

Abstract
Genomic information has already been applied to prokaryotic species definition and classification. However, the contribution of the genome sequence to prokaryotic genus delimitation has been less studied. To gain insights into genus definition for the prokaryotes, we attempted to reveal the genus-level genomic differences in the current prokaryotic classification system and to delineate the boundary of a genus on the basis of genomic information. The average nucleotide sequence identity between two genomes can be used for prokaryotic species delineation, but it is not suitable for genus demarcation. We used the percentage of conserved proteins (POCP) between two strains to estimate their evolutionary and phenotypic distance. A comprehensive genomic survey indicated that the POCP can serve as a robust genomic index for establishing the genus boundary for prokaryotic groups. Basically, two species belonging to the same genus would share at least half of their proteins. In a specific lineage, the genus and family/order ranks showed slight or no overlap in terms of POCP values. A prokaryotic genus can be defined as a group of species with all pairwise POCP values higher than 50%. Integration of whole-genome data into the current taxonomy system can provide comprehensive information for prokaryotic genus definition and delimitation.
INTRODUCTION
Since its introduction into the botanical classification by Carl Linnaeus in the 18th century, the binomial nomenclature has generally been accepted for the naming of plants, animals, and prokaryotes (1). The nature of the binomial name dictates that a species must also be assigned to a genus. In prokaryotic taxonomy practice, an unidentified strain can be classified as a member of an earlier named species or as a representative of a new species within an extant genus or can represent a member of a newly described and named genus. A great many studies have been devoted to prokaryotic species classification and definition, and several valuable species concepts have been proposed (2–5). For pragmatic purposes, there are generally accepted operational standards for species delimitation. The level of DNA-DNA hybridization (DDH) between strains being compared is such a standard and has been widely used in prokaryotic species demarcation (6–8). When two strains have 70% or greater hybridization to each other, these two strains can generally be considered to belong to the same species. If an unknown strain shows less than 70% DDH (on the basis of comparison of their 16S rRNA genes) to its most closely related species with standing in the nomenclature, the new strain can be classified as a novel species within an extant genus or can represent the first species of a novel genus to be described. Since 2005, more than 100 new prokaryotic genera have been established every year, and currently, there are approximately 2,100 genera with standing in the prokaryote nomenclature (List of Prokaryotic Names with Standing in Nomenclature [LPSN]; http://www.bacterio.cict.fr/) (9). In the prokaryotic genus classification procedure, 16S rRNA gene sequence analyses play a paramount role. A new proposed genus would have approximately 6% divergence in 16S rRNA gene sequence from its closest genus (10). Strains of the same genus normally form a monophyletic branch from closely related genera in the phylogenetic tree constructed on the basis of molecular markers, usually the16S rRNA gene. Phenotypic characteristics are also considered. Though no characteristics are unique to a genus, a newly defined genus would have unique combinations of phenotypic characteristics different from those of other related genera. Currently, if a newly isolated prokaryotic strain(s) can form a monophyletic group in the phylogenetic analysis, show an approximately 6% 16S rRNA gene sequence difference from the 16S rRNA gene sequence of its most closely related genus, and have unique phenotypic differences, the strain(s) can reasonably be proposed to represent a new genus.
With the rapid development and low cost of prokaryotic genome sequencing, the amount of whole-genome data in databases is increasing quickly (11). Full genomic information can provide extraordinary opportunities for prokaryotic taxonomy (12, 13). Genomic data have already been applied to bacterial species definition and classification on the basis of genomic parameters, such as average amino acid sequence identity (AAI) and average nucleotide sequence identity (ANI) (14, 15). The ANI of common genes between strains being compared is especially closely correlated with the level of DDH, and a 95 to 96% ANI value can serve as a genomic measure for prokaryotic species delineation (16, 17). However, there is currently no genomic standard for prokaryotic genus delineation.
