

Abstract
Although anaerobic glycerol metabolism in Enterococcus faecalis requires exogenous fumarate for NADH oxidation, E. faecalis strain W11 can metabolize glycerol in the absence of oxygen without exogenous fumarate. In this study, metabolic end product analyses and reporter assays probing the expression of enzymes involved in pyruvate metabolism were performed to investigate this fumarate-independent anaerobic metabolism of glycerol in W11. Under aerobic conditions, the metabolic end products of W11 cultured with glycerol were similar to those of W11 cultured with glucose. However, when W11 was cultured anaerobically, most of the glucose was converted to l-lactate, but glycerol was converted to ethanol and formate. During anaerobic culture with glycerol, the expression of the l-lactate dehydrogenase and pyruvate dehydrogenase E1αβ genes in W11 was downregulated, whereas the expression of the pyruvate formate-lyase (Pfl) and aldehyde/alcohol dehydrogenase genes was upregulated. These changes in the expression levels caused the change in the composition of end products. A pflB gene disruptant (Δpfl mutant) of W11 could barely utilize glycerol under anaerobic conditions, but the growth of the Δpfl mutant cultured with either glucose or dihydroxyacetone (DHA) under anaerobic conditions was the same as that of W11. Glucose metabolism and DHA generates one NADH molecule per pyruvate molecule, whereas glycerol metabolism in the dehydrogenation pathway generates two NADH molecules per pyruvate molecule. These findings demonstrate that NADH generated from anaerobic glycerol metabolism in the absence of fumarate is oxidized through the Pfl-ethanol fermentation pathway. Thus, Pfl is essential to avoid the accumulation of excess NADH during fumarate-independent anaerobic glycerol metabolism.
INTRODUCTION
Enterococci are the most common lactic acid bacteria in the human intestinal tract, and some strains are well-known opportunistic pathogens (1, 2). Physiological studies have shown that Enterococcus faecalis can use glycerol as a carbon source (3). Microorganisms utilize glycerol through a dehydrogenation pathway, a phosphorylation pathway, or both. The dehydrogenation pathway begins with the dehydrogenation of glycerol to produce dihydroxyacetone (DHA), which is catalyzed by glycerol dehydrogenase (GldA). The DHA produced is then phosphorylated by DHA kinase (4). In the phosphorylation pathway, glycerol is first phosphorylated to glycerol-3-phosphate (glycerol-3P) by a glycerol kinase and then oxidized to dihydroxyacetone phosphate by a glycerol-3P oxidase or a glycerol-3P dehydrogenase (4). In either pathway, the resulting dihydroxyacetone phosphate enters glycolysis. Lactic acid bacteria, such as several Lactobacillus species and Pediococcus pentosaceus, generally utilize glycerol through the phosphorylation pathway, rather than the dehydrogenation pathway (5, 6), although E. faecalis strains can utilize glycerol through either the dehydrogenation pathway or the phosphorylation pathway (7).
In some E. faecalis strains, glycerol metabolism has been reported. When strain l0C1 was cultured with glycerol under anaerobic conditions, it required exogenous fumarate for NADH oxidation and produced lactate and succinate (8, 9). A recent study of strain JH2-2 showed that NADH oxidase (Nox) was important for aerobic glycerol metabolism through the dehydrogenation pathway (10). These studies suggest that NADH oxidation is critical for glycerol metabolism. E. faecalis strain W11, which was isolated from salt-preserved wakame, is one of the lactic acid bacteria present in our laboratory's strain collection. When W11 was cultured with glycerol, it could utilize glycerol in the absence of exogenous fumarate, under either aerobic or anaerobic conditions. This suggests that W11 is equipped with an unknown NADH oxidation system that supports anaerobic glycerol metabolism, although the details of fumarate-independent anaerobic glycerol metabolism in W11 remained unclear.
E. faecalis can convert pyruvate to a variety of end products, including acetate, acetoin, ethanol, and/or d/l-lactate (3). The composition of the metabolic end products is affected by culture conditions such as aeration and carbon sources (11, 12). Acetyl coenzyme A (acetyl-CoA) can be produced from pyruvate by pyruvate dehydrogenase (Pdh) or by pyruvate formate-lyase (Pfl), which is one of the main pyruvate metabolic reactions (3). The Pdh reaction generates NADH and CO2, in addition to acetyl-CoA, whereas the Pfl reaction generates formate (13, 14). Pfl, which is encoded by pflB, is synthesized in an inactive (nonradical) form and is then posttranslationally converted to an active (radical) form by accepting an electron from the Pfl-activating enzyme (15). This active form of Pfl is easily inactivated by oxygen (16). Thus, Pfl is known as an enzyme of anaerobic metabolism and has been investigated mainly during anaerobic glucose metabolism (17, 18). In contrast, the actions of Pfl during the aerobic/anaerobic metabolism of glycerol have not been sufficiently investigated.
In this study, we investigated glycerol metabolism by W11. W11 utilized glycerol through the dehydrogenation pathway using an NAD+-dependent glycerol dehydrogenase. W11 cultured with glycerol under anaerobic conditions produced large amounts of ethanol and formate compared to production yields under aerobic conditions. These differences were caused by changes in the expression of pyruvate metabolism genes. The maximum optical density at 600 nm (OD600) attained by a pflB gene disruptant (Δpfl mutant) cultured with glycerol under anaerobic conditions decreased less than 4-fold compared with that of W11, but the growth rate and maximum culture density of the Δpfl mutant cultured with glycerol under aerobic conditions were the same as those of W11. Measurement of the NADH/NAD+ ratio in W11 showed that glycerol metabolism generated more NADH than glucose and DHA metabolism. These findings indicate that Pfl diverts acetyl-CoA to ethanol fermentation, during which two NADH molecules are oxidized, which is critical to avoid excess NADH accumulation in the absence of fumarate.
MATERIALS AND METHODS
Strains and media.
The bacterial strains and plasmids used in this study are listed in Table 1. W11 has been deposited in the International Patent Organism Depositary as Enterococcus faecalis FERM P-21943. Man, Rogosa, and Sharpe (MRS) broth (Becton, Dickinson, MD, USA), modified MRS (M-MRS) broth (1.6% nutrient broth [Becton, Dickinson], 0.5% yeast extract [Becton, Dickinson], 0.1% polysorbate 80, 0.2% ammonium citrate, 0.5% CH3COONa·3H2O, 0.01% MgCl2·6H2O, 0.005% MnSO4·7H2O, 0.2 M K2HPO4, pH 7.0), M17 broth (Becton, Dickinson), Luria-Bertani (LB) medium (1% tryptone [Becton, Dickinson], 0.5% yeast extract [Becton, Dickinson], 0.5% NaCl), and Super Optimal broth with catabolite repression medium (2% tryptone [Becton, Dickinson], 0.5% yeast extract [Becton, Dickinson], 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2·6H2O, 10 mM MgSO4·7H2O, 20 mM glucose, pH 7.0) were used.
