4,000,000 to 15,000,000 250-bp PE reads for each sample were aligned to the C. perfringens SM101 genome (NC_008262.1), which was used as a reference, with Geneious software, version 6.1.2 (Biomatters). Variants consisting of single base substitutions and insertions and deletions were called if they occurred in >90% of the aligned reads with 40% to 60% representation in forward and reverse orientations and >100× coverage. Thirty-one variants from the reference were found in every sample (see Table S1 in the supplemental material). These were presumed to be either variants in the current parental laboratory strains or errors in the original reference assembly. Therefore, the analysis of genotypic variants associated with phenotypic changes focused on the remaining base substitutions or indels that were unique to individual samples. The positions of all variants are reported relative to the SM101 reference genome.
Plasmid construction.
DNA manipulation was performed using standard protocols (12). Two plasmids, pHLL59 and pHLL61, were constructed to complement mutations in CPR_1831 and CPR_2104, respectively. Primers OHL141 and OHL142 were used to amplify the coding sequence of CPR_1831 and its own ribosome binding site. A SalI site was included in primer OHL141, while a BamHI site was included in OHL142. The PCR product was ligated to pGEM-T Easy (Promega) to create plasmid pHLL58, and the gene was then cloned into pKRAH1 (13) to create plasmid pHLL59. The CPR_2104 complementing plasmid, pHLL61, was constructed in the same manner using primers OKM13 and OKM14. E. coli and C. perfringens were transformed by electroporation as described previously (13). All constructs were verified by DNA sequencing.
Colony and cell morphologies and video microscopy.
Five microliters of log-phase liquid culture was spotted onto BHI plates (supplemented with chloramphenicol and lactose if needed) and incubated in the anaerobic chamber for 40 h. Images of colonies were obtained using a Bio-Rad Gel Doc XR imager with Applied One software, version 4.6.5. For video microscopy, bacteria grown overnight on a BHI agar plate were inoculated into a tissue culture flask coated with a thin layer of BHI agar medium (4) and were incubated for 30 min in an anaerobic chamber. The cap on the flask was then closed, and the flask was removed from the chamber and was imaged in an Olympus IX81 upright microscope linked to a Hamamatsu model C4742 charge-coupled device (CCD) camera. SlideBook software, version 5.1 (Intelligent Imaging Innovations), was used to compile time lapse videos and to measure the lengths of cephalexin-treated cells. For all other conditions and strains, cell lengths were measured by using phase-contrast images obtained from an Olympus IX81 upright microscope linked to a DeltaVision imaging software package (Applied Precision). MicrobeTracker software (14) linked to a MATLAB (MathWorks) platform was used to measure the cell lengths and widths of ∼1,000 to ∼6,000 individual bacteria, depending on the strain, in the phase-contrast images.
Sporulation assay.
To determine if the CPR_1831 gene plays a role in sporulation, we performed sporulation assays with C. perfringens SM101 and SM124 as described previously (15). Briefly, cells were inoculated into 4 ml BHI liquid medium and were incubated for 7 h. Then 40 μl of the BHI culture was transferred to 4 ml of FTG medium and was incubated for 18 h. In triplicate, 40 μl of the FTG culture was transferred to 4 ml of DSSM and was incubated for 24 h. Cultures were divided in half. One half was incubated at 75°C for 10 min to kill vegetative cells, and one half was left untreated. Both were then serially diluted in Dulbecco's phosphate-buffered saline (DPBS) and were plated on 0.5× strength BHI agar plates, and the number of CFU was determined.
Measurement of NAM levels as an indicator of PG.
C. perfringens cell suspensions were made in triplicate by scraping bacteria from the edges of colonies grown overnight on BHI plates, suspending them in DPBS, and washing once with 1 mM MgCl2. Cells at a known OD600 were lyophilized, acid hydrolyzed, and subjected to amino sugar analysis as described previously (16, 17). Hydrolysis and analysis of known N-acetyl-muramic acid (NAM) standards allowed determination of the amount of muramic acid per OD600 unit. This amount was then used to calculate the amount of peptidoglycan (PG) in units of nmol NAM/μm2 of cell surface. This was done by first measuring the number of cells per ml, using a hemocytometer, in a suspension of bacteria at a known OD600 value to calculate the number of bacteria per OD600 unit (see Table S2 in the supplemental material). This number was then multiplied by the surface area of an average bacterium (see Fig. 2B and Table S2 in the supplemental material) on the assumption that the bacteria were cylindrical and that thus, the formula for the surface area of a cylinder (A = 2πr2 + 2πrl, where l is the length of an average cell) applied. This product gave the total surface area of all the bacteria in the suspension in units of μm2 of cell surface/ml. After correction for the actual OD600 reading of the sample, the number of nmol NAM/ml was then divided by the μm2 of cell surface/ml to give nmol NAM/μm2 of cell surface.