ANI can describe the genetic relationships between strains belonging to the same species. However, the genus-level genomic differences for existing taxonomy systems have been studied less. A previous study revealed that adjacent ranks in the prokaryotic taxonomy showed extensive overlap in terms of AAI values, implying a continuum of genetic diversity in prokaryotic organisms (14). In this case, the classification of genus would be artificial. The study performed by Konstantinidis and Tiedje used a limited number of strains from the Bacteria and Archaea domains, and one genus-level AAI value for one strain was compared to another genus-level AAI for a strain of a genus from a different phylum, but this comparison provided misleading results (14). In this study, with a dramatically increased number of sequenced strains, we systematically investigated genus-level genetic diversity by comparing the genomic contents of strains from the same and different genera of a specific family/order; i.e., strains from different orders were not compared.
Integration of whole-genome data into the current taxonomy system can provide comprehensive information that will lead toward a definition of a prokaryotic genus and the delimitation of prokaryotic genera. Based on the study of genus-level genetic diversity, we attempt to establish a prokaryotic genus boundary based on genomic information. Here we propose a criterion for prokaryotic genus delimitation and contribute to the definition of genus from a genomic point of view.
MATERIALS AND METHODS
Strain selection and filtration.
The sequences of all completely sequenced Archaea and Bacteria available up to December 2013 were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/). Only the strains with validly published genus and species names were selected for analyses. The taxonomic position of a selected strain was originally obtained from an NCBI GenBank file and then checked and verified by analysis of its taxonomic position in LPSN and the Ribosomal Database Project (RDP) (18). Genera with at least 6 different sequenced species (5 species in the genus Bacteroides) were retained for interspecies analyses. The ANI value was used to sieve the species. Two species with ANIs of >94% were considered one species. The family/order to which the retained genus belonged had to have at least 6 different genera with sequenced species. The formation of a genus was validated by 16S rRNA gene phylogenetic analysis. The phylogenetic tree was built using the neighbor-joining method embodied in the MEGA5 program with the Tamura 3-parameter model and 1,000 bootstrap replications. The species from one genus had to form an isolated branch in the phylogenetic tree. Most genera were classified properly. Only the classifications of the genera Bacillus and Lactobacillus were modified in this study. Seventeen genera that could simultaneously satisfy these two criteria were found. These 17 genera were grouped into 12 families/orders. For interspecies analyses, the type strain of a species was preferentially selected if several strains of the same species were sequenced. If the type strain of a species was not sequenced, the strain with the largest genome size was selected. For intergenera analyses, only one representative species of a genus was selected, and the selection scope was extended to the order level to increase the sample number if necessary. The type species of a genus was preferentially selected if several species of the same genus were sequenced. If the type species of a genus was not sequenced, the species with the largest genome size was selected. The strains selected for this study are listed in Table S1 in the supplemental material.
Average nucleotide sequence identity and 16S rRNA gene sequence identity calculation.
The ANI between a pair of genomes was calculated by use of jSpecies software with the BLAST algorithm (17). The 16S rRNA gene sequence(s) was retrieved from the full genome sequence on the basis of gene annotation. The 16S rRNA gene sequence identity between strains was calculated by use of all copies of the 16S rRNA gene of the strains, and the average identity value was used as the 16S rRNA gene sequence identity between two strains. The 16S rRNA gene sequence identity was determined according to the methods described previously (19). Briefly, two 16S rRNA sequences were globally aligned, and identity was calculated without considering gaps in the alignment.
POCP.
The conserved proteins between a pair of genomes were determined by aligning all the protein sequences of one genome (query genome) with all the protein sequences of another genome using the BLASTP program (20). Proteins from the query genome were considered conserved when they had a BLAST match with an E value of less than 1e−5, a sequence identity of more than 40%, and an alignable region of the query protein sequence of more than 50%. For a pair of genomes, each genome was used as the query genome to perform the BLASTP search. The number of conserved proteins in each genome of strains being compared was slightly different because of the existence of duplicate genes (paralogs). The percentage of conserved proteins (POCP) between two genomes was calculated as [(C1 + C2)/(T1 + T2)] · 100%, where C1 and C2 represent the conserved number of proteins in the two genomes being compared, respectively, and T1 and T2 represent the total number of proteins in the two genomes being compared, respectively. In theory, the POCP value can vary from 0% to 100%, depending on the similarity of the protein contents of two genomes.