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Characteristic(s)a | Source |
|---|---|---|
| Strains | ||
| Enterococcus faecalis W11 | Wild type | IPOD |
| Enterococcus faecalis Δpfl mutant | ΔpflB::Tcr | This study |
| Escherichia coli DH10B | F− mcrA Δ(mrr-hsdRMS-mcrBC) Φ80dlacZΔM15 ΔlacX74 deoR recA1 araD139 Δ(ara leu)7697 galU galK λ− rpsL endA1 nupG | Laboratory stock |
| Plasmids | ||
| pHSG396 | Cloning vector, Cmr | TaKaRa |
| pHY300PLK | Shuttle vector for E. coli and Bacillus subtilis, Apr Tcr | TaKaRa |
| pAM401 | Shuttle vector for E. coli and E. faecalis, Cmr Tcr | ATCC |
| pAM-lacZ | Control plasmid of reporter assay for E. faecalis, Cmr | This study |
| pAM-PldhL1-lacZ | Reporter assay plasmid of ldhL1 gene using β-galactosidase for E. faecalis, Cmr | This study |
| pAM-PpdhAB-lacZ | Reporter assay plasmid of pdhAB gene using β-galactosidase for E. faecalis, Cmr | This study |
| pAM-PadhE-lacZ | Reporter assay plasmid of adhE gene using β-galactosidase for E. faecalis, Cmr | This study |
| pAM-PpflB-lacZ | Reporter assay plasmid of pflB gene using β-galactosidase for E. faecalis, Cmr | This study |
Ap, ampicillin; Cm, chloramphenicol; Tc, tetracycline.
Culture conditions.
W11 was precultured in 20-ml test tubes containing 5 ml of MRS broth at 120 rpm and 30°C. To replace the carbon source contained in the MRS medium (Becton, Dickinson), the main cultures were performed in M-MRS broth containing each carbon source. For culture under aerobic conditions, precultures (1 ml) were transferred to 100 ml of M-MRS broth in a 500-ml Erlenmeyer flask and then cultured at 120 rpm and 30°C. For culture under anaerobic conditions, the headspace air in the flasks was replaced with nitrogen gas by purging for 15 min, and the flasks were then sealed with butyl rubber stoppers. To construct plasmids, Escherichia coli DH10B was cultured in LB medium at 37°C with vigorous agitation in the presence of appropriate antibiotics (chloramphenicol, 20 mg liter−1; tetracycline sulfate, 10 mg liter−1).
Disruption of the pflB gene.
The construction of a suicide vector for the pflB gene is shown in Fig. 1A. The pflB-pflA genes, amplified by PCR using KOD-FX polymerase (Toyobo Co. Ltd., Osaka, Japan) and the primers listed in Table 2, were digested with XbaI and HindIII and cloned into XbaI-HindIII-digested pHSG396 (TaKaRa Bio Inc., Shiga, Japan). The tetracycline resistance gene, amplified from pHY300PLK (TaKaRa) by PCR using the primers listed in Table 2, was digested with SalI, cloned into the same restriction site of the resulting plasmid, and inserted into W11 using an electroporation method. The gene disruption was confirmed by PCR (Fig. 1B), and the resulting strain was designated the Δpfl mutant.
FIG 1.

Disruption of the pflB gene in E. faecalis W11. (A) Strategy for homologous recombination into pflB loci to construct a pflB gene disruptant. The small arrows inserted show primer sites used to confirm the pflB gene disruption by PCR. (B) Confirmation of the pflB gene disruption using PCR. Lane 1, W11; lane 2, Δpfl mutant. DNA size markers (M) are shown in kb.
TABLE 2.
Primers used in this study
| Category and primer name | Sequence (5′ to 3′)a |
|---|---|
| Construction of suicide plasmid for pflB gene | |
| EF delta pfl XbaI F | GCGTCTAGACGCTTGCATCTAAAAGGTTATGTG |
| EF delta pfl HindIII R | CTCCAAGCTTGGAGGCTCAATTCCTTCTAATGG |
| pHY300PLK tet P (+) SalI | FCCGGTCGACCGGGCCATATTGTTGTATAAGTG |
| pHY300PLK tet R | GCTTCTAGAGATCTGCAGGTCG |
| Confirmation of pflB gene disruption | |
| EF delta pfl check F | GGGCACACAAGAAGTTATTCAGTC |
| EF delta pfl check R | GGCATATTGAGTTTCTTCACTTGG |
| Construction of reporter assay and plasmids | |
| EF ldhL1 Pro XbaI F | AAGTCTAGACTTTCATTCTCCTTGGATTATAAGC |
| EF ldhL1 Pro SalI R | ATGGTCGACCATGTGTACCATTCCTTCCTCTAC |
| EF pdhAB Pro XbaI F | TGCTCTAGAGCAATGAATGTTCGTGATAAAACC |
| EF pdhAB Pro SalI R | ATGGTCGACCATTTTGTCACACAATCCTCTCTATTACG |
| EF pflB Pro XbaI F | CCCTCTAGAGGGCACACAAGAAGTTATTCAGTC |
| EF pflB Pro SalI R | ATGGTCGACCATGAAGTGTTTGCCTCCTTAGTT |
| EF adhE Pro F | GGTTAAGCCGAATGAATTTTTTGAC |
| EF adhE Pro SalI R | ATGGTCGACCATGTTCATCCTCCTAAGAGTTTCATT |
| beta-gal SalI F | GACGTCGACGTCGTTTTACAACGTCGTGACTG |
| beta-gal SD ATG-fuse XbaI F | GTTTCTAGAAACTAAGGAGGCAAACACTTCATG |
| beta-gal SphI R | AGGGCATGCCCTGCCCGGTTATTATTATTTTTGA |
| RT-PCR | |
| EF RT-PCR 16S F | GTGTCGTTGATGGATGGACC |
| EF RT-PCR 16S R | TCTACGCATTTCACCGCTACAC |
| EF RT-PCR pflA F | CCGTTTTATCGTATTTACACAAGGG |
| EF RT-PCR pflA R | GGCATCCAGACGGATTAAATATTC |
| EF RT-PCR pflB F | GAGAACAAGCGTTGACACTCG |
| EF RT-PCR pflB R | CCATATAATGCGATACGACGATAGTC |
| EF RT-PCR glpF1 F | CGAACAAAATACGCTGGTTCTG |
| EF RT-PCR glpF1 R | CAAACAAGCAAGCCAACAGC |
| EF RT-PCR glpF2 F | GGTTGGGTCGTTATTGCTTTAGG |
| EF RT-PCR glpF2 R | ATTGGCTGGATAGTTACGGACAG |
Underlined nucleotides indicate restriction sites.