FIG 2.

(A) Representative images showing cells of different strains scraped off plates. For strains SM124(pHLL59), SM124(pKRAH1), SM127(pHLL61), and SM127(pKRAH1), 0.5 mM lactose and 20 μg/ml chloramphenicol were added to the medium. Bars, 10 μm. (B) Lengths of bacteria isolated from plates grown under the same conditions as those used for panel A. Values are means and SEM. Brackets indicate strains that differ significantly in length. P values were obtained using the Student double-tailed t test.
Cell volume produced by colonies of each strain.
The cell volume produced by individual colonies was measured by inoculating duplicate BHI agar plates in four spots with 10 μl of log-phase cells from a liquid BHI culture. The plates were incubated at 37°C in the anaerobic chamber for 40 h. The cells were scraped off the plate with a plastic loop, and the remaining cells were recovered by washing the plate with 3 ml of DPBS. The cell suspension was pelleted by centrifugation and was resuspended in 8 ml of DPBS, and the OD600 was measured. This value was then used to calculate the total volume by using the formula for the volume of a cylinder and the length of an average cell from each strain.
Sensitivity to osmotic stress.
Liquid-culture cells in log phase were pelleted by centrifugation, washed once with ultrapure water, then resuspended in water, and incubated at room temperature for 30 min, followed by serial dilution and plating on BHI. Three individual replicates were tested for each strain. As a control, the same liquid culture for each strain was subjected to serial dilution and plating, but without incubation in water. The number of CFU was determined after overnight incubation at 37°C.
Cephalexin-induced filamentous growth.
C. perfringens SM101 and SM102 cells were struck out on BHI plates supplemented with increasing concentrations of cephalexin and were incubated overnight at 37°C. Light microscopy images indicated that all of the cephalexin concentrations induced filamentous growth, but only the cells from the plate with the lowest concentration of cephalexin (0.1 μg/ml) failed to exhibit significant autolysis. To determine whether cephalexin-induced filamentous growth can cause the spreading colony phenotype in wild-type strains SM101 and SM102, 5 μl of liquid culture in log phase was spotted onto BHI plates supplemented with 0.1 μg/ml cephalexin. The culture was incubated for 40 h at 37°C, and images of colonies were obtained as described above.
RESULTS
Spontaneous hypermotile variants arise in C. perfringens strains SM101 and SM102.
Strains SM101 and SM102 are highly electroporation competent derivatives of the acute food poisoning strain NCTC 8798 (18). When cultured on BHI with 1% agar, strains SM101 and SM102 exhibit normal gliding motility, which can be seen as the 1- to 2-mm spreading zone beyond the site of inoculation (Fig. 1B and C). At random points, rapidly migrating zones of bacteria moving away from the initial zone of inoculation can be seen (Fig. 1B and C, arrows). The parent strain of SM101 and SM102, NCTC 8798, also shows these hypermotile derivatives (Fig. 1A), suggesting that SM101 and SM102 are not different in that respect. When suspensions of hypermotile strains SM124 and SM127, derived from strains SM101 and SM102, respectively, were spotted onto plates, they maintained their rapidly migrating phenotype (Fig. 1D and G). Closer examination showed that the SM124 and SM127 colonies, while spreading rapidly, were thinner than the thick flares on the outer edges of the slower-moving parental strains, which had a mucoid appearance (compare the centers of Fig. 1B and C to those of Fig. 1D and G). This suggested a genetic rather than a physiological basis for the dramatic change in motility.
Examination of cells scraped off BHI agar plates indicated that strains SM124 and SM127 were significantly longer than their respective parent strains, SM101 and SM102 (Fig. 2A). Measurements of cell lengths using phase-contrast images of cells and MicrobeTracker software indicated that SM124 cells were 1.75 times longer than SM101 cells, while SM127 cells were 2.4 times longer than SM102 cells (Fig. 2B), but there were no differences in bacterial widths (data not shown).