RESULTS AND DISCUSSION
Strain information.
In order to delimit the prokaryotic genus boundary from a genomic point of view, genomic data for species from the same genus and species from different genera of the same family containing the multispecies genus should be analyzed and compared. For the analyses of the different genera, in many cases, the selected genomes were extended to the order level to increase the sample number. The strain selection and filtration strategies were detailed in Materials and Methods. The classifications of the genera Bacillus, Brucella, and Lactobacillus were modified on the basis of genomic and phylogenetic analyses. The genus Brucella, which belongs to the order Rhizobiales, has 8 different named species. However, the ANI analyses showed that any two species had ANI values greater than 99%. Therefore, the 8 species were considered one species and only the type species was picked out. The genus Bacillus, which belongs to the order Bacillales, has 17 named species for which the complete genomes have been sequenced. However, these 17 species were not monophyletic in the 16S rRNA phylogenetic tree. Only the type species Bacillus subtilis and 4 closely related species were retained to represent the genus Bacillus (see Fig. S1 in the supplemental material). The genus Lactobacillus, which belongs to the order Lactobacillales, has 19 named species for which complete genome sequences are available. These 19 species were also not monophyletic in the 16S rRNA phylogenetic tree. In addition, the pairwise 16S rRNA gene sequence identity of the Lactobacillus species varied widely from 86% to 99%. This shows that the current classification of the genus Lactobacillus needs modification. Therefore, the type species Lactobacillus delbrueckii and 5 closely related species were retained to represent this genus (see Fig. S2 in the supplemental material). After filtration, 235 archaeal and bacterial species from 8 phyla, 12 orders, and 97 genera were analyzed in total (Table 1; see Table S1 in the supplemental material). In this study, the term “interspecies” refers to two species from the same genus and the term “intergenera” refers to two species from different genera of the same family/order. There were 17 genera grouped into 12 families/orders for interspecies analyses. Their taxonomic positions are listed in Table S2 in the supplemental material. Sixteen of the 17 genera were from the Bacteria domain; 1 genus was from the Archaea domain. As anticipated, a large number of the 16 bacterial genera resided in the well-studied phyla Firmicutes (6 genera) and Proteobacteria (5 genera).
TABLE 1.
Taxonomy statistics of strains selected for this study
| Domain | No. of: |
|||
|---|---|---|---|---|
| Phyla | Orders | Genera | Species | |
| Archaea | 1 | 1 | 5 | 10 |
| Bacteria | 7 | 11 | 92 | 225 |
| Total | 8 | 12 | 97 | 235 |
ANI is not suitable for genus delimitation.
The ANI value represents the sequence identities of the conserved regions between two genomes and can be used to measure the genetic distance between strains being compared (15). As ANI is a pragmatic genomic standard for prokaryotic species delimitation and as we intended to base the prokaryotic genus definition on genomic information, we first investigated whether ANI can be used for prokaryotic genus delimitation as well. ANI comparisons were performed to investigate the interspecies and intergenera genetic distances within the 12 families/orders (Fig. 1; see Fig. S3 in the supplemental material). It could be seen that the intergenera ANI values were very close to and partially overlapped the interspecies ANI values. As an example, for the order Alteromonadales, the intergenera ANI values ranged from 63% to 68% and the interspecies ANI values ranged from 68% to 82%. In addition, the majority (72%) of the interspecies ANI values resided in the narrow region of 68 to 72% (see Fig. S3A in the supplemental material). This indicates that the resolution of the ANI is not suitable for genus delimitation. In addition, because the ANI calculation algorithm considers only highly similar sequences, when two strains have a distant genetic relationship (for example, when the strains are from different genera), only a slight proportion (≈10%) of the whole-genome DNA sequence is used for ANI calculation and the large majority of DNA information is abandoned. In summary, ANI is not suitable for genus-level comparisons and cannot be used for genus delimitation on the basis of our current data.