Transformation of E. faecalis.
Electrocompetent cells of W11 were prepared as previously described (19). W11 precultured in MRS broth was transferred to M17SG (M17 broth containing 0.5 M sucrose and 6% glycine). After being cultured for 21 h under aerobic conditions, the cells were collected by centrifugation at 5,000 × g and 4°C for 10 min and washed twice with ice-cold 10% glycerol containing 0.5 M sucrose. The cells and DNA were pulsed in 0.1-cm-gap electroporation cuvettes (BEX Co., Tokyo, Japan) at 1.25 kV for 5 ms using a Gene Pulser II (Bio-Rad, Hercules, CA), mixed with 1 ml ice-cold M17SG, and then incubated for 2 h at 30°C. Transformants were selected after incubation for 3 days on M17S (M17 broth containing 0.5 M sucrose) agar plates containing 10 mg of tetracycline sulfate liter−1 or 10 mg of chloramphenicol liter−1.
Reporter assays.
The Escherichia coli β-galactosidase gene (lacZ) lacking a translation start codon was amplified by PCR, digested with SalI and SphI, and cloned into SalI-SphI-digested pHSG396 to generate pHSG396-lacZ. The 5′ regions of the ldhL1 (334-bp), pdhAB (530-bp), adhE (254-bp), and pflB (443-bp) genes in W11 were amplified by PCR to clone those gene's promoter sequences (called PldhL1, PpdhAB, PadhE, and PpflB, respectively). These regions were selected because the translation start codons of the ldhL1, pdhAB, adhE, and pflB genes were more than 250 bp away from the those of the nearest upstream open reading frames. Each amplified gene fragment, including its promoter sequence, was digested with XbaI and SalI and inserted into the same restriction sites in pHSG396-lacZ. The resulting plasmids were digested with XbaI and SphI, and the PldhL1-lacZ, PpdhAB-lacZ, PadhE-lacZ, and PpflB-lacZ gene fragments were cloned into XbaI-SphI-digested pAM401 to generate pAM-PldhL1-lacZ, pAM-PpdhAB-lacZ, pAM-PadhE-lacZ, and pAM-PpflB-lacZ. The negative-control plasmid, pAM-lacZ, was constructed as follows. The lacZ gene with a start codon and a Shine-Dalgarno sequence was amplified by PCR using pAM-PpflB-lacZ as a template, digested with XbaI and SphI, and inserted into the same restriction sites in pAM401. Table 2 lists the primers used. The plasmids were inserted into W11 by the electroporation method, and the expression of each gene was assayed according to the β-galactosidase activity, as described previously (20).
Enzyme assays.
E. faecalis strains were cultured in 100 ml of culture medium in 500-ml Erlenmeyer flasks under each condition. The cells collected by centrifugation (15,000 × g, 15 min, 4°C) were washed with 100 mM phosphate buffer (pH 7.0), resuspended in the same buffer, and disrupted by sonication (three cycles of 2 min, on/off at 1-s intervals, at 50 W) on ice. After centrifugation at 20,000 × g and 4°C for 15 min, the supernatant was filtered using a membrane filter (0.44 μm) and then used as a cell extract. Glycerol dehydrogenase (GldA) activity was assayed in 100 mM carbonate buffer (pH 9.0) containing 1 mM NAD+ and 100 mM glycerol. The reaction was initiated with the addition of samples. Glycerol kinase (GlpK) activity was measured using a coupled enzyme assay with glycerol 3-phosphate dehydrogenase (21). The GlpK activity was assayed in 100 mM glycine-hydrazine buffer (pH 8.8) containing 1.5 mM ATP, 4 mM MgCl2, 0.5 mM NAD+, 2 U of glycerol 3-phosphate dehydrogenase (Sigma-Aldrich, St. Louis, MO), and 1.5 mM glycerol. These reactions were monitored at room temperature (25°C) by measuring the absorbance at 340 nm.
Metabolite analysis.
E. faecalis strains were cultured in 100 ml of culture medium in 500-ml Erlenmeyer flasks under each condition. To measure the NADH/NAD+ ratio, cells collected by centrifugation (15,000 × g, 1 min, 4°C) were washed with cold phosphate-buffered saline (PBS), resuspended in extraction buffer, and disrupted by sonication as described above. After centrifugation at 20,000 × g (4°C for 5 min), the supernatant was ultrafiltered (10 kDa) using an Amicon Ultra-0.5 centrifugal filter unit with an Ultracel-10 membrane (EMD Millipore Co., Billerica, MA, USA) and then stored at −80°C. The NADH/NAD+ ratio was measured using an NAD/NADH quantitation kit (Bio Vision, Inc., CA, USA) according to the manufacturer's instructions. Acetaldehyde, acetate, ethanol, formate, glucose, glycerol, and d/l-lactate in the culture media were quantified using the F-kit (Roche Diagnostics, Basel, Switzerland) according to the manufacturer's instructions, and acetoin was quantified as previously described (22). DHA was detected in a split mode by using a GC/MS-QP2010 Plus gas chromatograph mass spectrometer (Shimadzu Co., Kyoto, Japan).
Reverse transcription (RT)-PCR.
Total RNA was purified using the RNeasy Protect Bacteria minikit (Qiagen, Hilden, Germany) with the RNase-free DNase set (Qiagen), and cDNA was synthesized using the QuantiTect Rev transcription kit (Qiagen). The specific amplicons for the 16S rRNA, pflA, pflB, glpF1, and glpF2 genes were obtained by PCR using cDNA as a template, the primers described in Table 2, and KOD-Plus DNA polymerase (Toyobo).
RESULTS
Growth under aerobic/anaerobic conditions.
W11 could utilize glucose and glycerol without exogenous fumarate under either aerobic or anaerobic conditions (Fig. 2). With either carbon source, the maximum culture density (OD600) achieved under anaerobic conditions was approximately 20% lower than the density achieved under aerobic conditions (Fig. 2). NAD+-dependent glycerol dehydrogenase (GldA) activity, but not glycerol kinase (GlpK) activity, was detected in W11 cultured with glycerol (Table 3). These data indicate that W11 utilizes glycerol only by means of the dehydrogenation pathway and alters the metabolic pathway in the presence or absence of oxygen, as seen in glucose metabolism (23).
FIG 2.