Video microscopy of cells taken from the edge of a colony on a plate and spread on BHI with 1% agar in tissue culture flasks indicated that strains SM124 and SM127 made long, thin filaments of cells attached end to end and that each individual cell was longer, on average, than cells of strains SM101 and SM102 (compare the cells in Video S1 to those in Video S3 and the cells in Video S2 to those in Video S4 in the supplemental material). The long, thin filaments in strains SM124 and SM127 also appear to move in a straighter, less curvilinear fashion than those of strains SM101 and SM102, leading to greater extension away from the source colony (see Videos S1 to S4 in the supplemental material). Taken together, these microscopic details are consistent with the macroscopic appearance of the colonies on agar plates (Fig. 1).
Identification of genetic differences between the parental and hypermotile strains.
Because the hypermotile strains appeared to have undergone a gain-of-function phenotypic change that was genetically stable, we wanted to determine if this was due to changes in the sequence of the genome, such as SNPs, inversions, or deletions. Therefore, we isolated chromosomal DNA from each parental strain and its hypermotile derivative and used next-generation sequencing methods to identify changes. The sequence for strain SM101 has been determined previously by using the Sanger method on chromosomal DNA isolated in our laboratory (3). Comparison of the DNA sequences for strain SM101 from this report to the previously published sequence showed that 31 SNPs and short sequence differences were present (see Table S1 in the supplemental material). These could be due to actual changes in the genomes or to sequencing errors, but it is difficult to differentiate between these two scenarios. Ten SNPs were identified between strains SM101 and SM124 (Table 2) and 6 between strains SM102 and SM127 (Table 3). The two plasmids and the episomal phage chromosome found in strain SM101 (3) were present in all strains, but none contained SNPs. Also, none of the SNPs in one pair of strains were located in, or adjacent to, the same gene as the SNPs in the other pair (Tables 2 and 3), suggesting that different genetic changes were involved in producing the hypermotile phenotype in SM124 and SM127. Interestingly, 12 of the 16 SNPs involved GC-to-AT mutations, even though strain SM101 is only ∼28% GC (3). Although the sample is small, this finding suggests an increased mutation rate bias in the replication of GC bases, a phenomenon seen in many organisms (19).
TABLE 2.
Location of SNPs between the parental strain SM101 and its hypermotile derivative strain SM124
| Gene | Annotated protein functiona | Locationb | Base change | Codon changec | Mutation type |
|---|---|---|---|---|---|
| CPR_0055 | DNA polymerase III subunits gamma and tau | 61664 | T to G | AAT to AAG (Asn to Lys) | Substitution |
| CPR_0185–CPR_0186 | Between cobalt transporter subunit and hypothetical protein | 227182 | C to T | ||
| CPR_0458 | N-Acetylmannosaminyl transferase | 544247 | C to A | GCT to GAT (Ala to Asp) | Substitution |
| CPR_0547–CPR_0548 | Between ISCpe2 transposase and phosphate transport regulator CPR_0548 | 648489 | G to T | ||
| CPR_0911 | Lipoprotein, putative CDS | 1043513 | T to G | AAT to AAG (Asn to Lys) | Substitution |
| CPR_1135 | Transcriptional regulator of fatty acid biosynthesis FabT | 1281879 | G to T | GTT to TTT (Val to Phe) | Substitution |
| CPR_1215 | Activator of (R)-2-hydroxyglutaryl-CoA dehydratase | 1365085 | C to A | GAG to GAT (Glu to Asp) | Substitution |
| CPR_1831 | Cell division protein FtsI (peptidoglycan synthetase) | 2023347 | C to A | GAA to TAA (Glu to stop codon) | Truncation |
| CPR_2039 | Topoisomerase IV subunit A | 2253090 | T to G | AAA to CAA (Lys to Gln) | Substitution |
| CPR_2578 | Hypothetical protein | 2808164 | C to A | TAC to TAA (Tyr to stop codon) | Truncation |
CDS, coding sequence; CoA, coenzyme A.
Location in published sequence of strain SM101 (3).
May differ from base change if coding sequence is on reverse strand.
TABLE 3.
Location of SNPs between the parental strain SM102 and its hypermotile derivative strain SM127
| Gene | Annotated protein functiona | Locationb | Base change | Codon changec | Mutation type |
|---|---|---|---|---|---|
| CPR_0456 | UDP-glucose dehydrogenase | 542746 | G to T | GAG to TAG (Glu to stop codon) | Truncation |
| CPR_0742–CPR_0743 | Between two proton/glutamate symporters and HAD hydrolase | 868274 | A to C | ||
| CPR_0456 | Putative membrane protein CDS | 1450256 | G to T | GAC to GAA (Asp to Glu) | Substitution |
| CPR_1884 | Voltage-gated chloride channel family protein | 2078621 | G to T | GCT to GAT (Ala to Asp) | Substitution |
| CPR_2104 | Cell division topological specificity factor MinE | 2331046 | C to A | GGG to TGG (Gly to Trp) | Substitution |
| CPR_2491–CPR_2492 | Between a foldase protein PrsA precursor and SpoVT, an AbrB family transcriptional regulator | 2705576 | G to T |
HAD, haloacid dehalogenase; CDS, coding sequence.