FIG 1.
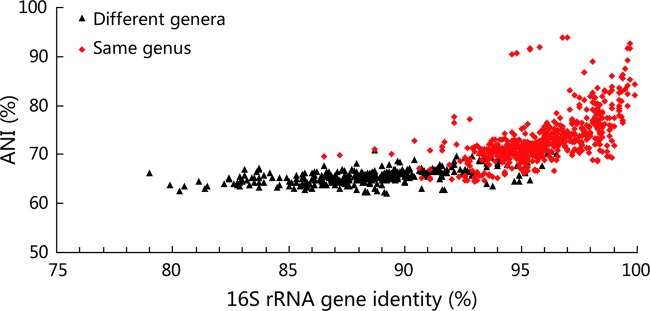
Relationships between ANI and 16S rRNA gene identity for pairs of genomes from different genera and from the same genus. Black triangles, intergenera comparisons in every order; red diamonds, interspecies comparisons.
POCP and genus-level genomic diversity.
We propose that the POCP between genomes being compared can be used to infer the genetic and phenotypic relatedness between a pair of species. The POCP was conceived on the basis of the notion that proteins are responsible for execution of versatile cellular functional processes. All the morphological, physiological, and biochemical traits of a prokaryotic strain have their foundation in proteins. The genomic DNA sequences also accumulate discrepancies, but many nucleotide substitutions usually cause nonsynonymous amino acid substitutions (21–23) which neither alter protein function nor change the strain phenotype. The POCP value is calculated using all the proteins of the genomes being compared. Therefore, the POCP value could be suitable for evaluating the evolutionary distance and phenotypic difference between two strains.
The interspecies and intergenera POCP analyses were performed for 12 families/orders. The interspecies POCP value ranged from 43% to 97%, indicating the high genus-level genomic diversity. The average interspecies and intergenera POCP values were 63% and 36%, respectively (Fig. 2). The difference between these two values was statistically significant (P < 0.01, two-sample t test). Though there were some variations in interspecies POCP values between different genera, in most cases, the interspecies and intergenera POCP values were evenly distributed over a wide region and could be clearly separated from each other (Fig. 3; see Fig. S4 in the supplemental material). The overlap of the ranks genus and family/order was much less in terms of POCP than ANI. Again, considering the order Alteromonadales as an example, the intergenera POCP values ranged from 25% to 55%, while the interspecies POCP values ranged from 56% to 87% (see Fig. S4A in the supplemental material). Thus, the value of 55% can be used to separate the POCP values of the two groups (interspecies and intergenera) and to delimit the genus boundary for the order Alteromonadales. The POCP and 16S rRNA gene identity of the order Alteromonadales showed a linear relationship with a slope of 4.24 (R2 = 0.89), while ANI and 16S rRNA gene identity could fit a linear relationship with a slope of only 0.92 (R2 = 0.83). This indicates that the resolution of POCP is better than that of ANI for genus delimitation on the basis of our data set.
FIG 2.