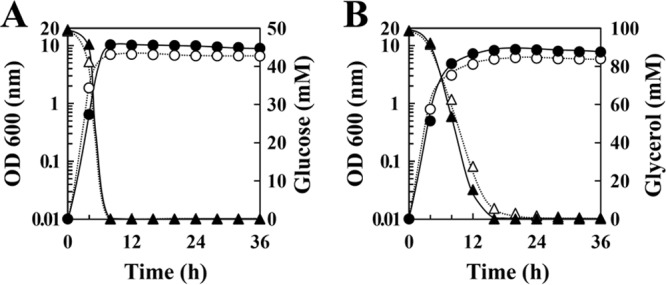
Glucose and glycerol metabolism in E. faecalis W11. W11 was cultured in M-MRS broth containing either 50 mM glucose (A) or 100 mM glycerol (B) under aerobic (●, ▲) or anaerobic (○, △) conditions. ● and ○, optical density at 600 nm; ▲ and △, concentration of the carbon source in the culture medium. The results are representative, and the experiment was performed at least three times.
TABLE 3.
Specific activities of GldA and GlpK in E. faecalis W11a
| Enzyme | Sp act (μmol mg protein−1 min−1) |
|||
|---|---|---|---|---|
| Glucose |
Glycerol |
|||
| +O2 | −O2 | +O2 | −O2 | |
| GldA | 0.06 ± 0.02 | 0.03 ± 0.01 | 0.67 ± 0.12 | 0.99 ± 0.10 |
| GlpK | ND | ND | ND | ND |
W11 was cultured in M-MRS containing either 50 mM glucose or 100 mM glycerol for 8 h (glucose) or 16 h (glycerol) under aerobic (+O2) or anaerobic (−O2) conditions. The results are the means of results from three experiments ± standard errors. ND, not detected.
End products from glucose and glycerol.
W11 was cultured in M-MRS broth, and the end products in the culture media were measured. Under aerobic conditions, W11 cultured with glucose or glycerol accumulated 18.5 mM l-lactate and 35.1 mM acetate (glucose) or 7.7 mM l-lactate and 55.0 mM acetate (glycerol) in culture media (Table 4). The amounts of acetoin were not much different between the glucose and glycerol conditions. Other pyruvate derivatives, such as acetaldehyde, ethanol, formate, and d-lactate, were not detected (Table 4). These data indicated that pyruvate from glycolysis was converted to acetate, acetoin, and l-lactate under aerobic conditions whether the carbon source was glucose or glycerol. In contrast, when W11 was cultured under anaerobic conditions, most of the glucose was converted to l-lactate (78.8 mM), but glycerol was converted to ethanol (61.3 mM) and formate (62.1 mM) instead of l-lactate (Table 4). These results suggest that anaerobic glycerol metabolism converted pyruvate to ethanol rather than to l-lactate via the acetyl-CoA produced by Pfl.
TABLE 4.
Metabolite profiles of E. faecalis strains cultured under each conditiona
| Strain | Carbon source | O2 | Concn of consumed carbon source (mM) | Metabolite concn (mM) |
Carbon balance (%)b | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AAL | Acetate | Acetoin | Ethanol | Formate | d-Lactate | l-Lactate | |||||
| W11 | Glucose | + | 50 | <0.1 | 35.1 | 19.1 | <0.1 | 0.2 | 0.1 | 18.5 | 92 |
| − | 50 | <0.1 | <0.1 | 0.3 | 6.2 | 11.7 | 0.1 | 78.8 | 88 | ||
| Δpfl mutant | Glucose | + | 50 | <0.1 | 37.1 | 15.6 | <0.1 | 0.2 | 0.1 | 36.9 | 105 |
| − | 50 | <0.1 | 0.9 | 0.2 | <0.1 | 0.2 | 0.1 | 90.1 | 92 | ||
| W11 | Glycerol | + | 100 | <0.1 | 55.0 | 12.9 | <0.1 | 0.1 | 0.2 | 7.7 | 89 |
| − | 100 | <0.1 | <0.1 | 0.3 | 61.3 | 62.1 | 0.2 | 33.8 | 96 | ||
| Δpfl mutant | Glycerol | + | 100 | <0.1 | 75.2 | 13.0 | 1.1 | <0.1 | 0.1 | 6.6 | 109 |
| − | 36 | <0.1 | 2.6 | 0.2 | 5.4 | 0.2 | 0.2 | 34.3 | 119 | ||
| W11 | DHA | + | 100 | <0.1 | 79.8 | 4.3 | <0.1 | 1.4 | 1.6 | 0.3 | 90 |
| − | 100 | <0.1 | 24.3 | 0.5 | 10.7 | 26.7 | 1.6 | 45.6 | 83 | ||
| Δpfl mutant | DHA | + | 100 | <0.1 | 78.5 | 1.9 | <0.1 | <0.1 | 1.3 | 3.6 | 87 |
| − | 100 | <0.1 | 17.8 | 0.2 | 11.6 | 0.1 | 1.2 | 58.8 | 90 | ||
E. faecalis strains were cultured in M-MRS broth containing 50 mM glucose, 100 mM glycerol, or 100 mM DHA for 36 h under aerobic (+) and anaerobic (−) conditions. M-MRS broth contains 36 mM acetate, 0.5 mM ethanol, 0.1 mM formate, 0.3 mM d-lactate, and 11.1 mM l-lactate; metabolite concentrations are presented exclusive of these initial amounts. The results are the means of results from three experiments. The standard errors were all <10%. AAL, acetaldehyde.
The concentrations of CO2 (mM) generated by the Pdh and AlsS reactions were calculated as follows. CO2 (Pdh) = (AAL [mM] + acetate [mM] + ethanol [mM]) − formate (mM). CO2 (AlsS) = acetoin (mM).
The influence of gene expression of enzymes on pyruvate metabolism.
The influence of gene expression on pyruvate metabolism in W11 was measured using β-galactosidase (LacZ)-based reporter assays. The activity of LacZ expressed from PldhL1 in W11 cultured with glucose was 1.27 ± 0.11 μmol mg protein−1 min−1 under aerobic conditions. A similar level was measured under anaerobic conditions (Fig. 3A). However, when W11 was cultured with glycerol, the activity of LacZ expressed from PldhL1 under anaerobic conditions decreased 2.8-fold, although the activity under aerobic conditions was at the same level as that in W11 cultured with glucose (Fig. 3A). Similarly, the activity of LacZ expressed from PpdhAB under anaerobic conditions was 1.8- to 3.0-fold lower than the activity measured under aerobic conditions (Fig. 3B). In contrast, the activity of LacZ expressed from PadhE was increased by the anaerobic culture with glycerol (Fig. 3C). Although the activity of LacZ expressed from PpflB was most increased under the same conditions as the activity of LacZ from PadhE, activity was detected from PpflB-LacZ under all conditions (Fig. 3D). The pflA and pflB genes were transcribed even if W11 was cultured under aerobic conditions (Fig. 3E and F). These findings indicate that the Pfl-ethanol fermentation pathway functions under anaerobic conditions using glycerol as a carbon source, which is consistent with the end products accumulated in the culture media (Table 4).
FIG 3.