Location in published sequence of strain SM101 (3).
May differ from base change if coding sequence is on reverse strand.
The one common denominator we could identify was that both lists of SNPs included genes associated with cell division: an ftsI homolog (CPR_1831) in the SM101-SM124 pair (Table 2) and a minE homolog (CPR_2104) in the SM102-SM127 pair (Table 3). Since we noted that cells from the hypermotile strains were longer than those from their parents (Fig. 2), we investigated whether these mutations were the cause of the hypermotility.
Complementation of cell division mutations.
The genes encoding the FtsI and MinE homologs were cloned into a plasmid vector, pKRAH1, which contains a lactose-inducible promoter for regulated gene expression (13). These plasmids, pHLL59 (FtsI) and pHLL61 (MinE), were transformed into strains SM124 and SM127, respectively. In the presence of BHI with 0.5 mM lactose, the colony morphologies of strains SM124(pHLL59) and SM127(pHLL61) were restored to those exhibited by their respective parental strains, SM101 and SM102 (Fig. 1E and H). The control strains containing the empty vector pKRAH1 did not show reversion to the parental colony morphology (Fig. 1F and I). The addition of 0.5 mM lactose to BHI plates did not affect the colony morphology of strain SM101, SM102, SM124, or SM127 (data not shown). Complementation of each mutation also led to a reduction in the average cell length, rendering it comparable to that of the parental strain, while the empty-vector controls did not (Fig. 2). Video microscopy of moving filaments in the complemented strains (see Videos S5 and S6 in the supplemental material) showed that the cells were shorter than those of the hypermotile strains and that the filaments were not as consistently long and straight (a low density of cells was filmed in these videos in order to highlight individual cell lengths and filament structure more clearly).
Other phenotypic changes conferred by the mutations in genes encoding the FtsI and MinE homologs.
The growth rates and growth yields of the parental, mutant, and complemented mutant strains were compared (see Fig. S1 in the supplemental material). There was a longer lag time for strains carrying plasmids but no differences in growth rate or yield for any strain except SM127(pKRAH1), which had a somewhat lower growth rate (see Fig. S1). Using light microscopy, we did note that strain SM127, containing the point mutation in the CPR_2104 gene (the only minE homolog in strain SM101), exhibited a higher level of cell lysis after it reached stationary phase than did any of the other strains, including SM127(pHLL61), but this was not sufficient to affect the growth yield to any great extent (see Fig. S1).
As a test to determine if hypermotility leads to higher cell reproduction efficiency on surfaces, we wanted to determine the total volume of cells produced by colonies of the hypermotile and parent strains. To do this, we measured the OD600 values of all the cells in quadruplicate colonies on duplicate plates, multiplied this by the number of bacteria per OD600 unit of 1.0, and used the formula for the volume of a cylinder with the length of an average bacterium from each strain (see Table S2 in the supplemental material). The hypermotile variant SM124 produced 10% greater cell volume than its parental strain, SM101, but strain SM127 produced only 66% as much cell volume as its parental strain, SM102 (see Table S2).
C. perfringens SM101 contains six genes encoding FtsI homologs (http://www.microbesonline.org/), including CPR_1831. A C-to-A transversion led to a nonsense mutation (Glu to a stop codon) at residue 63 out of 739 and truncation of the open reading frame at that site (Table 2). CPR_1831 is predicted to be the third gene in an 11-gene operon (Fig. 3A). The other annotated genes in the operon (murE, murF, etc.) appear to code for proteins involved in cell wall biosynthesis (Fig. 3A). Also, since the minE mutant strain SM127 produced significantly longer cells than its parent strain (Fig. 2B), it was possible that PG levels would be altered in this strain. To determine if this was the case, we measured the amount of PG (using NAM as an indicator) present in the cells of hypermotile, parental, and complemented hypermotile strains. We found similar levels of PG in cells from strains SM101 and SM124 (see Fig. S2 in the supplemental material), suggesting that the CPR_1831 gene product is not involved in PG synthesis for the entire cell. In contrast, strain SM127 had twice as much PG per unit of cell surface area as its parental strain, but this level was reduced to that of the parent strain when the minE mutation was complemented (see Fig. S2).