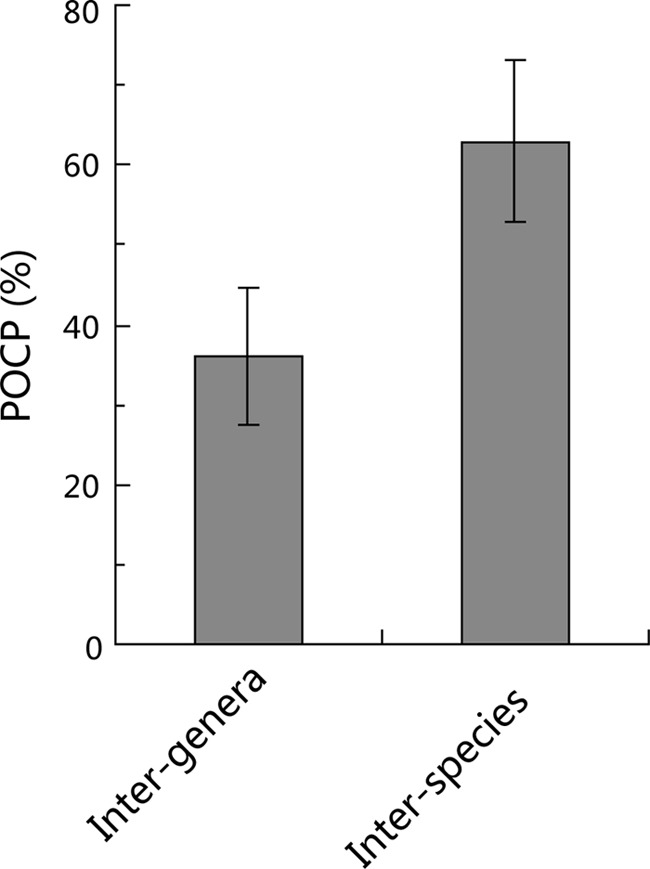
Average interspecies and intergenera POCP values. The bars represents standard deviations.
FIG 3.
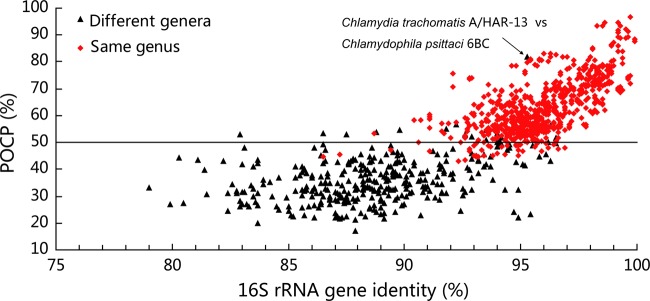
Relationships between POCP and 16S rRNA gene identity for pairs of genomes from different genera and from the same genus. Black triangles, intergenera comparisons in every order; red diamonds, interspecies comparisons.
A POCP standard for a prokaryotic genus boundary.
Generally, a POCP value of 50% can be proposed as a genus boundary for prokaryotic lineages being analyzed (Fig. 3). At first glance, there are some exceptions. An obvious exception was the intergenera comparison of Chlamydia trachomatis A/HAR-13 and Chlamydophila psittaci 6BC from the order Chlamydiales. These two species from different genera had a POCP value of 82% (Fig. 3). According to this high POCP value, these two species should be within the same genus. Coincident with this POCP result, it is proposed that a single genus, Chlamydia, should be used to encompass the species of the genera Chlamydia and Chlamydophila (24; see the notes for the genus Chlamydophila in LPSN). In the order Campylobacterales, the intergenera exception was the pair of strains Sulfurimonas autotrophica DSM 16294 and Sulfuricurvum kujiense DSM 16994, for which the POCP value was 55%. They can be classified into a single genus according to the POCP genus classification standard. Consistent with this conclusion, these two strains have similar genome sizes (2.2 Mb and 2.8 Mb, respectively) (25, 26). Both strains are facultatively anaerobic, can oxidize sulfur, and have the ability to grow autotrophically (27, 28). The exceptions with low POCP values in the genus Helicobacter resulted from the comparison of the strains Helicobacter cinaedi PAGU611, which has the largest genome, with other strains with smaller genomes (see Fig. S4D and Table S1 in the supplemental material). Therefore, genomic information, especially genome size, should be taken into consideration for prokaryotic taxonomy in the future. The POCP concept takes the genome size into consideration, which will decrease its robustness. However, consideration of genome size in taxonomy would correspond to the biological and evolutionary traits of microorganisms. If two strains have a great discrepancy in genome size, they must have taken different evolutionary pathways and face different environmental pressures. They should be regarded as different species or different genera, even though their ANI or AAI values may be high. By removing these exceptions, the POCP of 50% would be a proper genomic parameter for delimiting the prokaryotic genus boundary. This means that if two strains belong to the same genus, they would share at least half of their protein repertoires, showing the high genomic diversity at the prokaryotic genus level.