The expression of enzymes in pyruvate metabolism. Reporter assays using the promoters of the genes encoding l-lactate dehydrogenase (LdhL1) (A), pyruvate dehydrogenase subunit E1αβ (PdhAB) (B), aldehyde/alcohol dehydrogenase (AdhE) (C), or pyruvate formate-lyase (PflB) (D). W11 cells harboring plasmid pAM-lacZ, pAM-PldhL1-lacZ, pAM-PpdhAB-lacZ, pAM-PadhE-lacZ, or pAM-PpflB-lacZ were cultured in M-MRS broth containing either 50 mM glucose (Glc) or 100 mM glycerol (Gly) for 8 h (glucose) or 16 h (glycerol) under aerobic (red bars) and anaerobic (blue bars) conditions. The LacZ activity in W11 cells harboring control plasmid pAM-lacZ was less than 0.01 μmol mg−1 protein min−1. The data shown are the means of results from three experiments. Bars indicate standard errors. P < 0.05. (E) Colorimetric reporter assay for the pflB gene promoter. W11 bacteria harboring pAM-lacZ (LacZ) or pAM-PpflB-lacZ (PpflB-LacZ) were streaked on M-MRS plates containing 5 mM 5-bromo-4-chloro-3-indolyl-β-d-galactoside and incubated for 48 h under aerobic conditions. The added carbon source was either 50 mM glucose (Glc) or 100 mM glycerol (Gly). (F) RT-PCR to detect the pflA and pflB transcripts in W11. The strain was cultured in M-MRS broth containing 100 mM glycerol for 16 h under aerobic (+O2) or anaerobic (−O2) conditions. DNA size markers (M) are shown in kb.
Growth and redox balance of the Δpfl mutant.
Regardless of the carbon source, the metabolic end products of the Δpfl mutant cultured under aerobic conditions comprised acetate, acetoin, and l-lactate (Table 4). In contrast, the media from the Δpfl mutant cultured under anaerobic conditions accumulated little formate or ethanol (Table 4), indicating that Pfl is important for anaerobic metabolism to produce acetyl-CoA.
The maximum culture density (OD600) of the Δpfl mutant cultured with glucose was the same as that of W11, regardless of the presence or absence of oxygen (Fig. 4A), whereas anaerobic culture with glycerol decreased the maximum culture density of the Δpfl mutant 4-fold compared with that of W11 (Fig. 4B). This growth decrease was restored by the addition of exogenous fumarate (Fig. 4C), indicating that W11 could oxidize NADH through fumarate reduction, although the pfl gene was disrupted. We measured the NADH/NAD+ ratio and found that this ratio in W11 cells cultured with glycerol was 5.8- to 8.1-fold higher than that of W11 cultured with glucose (Fig. 5A). These ratios are consistent with the difference in NADH generation rates between glucose and glycerol metabolic pathways (3, 4). In contrast, when the Δpfl mutant was cultured with glycerol under anaerobic conditions, the maximum culture density and the NADH/NAD+ ratio of the Δpfl mutant cells were similar to those cultured without a carbon source (Fig. 5B). These results indicate that when fumarate is absent under anaerobic conditions, the Δpfl mutant downregulates glycerol metabolism and grows by glycerol-independent metabolism, although intracellular NAD+ concentrations are sufficient.
FIG 4.
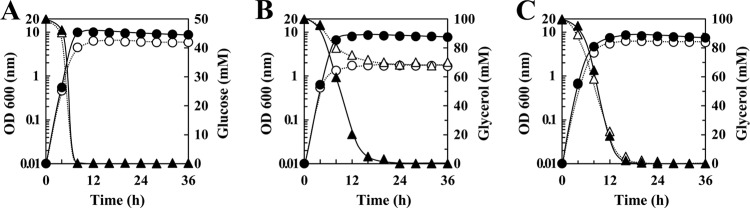
Glucose and glycerol metabolism in the E. faecalis Δpfl mutant. The Δpfl mutant was cultured in M-MRS broth containing 50 mM glucose (A), 100 mM glycerol (B), or 100 mM glycerol with 50 mM fumarate (C) under aerobic (●, ▲) or anaerobic (○, △) conditions. ● and ○, optical densities at 600 nm; ▲ and △, concentrations of the carbon source in the culture medium. The results are representative, and the experiment was performed at least three times.
FIG 5.
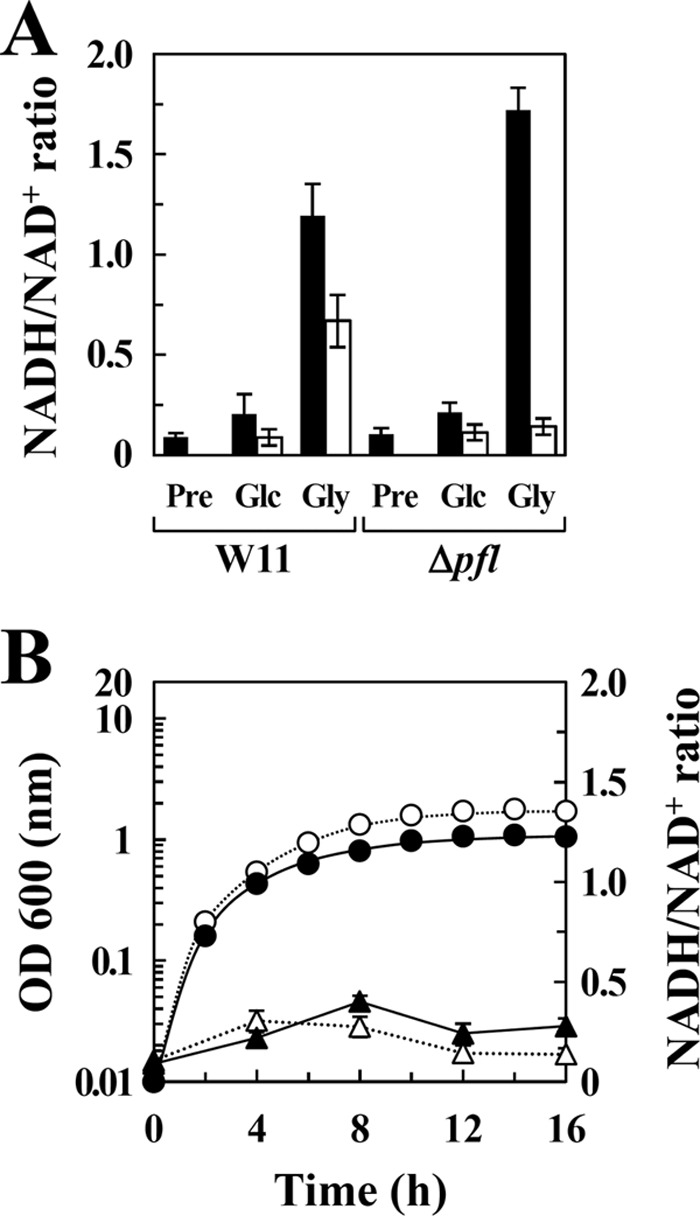
NADH/NAD+ ratios in E. faecalis strains. (A) The NADH/NAD+ ratios in W11 and Δpfl mutant cells cultured in M-MRS broth containing either 50 mM glucose (Glc) or 100 mM glycerol (Gly) for 8 h (glucose) or 16 h (glycerol) under aerobic (filled bars) or anaerobic (unfilled bars) conditions. Initial (time zero) NADH/NAD+ ratios in strains were measured using precultured cells (Pre). (B) Growth (●, ○) and NADH/NAD+ ratio (▲, △) of Δpfl mutant cells cultured in M-MRS broth (●, ▲) and M-MRS broth containing 100 mM glycerol (○, △) under anaerobic conditions. Data are the means of results from three experiments. Bars indicate standard errors. P < 0.05.