FIG 3.
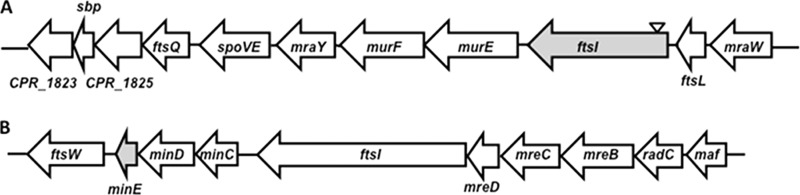
Locations of genes associated with hypermotility (shaded) in C. perfringens strains SM101 and SM102. The annotated putative gene function is shown. If the function is unknown, the gene is given its genome assignment name. (A) CPR_1831. The inverted triangle indicates the location of the nonsense mutation. (B) minE. This gene appears to be the third gene in a minCDE operon, based on the criteria used in the website MicrobesOnline (http://www.microbesonline.org/).
Like the other FtsI homologs in C. perfringens, the product of CPR_1831 is annotated as being in the stage V sporulation protein D (SpoVD) subfamily of penicillin-binding proteins (PBPs) (also called PBP-2B). Since CPR_1831 may encode a SpoVD homolog, we tested the effect of the mutation on sporulation. Strains SM101 and SM124 were grown in DSSM, and the percentages of sporulation were measured, but no significant difference was seen between the two strains (101% ± 53.5% for SM101 and 55.1% ± 38.1% for SM124; P, 0.293 by a two-tailed Student t test). These results suggest that CPR_1831 does not function as a sporulation-associated SpoVD protein in C. perfringens strain SM101.
Since the CPR_1831 and minE mutations leading to increased cell length likely have effects on division septum formation and perhaps on other cell wall-related functions, we tested the parental and hypermotile strains by incubation in distilled water for 30 min to determine if the hypermotile strains were more sensitive to osmotic stress. However, neither of the hypermotile strains was more sensitive to this treatment than the parental strains (data not shown).
The antibiotic cephalexin increases cell length but does not lead to hypermotility.
One feature that the CPR_1831 and minE mutants had in common was increased cell length (Fig. 2). Since it was possible that increased cell length alone was sufficient to induce hypermotility, we looked for a method to induce the formation of long cells artificially. The antibiotic cephalexin has been shown to inhibit FtsI function (20) and, at sublethal doses, to inhibit the completion of septum formation and produce C. perfringens cells of increased length (data not shown). To determine if cephalexin-induced production of longer cells can also cause the hypermotility phenotype in the wild-type strains of C. perfringens, we spotted 5 μl of cells onto BHI plates supplemented with 0.1 μg/ml cephalexin, a concentration that induced the formation of long cells (means ± standard errors of the means [SEM], 13.9 ± 1.1 μm for SM101 and 13.4 ± 1.6 μm for SM102) but not extensive lysis (see Fig. S3A and B in the supplemental material). However, colonies exposed to this concentration did not show the characteristic hypermotility on agar plates (see Fig. S3C and D in the supplemental material). This suggests that increased cell length due to cephalexin exposure was not sufficient for hypermotility.
DISCUSSION
While experimenting with C. perfringens strain SM101, we noticed that rapidly migrating offshoots of cells arose from the edge of a colony at seemingly random times and locations. Neither of the other two strains of C. perfringens with complete genome sequences, strains 13 and ATCC 13124, appears to do this, although all three strains did exhibit the typical thicker flares that move away from a colony, as described previously (4, 6). Why SM101 is unique among these strains in producing this phenotype is unknown, but one possible explanation is that strain SM101 has significantly stronger cell-to-cell connections, which allow long filaments of cells to remain attached without breaking apart as they experience shear forces while moving through a viscous medium such as that found on an agar surface (21).
We identified two mutations that were responsible for the hypermotility phenotype, one in CPR_1831, encoding an FtsI homolog, and the other in a gene encoding a MinE homolog. This was accomplished by successfully restoring wild-type motility and cell size by complementation of the genes with mutations. However, since there are several other SNPs in each strain, it is possible that some of these SNPs, even though they are not directly responsible for the hypermotile phenotype, enable the mutant strains to survive with the defects that cause hypermotility. Since it is unlikely that complementation of each SNP would show an obvious phenotypic difference, we did not carry out these experiments.