Application of the POCP standard to the order Clostridiales.
The genus Clostridium belongs to the order Clostridiales and now contains 203 named species and 19 species for which complete genome sequences are available (see Table S3 in the supplemental material). It is reported that there is considerable diversity between species within the genus Clostridium and this genus needs extensive revision (29). With so many sequenced genomes in the order Clostridiales, the POCP concept was applied to the order Clostridiales to evaluate its practicability. All intergenera POCP values were less than 50%, indicating that the genus classification of this order is rational. However, there are serious problems with the classification of the genera Clostridium and Desulfotomaculum (with 6 sequenced species), especially the genus Clostridium (see Fig. S5 in the supplemental material). The interspecies POCP values of the genus Clostridium ranged from 23% to 64%, indicating the huge genus-level diversity in this genus. Consistent with the POCP results, the pairwise 16S rRNA gene identity of Clostridium species varied widely from 82% to 98%. The 16S rRNA gene phylogenetic analysis also showed that the species of Clostridium were not monophyletic in the phylogenetic tree (see Fig. S6 in the supplemental material). All these results indicated that the genus Clostridium should be divided into several genera. Using the 50% POCP standard, each of the pairs of species Clostridium beijerinckii and C. saccharobutylicum, C. clariflavum and C. thermocellum, and C. kluyveri and C. ljungdahlii should be within the same genus. This classification is concordant with the fact that the 16S rRNA gene identities of all three pairs were larger than 93%. The other species should then be classified as different genera. For the genus Desulfotomaculum, the species Desulfotomaculum carboxydivorans, D. reducens, and D. ruminis should be classified as one genus according to their pairwise POCP values (>62%) and 16S rRNA gene identities (>93%). These data show that the POCP concept can be applied to prokaryotic genus classification and the results of POCP analyses are consistent with the results of 16S rRNA gene identity and phylogeny analyses. Of course, this is just the genomic classification of the genera Clostridium and Desulfotomaculum. Formal reclassification of these two genera must also consider other phenotypic and genotypic characteristics. However, we believe that POCP analyses would provide valuable information for future reclassification.
Prokaryotic genus definition from a genomic point of view.
The data presented in this study show that POCP can be used as a genomic index for prokaryotic genus delimitation. If a strain belongs to a specific genus, the POCP between this strain and any other species of the same genus should be higher than the POCP between this strain and any species from a different genus. From a genomic point of view, a prokaryotic genus can be defined as a group of species for which all pairwise POCP values are higher than approximately 50%. With more and more prokaryotic strains being sequenced, we anticipate that the POCP concept can be used for prokaryotic genus classification in the near future.
Conclusion.
Currently, the 16S rRNA gene sequence is the preliminary and crucial reference for the establishment of new prokaryotic genera. POCP performed better than 16S rRNA gene identity for genus classification in some cases. POCP can serve as a genomic standard for genus demarcation. Integration of whole-genome information into the current taxonomy system will contribute substantially to advancing prokaryotic genus delimitation. With the revolution of high-throughput bacterial genome sequencing (30), the era of genomic taxonomy is on the horizon.
Supplementary Material
ACKNOWLEDGMENTS
The work was supported by the National Natural Science Foundation of China (31025001, 31290231, 31370056, 91228210), the Hi-Tech Research and Development Program of China (2012AA092103, 2012AA092105), and the COMRA Program (DY125-15-T-05).
Footnotes
Published ahead of print 4 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01688-14.