Metabolism of DHA.
DHA is produced by dehydrogenation of glycerol, which is catalyzed by GldA and accompanied by NAD+ reduction in the dehydrogenation pathway. In accordance, DHA metabolism generates one NADH molecule per pyruvate molecule, which is the same rate as that in glucose metabolism. Regardless of the presence or absence of oxygen, the growth rate, maximum culture density (OD600), and NADH/NAD+ ratio of Δpfl mutant cells cultured with DHA were the same as those seen with W11 (Fig. 6A and B), indicating that the downregulation of anaerobic glycerol metabolism seen in the Δpfl mutant is specific to glycerol.
FIG 6.
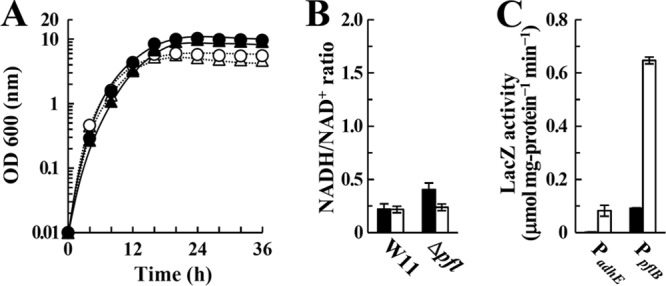
DHA metabolism in E. faecalis strains. (A) Growth of W11 (●, ○) and the Δpfl mutant (▲, △) strain cultured in M-MRS broth containing 100 mM DHA under aerobic (●, ▲) and anaerobic (○, △) conditions. The results are representative, and the experiment was performed at least three times. (B) The NADH/NAD+ ratios in W11 and Δpfl mutant cells cultured in M-MRS broth containing 100 mM DHA for 16 h under aerobic (filled bars) or anaerobic (unfilled bars) conditions. (C) Reporter assays of the aldehyde/alcohol dehydrogenase (AdhE) and pyruvate formate-lyase (PflB) gene promoters. W11 harboring pAM-lacZ, pAM-PadhE-lacZ, or pAM-PpflB-lacZ was cultured in M-MRS broth containing 100 mM DHA for 16 h under aerobic (filled bars) or anaerobic (unfilled bars) conditions. The LacZ activity in W11 harboring control plasmid pAM-lacZ was less than 0.01 μmol mg−1 protein min−1. Bars indicate standard errors. P < 0.05.
When W11 was cultured with DHA, the activity of LacZ expressed from PpflB under aerobic conditions was 0.09 ± <0.01 μmol mg protein−1 min−1. Under anaerobic conditions, the activity increased to 0.65 ± 0.01 μmol mg protein−1 min−1 (Fig. 6C). The activity of LacZ expressed from PadhE similarly increased under anaerobic conditions, although it was less than half that of W11 cultured with glycerol (Fig. 6C). The metabolic end products produced from DHA under anaerobic conditions contained more acetate (24.3 mM) and l-lactate (45.6 mM) than those produced during anaerobic glycerol metabolism, whereas the amounts of ethanol and formate were decreased to 10.7 mM and 26.7 mM, respectively (Table 4). The Δpfl mutant cultured with DHA under anaerobic conditions produced more l-lactate (58.8 mM) (instead of acetate [17.8 mM]) than W11 (Table 4). However, the amount of ethanol produced from DHA under anaerobic conditions was the same as that of W11 (Table 4), suggesting that acetyl-CoA could be produced by both Pfl and Pdh during DHA metabolism. These data indicate that Pfl is not essential for anaerobic DHA metabolism and support the conclusion that the Pfl-ethanol fermentation pathway works under excess NADH generation conditions, such as anaerobic glycerol metabolism.
DISCUSSION
In this study, we focused on glycerol metabolism in E. faecalis strain W11. Many microorganisms, including lactic acid bacteria, utilize glycerol in a phosphorylation pathway rather than in a dehydrogenation pathway (4, 5, 6, 24, 25, 26). In contrast, W11 utilized glycerol efficiently in only the dehydrogenation pathway (Table 3). The activity of GldA, which is the first enzyme in the dehydrogenation pathway, was detected in W11 cultured with glycerol but not in W11 cultured with glucose (Table 3). These results indicate that W11 has systems that respond to glycerol, one of which is fumarate-independent anaerobic glycerol metabolism.
The maximum culture density (OD600) of W11 cultured under anaerobic conditions was approximately 20% lower than the density attained under aerobic culture conditions (Fig. 2). Lactic acid bacteria generate ATP during the phosphoglycerate kinase, pyruvate kinase, and acetate kinase (Pck) reactions (3, 27). When W11 was cultured under anaerobic conditions, pyruvate was not converted to acetate but was instead converted to l-lactate (when cultured with glucose) and ethanol (when cultured with glycerol) (Table 4). Closing the acetate production pathway halted the Pck reaction and resulted in decreased growth.
The glycerol metabolic pathways in W11 proposed in this study are shown in Fig. 7. Glycerol metabolism through the dehydrogenation pathway generates two NADH molecules per pyruvate molecule. Therefore, W11 utilizing glycerol under anaerobic conditions selected ethanol fermentation to oxidize the two NADH molecules (Table 4). Acetyl-CoA, which is the substrate for AdhE, can be produced from pyruvate by either Pdh or Pfl, but the anaerobic culture media of W11 included formate in the same amount as ethanol (Table 4). Although ldhL1 gene expression during anaerobic glycerol metabolism was 2.8-fold lower than during aerobic glycerol metabolism (Fig. 3A), W11 cultured with glycerol under anaerobic conditions produced 4.4-fold more l-lactate than it did under aerobic conditions (Table 4). These results indicate that Pdh-dependent acetyl-CoA production in W11 is strongly downregulated during anaerobic glycerol metabolism. This prevents the production of unnecessary NADH, because the Pdh reaction generates NADH together with acetyl-CoA. However, pdhAB gene expression in W11 was detected under anaerobic conditions (Fig. 3B), suggesting the possibility that Pdh activity in E. faecalis is regulated by other mechanisms, such as a lipoic acid activation/inactivation system, rather than by transcription control (28, 29, 30).