The exact function of the FtsI-like CPR_1831 gene product is unknown, but it seems likely to be involved in non-sporulation-associated PG biogenesis, based on the mutant's increased cell length, motility phenotype, predicted function, and genetic location (see Results). Given that there are six separate FtsI-like proteins in strain SM101, there is probably some redundancy or overlap in their functions, so that the mutation leading to loss of CPR_1831 gives a significant phenotype (i.e., hypermotility) but is not lethal to the cell.
In C. perfringens, the minE gene appears to be in an operon with minC and minD (Fig. 3B). The MinCDE system has been well characterized in several species of Gram-negative bacteria. In E. coli, MinE has been shown to oscillate from one end of the cell to the other and to act as a topological specificity factor for the MinCD complex, which inhibits cell division by disrupting FtsZ ring formation (22). Bacillus subtilis, a Gram-positive bacterium, lacks a MinE homolog and instead uses another protein, DivIVA, to localize the MinCD complex (23). Interestingly, C. perfringens and other Clostridium species, such as Clostridium botulinum and Clostridium difficile, have homologs of both MinE and DivIVA (CPR_1819 in C. perfringens strain SM101). Which of these proteins is used (or whether both are used) to specify the location of the MinCD complex in C. perfringens is unknown, but their presence does suggest differences between the clostridial mechanism and both the Bacillus and Gram-negative bacterial mechanisms for divisome formation. Considering the large number of important human and animal pathogens present in the Clostridia and the number of antibiotics that affect cell division, this subject appears to need more research.
The mutation in the strain SM127 minE gene resulted in a Gly71-to-Trp71 substitution. The first ∼30 residues of MinE are thought to function in the inhibition of the MinCD complex, while the rest of the protein is reported to provide the topological specificity in MinE oscillation (24). The structural prediction program FUGUE (25) was used to construct a model of the C. perfringens MinE protein based on the crystal structure of Helicobacter pylori MinE (29% sequence identity). The H. pylori MinE structure is disordered between residues L60 and S65. The model, derived from the FUGUE server (http://tardis.nibio.go.jp/fugue/prfsearch.html) with 99% confidence, suggests that the mutation at Gly71 occurs in an analogous region of the C. perfringens MinE protein (data not shown). Given the location of the mutation, it seems likely that the topological specificity function of MinE was more affected than MinCD inhibition, although there is no direct evidence yet to support this.
Strain SM127 does contain twice the amount of PG per μm2 of surface area as does its parental strain, SM102 (see Fig. S2 in the supplemental material). We are not certain about the reasons for this, but it is interesting that 1 OD600 unit of these cells contains just half the volume of its parent (see Table S2 in the supplemental material), suggesting that there may be a correlation between PG levels and the optical properties of a cell suspension.
A biomechanical model explaining why the hypermotility phenotype appears, by using strains SM101 and SM124 as examples, is shown in Fig. 4. The contours of the migration zones arising from SM101 and SM124 colonies are different, with a shorter, thicker zone for SM101 and a thinner, more dispersed zone for SM124 (Fig. 4, top). Although the growth rates and yields for the two strains are similar in liquid BHI cultures (see Fig. S1 in the supplemental material), smaller amounts of total cell volume are made by the hypermotile strains under plate growth conditions (see Table S2 in the supplemental material). This suggests that even though the strains that spread more rapidly can move away from a zone of nutrient depletion, allowing them access to more nutrients from the growth medium, they do not produce more cells in a given amount of time (Fig. 4, top, triangular gradients). The thicker flares seen with SM101 are significantly more curvilinear than those seen with the hypermotile strain SM124 (Fig. 4, center), which means that they bend back toward the colony before they migrate as great a distance. What accounts for the more linear migration pattern seen in the hypermotile strains? One feature is that, since the cells are longer, there are fewer cell junctions per unit length in the hypermotile strains. The rigidity of the cells in comparison to the cell-cell juncture points allows for less deflection from linearity of the filament over the same length and a more linear path of motion (Fig. 4, bottom).
FIG 4.

Model illustrating features of the hypermotility phenotype (strain SM124) in comparison to normal motility (strain SM101). Although not shown, similar features can be found in the comparison of strain SM127 to strain SM102. See the Discussion for a full description of the elements shown here.