REFERENCES
- 1.Sangster NC, Pope SE. 2000. Quid significat nomen? (What's in a name?). Int. J. Parasitol. 30:231–238. 10.1016/S0020-7519(00)00004-7 [DOI] [PubMed] [Google Scholar]
- 2.Rosselló-Mora R, Amann R. 2001. The species concept for prokaryotes. FEMS Microbiol. Rev. 25:39–67. 10.1111/j.1574-6976.2001.tb00571.x [DOI] [PubMed] [Google Scholar]
- 3.Konstantinidis KT, Ramette A, Tiedje JM. 2006. The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361:1929–1940. 10.1098/rstb.2006.1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Staley JT. 2006. The bacterial species dilemma and the genomic-phylogenetic species concept. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361:1899–1909. 10.1098/rstb.2006.1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doolittle WF, Zhaxybayeva O. 2009. On the origin of prokaryotic species. Genome Res. 19:744–756. 10.1101/gr.086645.108 [DOI] [PubMed] [Google Scholar]
- 6.Wayne LG, Brenner DJ, Colwell RR, Grimont PAD, Kandler O, Krichevsky MI, Moore LH, Moore WEC, Murray RGE, Stackebrandt E, Starr MP, Trüper HG. 1987. Report of the ad hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Bacteriol. 37:463–464. 10.1099/00207713-37-4-463 [DOI] [Google Scholar]
- 7.Stackebrandt E, Frederiksen W, Garrity GM, Grimont PA, Kämpfer P, Maiden MC, Nesme X, Rosselló-Móra R, Swings J, Truper HG, Vauterin L, Ward AC, Whitman WB. 2002. Report of the ad hoc Committee for the Re-Evaluation of the Species Definition in Bacteriology. Int. J. Syst. Evol. Microbiol. 52:1043–1047. 10.1099/ijs.0.02360-0 [DOI] [PubMed] [Google Scholar]
- 8.Tindall BJ, Rosselló-Móra R, Busse HJ, Ludwig W, Kämpfer P. 2010. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 60:249–266. 10.1099/ijs.0.016949-0 [DOI] [PubMed] [Google Scholar]
- 9.Parte AC. 2014. LPSN—List of Prokaryotic Names with Standing in Nomenclature. Nucleic Acids Res. 42:D613–D616. 10.1093/nar/gkt1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer KH, Ludwig W, Glockner FO, Rosselló-Móra R. 2008. The All-Species Living Tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst. Appl. Microbiol. 31:241–250. 10.1016/j.syapm.2008.07.001 [DOI] [PubMed] [Google Scholar]
- 11.Shendure J, Ji H. 2008. Next-generation DNA sequencing. Nat. Biotechnol. 26:1135–1145. 10.1038/nbt1486 [DOI] [PubMed] [Google Scholar]
- 12.Zhi XY, Zhao W, Li WJ, Zhao GP. 2012. Prokaryotic systematics in the genomics era. Antonie van Leeuwenhoek 101:21–34. 10.1007/s10482-011-9667-x [DOI] [PubMed] [Google Scholar]
- 13.Thompson CC, Chimetto L, Edwards RA, Swings J, Stackebrandt E, Thompson FL. 2013. Microbial genomic taxonomy. BMC Genomics 14:913. 10.1186/1471-2164-14-913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konstantinidis KT, Tiedje JM. 2005. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187:6258–6264. 10.1128/JB.187.18.6258-6264.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 102:2567–2572. 10.1073/pnas.0409727102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57:81–91. 10.1099/ijs.0.64483-0 [DOI] [PubMed] [Google Scholar]
- 17.Richter M, Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106:19126–19131. 10.1073/pnas.0906412106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145. 10.1093/nar/gkn879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H, Park SC, Jeon YS, Lee JH, Yi H, Won S, Chun J. 2012. Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int. J. Syst. Evol. Microbiol. 62:716–721. 10.1099/ijs.0.038075-0 [DOI] [PubMed] [Google Scholar]