FIG 7.

Proposed glycerol metabolic pathway in E. faecalis strain W11. Solid arrows indicate the aerobic glycerol metabolic pathway, and dashed arrows indicate the fumarate-independent anaerobic glycerol metabolic pathway. DHA, dihydroxyacetone; DHAP, dihydroxyacetone phosphate; GldA, glycerol dehydrogenase; DhaKL, dihydroxyacetone kinase; EIIADha, phosphoenolpyruvate-dependent phosphotransferase for DhaKL; Pdh, pyruvate dehydrogenase; Pfl, pyruvate formate-lyase; d/l-Ldh,d-lactate dehydrogenase and l-lactate dehydrogenase; AlsS, acetolactate synthase; AlsD, acetolactate decarboxylase; AdhE, alcohol/acetaldehyde dehydrogenase; Pta, phosphate acetyltransferase; Ack, acetate kinase.
When fumarate was absent, the Δpfl mutant cultured under anaerobic conditions downregulated glycerol metabolism (Fig. 4B and C). If the Δpfl mutant had continued glycerol utilization under these conditions, excess NADH could have accumulated inside the cell, because the lactic acid fermentation pathway cannot oxidize two NADH molecules per pyruvate molecule, as in the Pfl-ethanol fermentation pathway (Fig. 7). Hence, we propose that the downregulation of glycerol metabolism in the Δpfl mutant is a mechanism to avoid the reductive stress induced by excess NADH. In the dehydrogenation pathway, glycerol is transported into the cell by the glycerol facilitator protein (GlpF), dehydrogenated to DHA by GldA, and then phosphorylated by dihydroxyacetone kinase (DhaKL) (3, 4, 31). The activity of GldA and the presence of its coenzyme NAD+ were detected when the Δpfl mutant was cultured with glycerol under anaerobic conditions (Fig. 5A and B and 8A), which implies that GldA reactions in the dehydrogenation pathway were not affected by pflB disruption. In contrast, the expression of the glpF1 and glpF2 genes was significantly repressed in the Δpfl mutant with the passage of culture time, although sufficient glycerol remained in the culture media (Fig. 4B and 8B). Thus, the Δpfl mutant most likely stopped glycerol uptake through GlpF regulation; however, the mechanism by which Pfl disruption represses glycerol metabolism is currently unknown.
FIG 8.

(A) GldA activity in Δpfl mutant cells cultured in M-MRS broth (●) or M-MRS broth containing 100 mM glycerol (○). Data are the means of results from three experiments. Bars indicate standard errors. (B) RT-PCR to detect glpF1 and glpF2 transcripts in Δpfl mutant cells. The Δpfl mutant was cultured in M-MRS broth containing 100 mM glycerol under anaerobic conditions. DNA size markers (M) are shown in kb.
The expression of the adhE gene in W11 was induced by glycerol under anaerobic conditions but not under other conditions (Fig. 3B and 6C). The metabolism of glycerol through the dehydrogenation pathway generated more NADH than the metabolism of either glucose or DHA and resulted in the accumulation of NADH inside the cell (Fig. 5A and 6B). The cellular NAD(P)H/NAD(P)+ ratio, particularly at the NAD(P)H level, is an important redox sensor (32). This means that the expression of the adhE gene in W11 was induced by a high NADH/NAD+ ratio rather than anaerobic conditions, as seen in E. coli (33). In addition, the expression of the adhE gene was more precisely controlled than the expression of the pflB and pflA genes (Fig. 3C and D and 6C). If adhE gene expression is overinduced, AdhE would convert NADH to NAD+ beyond necessity, causing oxidative stress in the cell. The precise regulation of the adhE gene by the redox balance may be one of the systems that safely activates the Pfl-ethanol fermentation pathway. In contrast, the loose regulation of pflB and pflA gene expression may be the system that allows the cell to quickly shift acetyl-CoA production from the Pdh reaction to the Pfl reaction.
Our results demonstrate that the fumarate-independent anaerobic glycerol metabolic pathway in W11 is ethanol fermentation via Pfl. The pflB gene disruption mutant, which cannot produce acetyl-CoA without NADH generation, downregulated glycerol metabolism under anaerobic conditions. These data indicate that the Pfl-ethanol fermentation pathway is a critical oxidation pathway in the presence of excess NADH that cannot be oxidized through lactic acid fermentation. During anaerobic metabolism, the NADH oxidation process is particularly important because E. faecalis cannot use oxygen to oxidize NADH with NADH oxidase (Nox) (10). To constantly metabolize glycerol in the absence of fumarate, W11 may have evolved a unique system for glycerol utilization.
ACKNOWLEDGMENTS
We thank Noboru Takizawa for providing Enterococcus faecalis W11.
This study was partly supported by a research grant from the Japan Soap and Detergent Association.