Mendez et al. (8) have published images of C. perfringens strains in addition to SM101 that appear to have the hypermotile phenotype in the absence of glucose. However, it is difficult to determine if the published images represent the originally isolated forms of the strains or hypermotile variants that were selected in the laboratory. Therefore, in studying motility in C. perfringens strains, it is important to maintain separate laboratory stocks of both the normal variants and hypermotile variants if they arise naturally.
We have attempted to find revertants of the hypermotile strains with normal motility, but because of their rapid spreading, hypermotile bacteria tend to grow over and cover up any slower-moving variants that arise in their midst. To overcome this problem, we looked for revertants on plates using single cells to form colonies but did not see any after examining several thousand colonies (data not shown).
We selected for mutants that converted to the hypermotile phenotype simply by picking cells that were contained in the fast-moving flares. There were >109 bacteria in the inoculation zone from which the hypermotile cells arose, and only a few flares arose from each zone (Fig. 1A to C). If the hypermotile strains arise as a consequence of a single mutation, then the frequency with which these occur is low. In strain SM124 complemented using a multicopy plasmid carrying the wild-type CPR_1831 gene, a hypermotile variant can be seen at the edge of the inoculum site (Fig. 1E). Since it is unlikely that every copy of the complementing gene is mutated or that the cell lost all copies of the complementation plasmid in the presence of antibiotic selection, this phenomenon is likely due to a mutation in a second gene that leads to hypermotility, possibly minE. In addition, while the antibiotic cephalexin was able to produce elongated cells of C. perfringens, it did not duplicate the hypermotile phenotype (see Fig. S3 in the supplemental material). Taken together, these observations support a model in which specific, but relatively rare, spontaneous mutations lead to the hypermotility phenotype. An important question, then, is whether hypermotile variants arise in natural settings outside the laboratory, such as the soil or the intestinal tracts of animals and humans. The two hypermotile strains isolated in this study were not deficient in growth in rich media and were not overly sensitive to the environmental conditions we tested, such as osmotic stress and sporulation conditions for the FtsI mutant (see Results). Therefore, it is conceivable that hypermotile variants do arise naturally, confer a temporary selective advantage on their progeny, and then revert to the normal motility phenotype. This could be tested by immediately identifying fresh clinical and soil isolates that have the hypermotile phenotype and sequencing the genomes. Any strains with mutations in cell division genes could be complemented with the wild-type copy to restore the normal motility phenotype, as we have done in this study. Such studies are planned in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Virginia Bioinformatics Institute (VBI) Genomics Research Laboratory for the genome-sequencing results reported here and Timofey Arapov for assistance with the MicrobeTracker software.
This work was supported by the VBI and The Fralin Life Sciences Institute as well as by National Institutes of Health grant R21 AI088298 and NSF grant 1057871 to S.B.M.
Footnotes
Published ahead of print 18 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01614-14.
REFERENCES
- 1.Rood JI. 1998. Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 52:333–360. 10.1146/annurev.micro.52.1.333 [DOI] [PubMed] [Google Scholar]
- 2.Shimizu T, Ohtani K, Hirakawa H, Ohshima K, Yamashita A, Shiba T, Ogasawara N, Hattori M, Kuhara S, Hayashi H. 2002. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. U. S. A. 99:996–1001. 10.1073/pnas.022493799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myers GS, Rasko DA, Cheung JK, Ravel J, Seshadri R, Deboy RT, Ren Q, Varga J, Awad MM, Brinkac LM, Daugherty SC, Haft DH, Dodson RJ, Madupu R, Nelson WC, Rosovitz MJ, Sullivan SA, Khouri H, Dimitrov GI, Watkins KL, Mulligan S, Benton J, Radune D, Fisher DJ, Atkins HS, Hiscox T, Jost BH, Billington SJ, Songer JG, Mcclane BA, Titball RW, Rood JI, Melville SB, Paulsen IT. 2006. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 16:1031–1040. 10.1101/gr.5238106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varga JJ, Nguyen V, O'Brien DK, Rodgers K, Walker RA, Melville SB. 2006. Type IV pili-dependent gliding motility in the Gram-positive pathogen Clostridium perfringens and other Clostridia. Mol. Microbiol. 62:680–694. 10.1111/j.1365-2958.2006.05414.x [DOI] [PubMed] [Google Scholar]