- 20.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konstantinidis KT, Serres MH, Romine MF, Rodrigues JL, Auchtung J, McCue LA, Lipton MS, Obraztsova A, Giometti CS, Nealson KH, Fredrickson JK, Tiedje JM. 2009. Comparative systems biology across an evolutionary gradient within the Shewanella genus. Proc. Natl. Acad. Sci. U. S. A. 106:15909–15914. 10.1073/pnas.0902000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li WH, Wu CI, Luo CC. 1985. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 2:150–174 [DOI] [PubMed] [Google Scholar]
- 23.Muse SV. 1996. Estimating synonymous and nonsynonymous substitution rates. Mol. Biol. Evol. 13:105–114. 10.1093/oxfordjournals.molbev.a025549 [DOI] [PubMed] [Google Scholar]
- 24.Schachter J, Stephens RS, Timms P, Kuo C, Bavoil PM, Birkelund S, Boman J, Caldwell H, Campbell LA, Chernesky M, Christiansen G, Clarke IN, Gaydos C, Grayston JT, Hackstadt T, Hsia R, Kaltenboeck B, Leinonnen M, Ojcius D, McClarty G, Orfila J, Peeling R, Puolakkainen M, Quinn TC, Rank RG, Raulston J, Ridgeway GL, Saikku P, Stamm WE, Taylor-Robinson DT, Wang SP, Wyrick PB. 2001. Radical changes to chlamydial taxonomy are not necessary just yet. Int. J. Syst. Evol. Microbiol. 51:249. 10.1099/00207713-51-1-249 [DOI] [PubMed] [Google Scholar]
- 25.Sikorski J, Munk C, Lapidus A, Ngatchou Djao OD, Lucas S, Glavina Del Rio T, Nolan M, Tice H, Han C, Cheng JF, Tapia R, Goodwin L, Pitluck S, Liolios K, Ivanova N, Mavromatis K, Mikhailova N, Pati A, Sims D, Meincke L, Brettin T, Detter JC, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CD, Rohde M, Lang E, Spring S, Goker M, Woyke T, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk HP. 2010. Complete genome sequence of Sulfurimonas autotrophica type strain (OK10T). Stand. Genomic Sci. 3:194–202. 10.4056/sigs.1173118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han C, Kotsyurbenko O, Chertkov O, Held B, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, Tapia R, Goodwin LA, Pitluck S, Liolios K, Pagani I, Ivanova N, Mavromatis K, Mikhailova N, Pati A, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CD, Brambilla EM, Rohde M, Spring S, Sikorski J, Goker M, Woyke T, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk HP, Detter JC. 2012. Complete genome sequence of the sulfur compounds oxidizing chemolithoautotroph Sulfuricurvum kujiense type strain (YK-1T). Stand. Genomic Sci. 6:94–103. 10.4056/sigs.2456004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inagaki F, Takai K, Kobayashi H, Nealson KH, Horikoshi K. 2003. Sulfurimonas autotrophica gen. nov., sp. nov., a novel sulfur-oxidizing epsilon-proteobacterium isolated from hydrothermal sediments in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 53:1801–1805. 10.1099/ijs.0.02682-0 [DOI] [PubMed] [Google Scholar]
- 28.Kodama Y, Watanabe K. 2004. Sulfuricurvum kujiense gen. nov., sp. nov., a facultatively anaerobic, chemolithoautotrophic, sulfur-oxidizing bacterium isolated from an underground crude-oil storage cavity. Int. J. Syst. Evol. Microbiol. 54:2297–2300. 10.1099/ijs.0.63243-0 [DOI] [PubMed] [Google Scholar]
- 29.Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandez-Garayzabal J, Garcia P, Cai J, Hippe H, Farrow JA. 1994. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int. J. Syst. Bacteriol. 44:812–826. 10.1099/00207713-44-4-812 [DOI] [PubMed] [Google Scholar]
- 30.Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW, Robinson ER, Pallen MJ. 2012. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat. Rev. Microbiol. 10:599–606. 10.1038/nrmicro2850 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.