Footnotes
Published ahead of print 25 April 2014
REFERENCES
- 1.Murray BE. 1990. The life and times of the enterococcus. Clin. Microbiol. Rev. 3:46–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mundy LM, Sahm DF, Gilmore M. 2000. Relationships between enterococcal virulence and antimicrobial resistance. Clin. Microbiol. Rev. 13:513–522. 10.1128/CMR.13.4.513-522.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huycke MM. 2002. Physiology of enterococci, p 133–175 In Gilmore MS, Clewell DB, Courvalin P, Dunny GM, Murray BE, Rice LB. (ed), The enterococci: pathogenesis, molecular biology, and antibiotic resistance. ASM Press, Washington, DC [Google Scholar]
- 4.Lin EC. 1976. Glycerol dissimilation and its regulation in bacteria. Annu. Rev. Microbiol. 30:535–578. 10.1146/annurev.mi.30.100176.002535 [DOI] [PubMed] [Google Scholar]
- 5.Rivaldi JD, Sousa Silva MLC, Duarte LC, Ferreira AEN, Cordeiro C, de Almeida Felipe MDG, de Ponces Freire A, de Mancilha IM. 2013. Metabolism of biodiesel-derived glycerol in probiotic Lactobacillus strains. Appl. Microbiol. Biotechnol. 97:1735–1743. 10.1007/s00253-012-4621-z [DOI] [PubMed] [Google Scholar]
- 6.Pasteris SE, Strasser de Saad AM. 2005. Aerobic glycerol catabolism by Pediococcus pentosaceus isolated from wine. Food Microbiol. 22:399–407. 10.1016/j.fm.2004.10.001 [DOI] [PubMed] [Google Scholar]
- 7.Bizzini A, Zhao C, Budin-Verneuil A, Sauvageot N, Giard JC, Auffray Y, Hartke A. 2010. Glycerol is metabolized in a complex and strain-dependent manner in Enterococcus faecalis. J. Bacteriol. 192:779–785. 10.1128/JB.00959-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunsalus IC. 1947. Products of anaerobic glycerol fermentation by Streptococci faecalis. J. Bacteriol. 54:239–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs NJ, VanDemark PJ. 1960. Comparison of the mechanism of glycerol oxidation in aerobically and anaerobically grown Streptococcus faecalis. J. Bacteriol. 79:532–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sauvageot N, Ladjouzi R, Benachour A, Rincé A, Deutscher J, Hartke A. 2012. Aerobic glycerol dissimilation via the Enterococcus faecalis DhaK pathway depends on NADH oxidase and a phosphotransfer reaction from PEP to DhaK via EIIADha. Microbiology 158:2661–2666. 10.1099/mic.0.061663-0 [DOI] [PubMed] [Google Scholar]
- 11.Jönsson M, Saleihan Z, Nes IF, Holo H. 2009. Construction and characterization of three lactate dehydrogenase-negative Enterococcus faecalis V583 mutants. Appl. Environ. Microbiol. 75:4901–4903. 10.1128/AEM.00344-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarantinopoulos P, Kalantzopoulos G, Tsakalidou E. 2001. Citrate metabolism by Enterococcus faecalis FAIR-E 229. Appl. Environ. Microbiol. 67:5482–5487. 10.1128/AEM.67.12.5482-5487.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeaman SJ. 1989. The 2-oxo acid dehydrogenase complexes: recent advances. Biochem. J. 257:625–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knappe J, Blaschkowski HP, Gröbner P, Schmitt T. 1974. Pyruvate formate-lyase of Escherichia coli: the acetyl-enzyme intermediate. Eur. J. Biochem. 50:253–263. 10.1111/j.1432-1033.1974.tb03894.x [DOI] [PubMed] [Google Scholar]
- 15.Frey M, Rothe M, Wagner AF, Knappe J. 1994. Adenosylmethionine-dependent synthesis of the glycyl radical in pyruvate formate-lyase by abstraction of the glycine C-2 pro-S hydrogen atom. Studies of [2H]glycine-substituted enzyme and peptides homologous to the glycine 734 site. J. Biol. Chem. 269:12432–12437 [PubMed] [Google Scholar]
- 16.Zhang W, Wong KK, Magliozzo RS, Kozarich JW. 2001. Inactivation of pyruvate formate-lyase by dioxygen: defining the mechanistic interplay of glycine 734 and cysteine 419 by rapid freeze-quench EPR. Biochemistry 40:4123–4130. 10.1021/bi002589k [DOI] [PubMed] [Google Scholar]
- 17.Melchiorsen CR, Jokumsen KV, Villadsen J, Johnsen MG, Israelsen H, Arnau J. 2000. Synthesis and posttranslational regulation of pyruvate formate-lyase in Lactococcus lactis. J. Bacteriol. 182:4783–4788. 10.1128/JB.182.17.4783-4788.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knappe J, Sawers G. 1990. A radical-chemical route to acetyl-CoA: the anaerobically induced pyruvate formate-lyase system of Escherichia coli. FEMS Microbiol. Rev. 6:383–398 [DOI] [PubMed] [Google Scholar]
- 19.Cruz-Rodz AL, Gilmore MS. 1990. High efficiency introduction of plasmid DNA into glycine treated Enterococcus faecalis by electroporation. Mol. Gen. Genet. 224:152–154 [DOI] [PubMed] [Google Scholar]
- 20.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 21.Deutscher J, Sauerwald H. 1986. Stimulation of dihydroxyacetone and glycerol kinase activity in Streptococcus faecalis by phosphoenolpyruvate-dependent phosphorylation catalyzed by enzyme I and HPr of the phosphotransferase system. J. Bacteriol. 166:829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grundy FJ, Waters DA, Takova TY, Henkin TM. 1993. Identification of genes involved in utilization of acetate and acetoin in Bacillus subtilis. Mol. Microbiol. 10:259–271. 10.1111/j.1365-2958.1993.tb01952.x [DOI] [PubMed] [Google Scholar]
- 23.Condon S. 1987. Responses of lactic acid bacteria to oxygen. FEMS Microbiol. Rev. 46:269–280. 10.1111/j.1574-6968.1987.tb02465.x [DOI] [Google Scholar]
- 24.Gancedo C, Gancedo JM, Sols A. 1968. Glycerol metabolism in yeasts. Pathways of utilization and production. Eur. J. Biochem. 5:165–172 [DOI] [PubMed] [Google Scholar]
- 25.Hondmann DH, Busink R, Witteveen CF, Visser J. 1991. Glycerol catabolism in Aspergillus nidulans. J. Gen. Microbiol. 137:629–636. 10.1099/00221287-137-3-629 [DOI] [PubMed] [Google Scholar]
- 26.Zwaig N, Kistler WS, Lin EC. 1970. Glycerol kinase, the pacemaker for the dissimilation of glycerol in Escherichia coli. J. Bacteriol. 102:753–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gottschalk G. 1985. Bacterial metabolism, 2nd ed. Springer-Verlag, New York, NY [Google Scholar]
- 28.Jiang Y, Cronan JE. 2005. Expression cloning and demonstration of Enterococcus faecalis lipoamidase (pyruvate dehydrogenase inactivase) as a Ser-Ser-Lys triad amidohydrolase. J. Biol. Chem. 280:2244–2256. 10.1074/jbc.M408612200 [DOI] [PubMed] [Google Scholar]
- 29.Behal RH, Buxton DB, Robertson JG, Olson MS. 1993. Regulation of the pyruvate dehydrogenase multienzyme complex. Annu. Rev. Nutr. 13:497–520. 10.1146/annurev.nu.13.070193.002433 [DOI] [PubMed] [Google Scholar]
- 30.Reed LJ, Leach FR, Koike M. 1958. Studies on a lipoic acid-activating system. J. Biol. Chem. 232:123–142 [PubMed] [Google Scholar]
- 31.Fu D, Libson A, Miercke LJ, Weitzman C, Nollert P, Krucinski J, Stroud RM. 2000. Structure of a glycerol-conducting channel and the basis for its selectivity. Science 290:481–486. 10.1126/science.290.5491.481 [DOI] [PubMed] [Google Scholar]
- 32.Green J, Paget MS. 2004. Bacterial redox sensors. Nat. Rev. Microbiol. 2:954–966. 10.1038/nrmicro1022 [DOI] [PubMed] [Google Scholar]
- 33.Leonardo MR, Dailly Y, Clark DP. 1996. Role of NAD in regulating the adhE gene of Escherichia coli. J. Bacteriol. 178:6013–6018 [DOI] [PMC free article] [PubMed] [Google Scholar]