- 5.Melville S, Craig L. 2013. Type IV pili in Gram-positive bacteria. Microbiol. Mol. Biol. Rev. 77:323–341. 10.1128/MMBR.00063-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu H, Bouillaut L, Sonenshein AL, Melville SB. 2013. Use of a mariner-based transposon mutagenesis system to isolate Clostridium perfringens mutants deficient in gliding motility. J. Bacteriol. 195:629–636. 10.1128/JB.01288-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cowles KN, Gitai Z. 2010. Surface association and the MreB cytoskeleton regulate pilus production, localization and function in Pseudomonas aeruginosa. Mol. Microbiol. 76:1411–1426. 10.1111/j.1365-2958.2010.07132.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mendez M, Huang IH, Ohtani K, Grau R, Shimizu T, Sarker MR. 2008. Carbon catabolite repression of type IV pilus-dependent gliding motility in the anaerobic pathogen Clostridium perfringens. J. Bacteriol. 190:48–60. 10.1128/JB.01407-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varga J. 2006. The role of CcpA in regulating the carbon-starvation response of Clostridium perfringens. Ph.D. dissertation. Virginia Tech, Blacksburg, VA UNI Dissertation Service, Ann Arbor, MI [Google Scholar]
- 10.Melville SB, Labbe R, Sonenshein AL. 1994. Expression from the Clostridium perfringens cpe promoter in C. perfringens and Bacillus subtilis. Infect. Immun. 62:5550–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pospiech A, Neumann B. 1995. A versatile quick-prep of genomic DNA from gram-positive bacteria. Trends Genet. 11:217–218. 10.1016/S0168-9525(00)89052-6 [DOI] [PubMed] [Google Scholar]
- 12.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 13.Hartman AH, Liu H, Melville SB. 2011. Construction and characterization of a lactose-inducible promoter system for controlled gene expression in Clostridium perfringens. Appl. Environ. Microbiol. 77:471–478. 10.1128/AEM.01536-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C. 2011. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol. Microbiol. 80:612–627. 10.1111/j.1365-2958.2011.07579.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orsburn B, Melville SB, Popham DL. 2008. Factors contributing to heat resistance of Clostridium perfringens endospores. Appl. Environ. Microbiol. 74:3328–3335. 10.1128/AEM.02629-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.González-Castro MJ, López-Hernández J, Simal-Lozano J, Oruña-Concha MJ. 1997. Determination of amino acids in green beans by derivatization with phenylisothiocianate and high-performance liquid chromatography with ultraviolet detection. J. Chromatogr. Sci. 35:181–185. 10.1093/chromsci/35.4.181 [DOI] [Google Scholar]
- 17.Meador-Parton J, Popham DL. 2000. Structural analysis of Bacillus subtilis spore peptidoglycan during sporulation. J. Bacteriol. 182:4491–4499. 10.1128/JB.182.16.4491-4499.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, Melville SB. 1998. Identification and characterization of sporulation-dependent promoters upstream of the enterotoxin gene (cpe) of Clostridium perfringens. J. Bacteriol. 180:136–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu LY, Wang GZ, Ma BG, Zhang HY. 2011. Exploring the common molecular basis for the universal DNA mutation bias: revival of Löwdin mutation model. Biochem. Biophys. Res. Commun. 409:367–371. 10.1016/j.bbrc.2011.05.017 [DOI] [PubMed] [Google Scholar]
- 20.Eberhardt C, Kuerschner L, Weiss DS. 2003. Probing the catalytic activity of a cell division-specific transpeptidase in vivo with beta-lactams. J. Bacteriol. 185:3726–3734. 10.1128/JB.185.13.3726-3734.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson WB, Mealor AE, Stevens SE, Jr, Ospeck M. 2007. Measuring the force production of the hormogonia of Mastigocladus laminosus. Biophys. J. 93:699–703. 10.1529/biophysj.107.104067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lutkenhaus J. 2007. Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annu. Rev. Biochem. 76:539–562. 10.1146/annurev.biochem.75.103004.142652 [DOI] [PubMed] [Google Scholar]
- 23.Edwards DH, Errington J. 1997. The Bacillus subtilis DivIVA protein targets to the division septum and controls the site specificity of cell division. Mol. Microbiol. 24:905–915. 10.1046/j.1365-2958.1997.3811764.x [DOI] [PubMed] [Google Scholar]
- 24.Zhao CR, De Boer PA, Rothfield LI. 1995. Proper placement of the Escherichia coli division site requires two functions that are associated with different domains of the MinE protein. Proc. Natl. Acad. Sci. U. S. A. 92:4313–4317. 10.1073/pnas.92.10.4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Blundell TL, Mizuguchi K. 2001. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310:243–257. 10.1006/jmbi.2001.4762 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.